

Abstract
SVS‐1/SUSD2 is a novel gene, which inhibits growth and reverses tumorigenic phenotypes of cancer cells in vitro. Here we report identification of a mutant of SVS‐1, designated SVS‐1‐vWDm , in which conserved amino acids GLLG at positions 591–594 in von Willebrand factor type D (vWD) domain are replaced by AAAA. As observed by laser confocal microscope, intracellular localization of the mutant protein has changed such that both the N‐terminus and the C‐terminus of SVS‐1‐vWDm were localized in the inner surface of the plasma membrane, whereas the N‐terminus of SVS‐1 was localized in the outer surface of the plasma membrane. Additionally, SVS‐1‐vWDm was processed much less efficiently and in a slightly different manner. In in vitro studies, adenovirus‐mediated transduction of the SVS‐1‐vWDm gene induced growth suppression of HeLa cells in a dose‐dependent manner, as the wild‐type gene and inhibition of anchorage‐independent growth. Of great interest is the finding that the mutant protein, vWDm, but not the wild‐type one induced apoptosis, as observed by nuclear as well as DNA fragmentation. Activation of caspase‐3 and ‐9, but not caspase‐8 or ‐12, was also demonstrated in vWDm‐expressing cells. An inhibition of Akt phosphorylation, a major survival signaling component, also occurred in vWDm‐expressing HeLa cells. Together these data suggest that vWDm induces apoptosis by inactivation of survival signaling component Akt and activation of caspase cascade (mitochondrial pathway) in HeLa cells. We propose SVS‐1‐vWDm as an alternative gene for use in developing new therapeutic strategies for the treatment of cancer. (Cancer Sci 2007; 98: 909–915)
Abbreviations:
- CCP
complement control protein
- DMSO
dimethylsulfoxide
- ER
endoplasmic reticulum
- FITC
fluorescein isothiocyanate
- Luc
luciferase
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- SCR
short consensus repeat
- SDS‐PAGE
sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- SUSD2
sushi domain containing 2
- TRITC
tetramethylrhodamine isothiocyanate
- TSTA
two‐step‐transcription amplification
- vWDm
von Willebrand factor Type D domain mutant.
Knowledge of the molecular mechanism governing malignant transformation brings new opportunities for therapeutic intervention against cancer using novel approaches, one of which is gene therapy. This new discipline is based on the transfer of genetic materials to an organism with the aim of correcting a disease. The genetic alterations that give rise to malignant transformation of cells have been unraveled with increasing detail in the last two decades, and provide multiple candidate targets for gene therapy intervention. However, the genetic and epigenetic alterations that lead to an established tumor are complex and require a few selected genes playing significant roles in the malignant phenotype. Tumor suppressor genes control cell proliferation and apoptosis. It has been demonstrated that restoration of the tumor suppressor genes can revert the malignant phenotype of cells.( 1 , 2 ) The most prominent of the tumor suppressors studied is p53. More than half of human cancers contain mutations in p53,( 3 , 4 ) and more than 80% of which are missense mutations. Among these, most mutations are found in the region of the DNA binding domain, implying the impairment of transcription factor activity.( 3 , 4 , 5 ) Recent studies have demonstrated a wide variety in the transactivation of target genes by mutant p53 proteins. Some demonstrate wild‐type activity, and others show increased activity levels.( 6 , 7 , 8 , 9 ) Overexpression of tumor suppressor genes in cancer cells frequently leads to induction of apoptosis and inhibits tumor growth.( 10 , 11 , 12 , 13 )
In search for new tumor suppressor genes for application to cancer therapy, we found a novel gene, tentatively named SVS‐1/SUSD2, possessing characteristics inherent to tumor suppressor genes, encoding a protein of 820 amino acids, and localizing on the plasma membrane as a type I transmembrane protein.( 14 ) Overexpression of SVS‐1 protein induced rounding and detachment of cells from substratum in both human fibrosarcoma HT1080 cells and in cervical carcinoma HeLa cells. In HeLa cells, overexpression of SVS‐1 protein induced cell aggregation or compaction of rounded‐up cells and inhibited growth. In HT1080 cells overexpression of SVS‐1 protein retarded migration, and invasion of the cells through Matrigel in vitro.
In the present study we constructed a mutant of SVS‐1 by artificially modifying the von Willebrand factor type D (vWD) domain to assess the role of the domain in the biological activity of the protein. The mutant protein exhibited inhibitory activity on the anchorage‐dependent and anchorage‐independent growth. Striking is the finding that, in contrast to the wild‐type SVS‐1, vWDm protein induced apoptosis in HeLa cells, as evidenced by DNA degradation, activation of caspase‐3 and ‐9, and inactivation of Akt, an important member of the survival pathway of cells. These results strongly suggest that the vWDm mutant of SVS‐1 is a potential therapeutic agent for cancer.
Materials and Methods
Most of the materials and methods are the same as those described separately,( 14 ) and therefore principles and modifications are described in this section. Anti‐cleaved‐caspase‐3, anti‐cleaved‐caspase‐8, anti‐cleaved‐caspase‐9, anti‐phospho(Ser‐473)‐Akt and anti‐Akt were purchased from Cell Signaling (Beverly, MA, USA). Anti‐procaspase‐12 was purchased from Pro Sci (Poway, CA, USA), and Lipofectamine 2000 reagent from Life Technologies.
Construction of vectors. Von Willebrand factor type D domain mutant, vWDm, was constructed by substituting the conserved GLLG amino acids at position 591–594 by AAAA in the vWD domain of the SVS‐1 protein. The oligonucleotide fragment containing the mutation was prepared using PCR‐amplification on a mouse clone SUSD2 (Genebank: AK004703) and inserted into the mammalian expression vector pcDNA3‐FSVSV5his, which was constructed with an N‐terminal FLAG tag inserted between the signal peptide and the somatomedin B domain, and the C‐terminal V5 tag (pcDNA3‐vWDm). The vWDm fragment was excised from pcDNA3‐vWDm and inserted into the inducible pG5 vector (pG5‐vWDm).
Immunofluorescence staining. Cells fixed with paraformaldehyde and permeabilized with Triton X‐100 were stained with anti‐FLAG or anti‐V5 antibodies followed by TRITC‐ or FITC‐labeled secondary antibodies as described separately.( 14 )
Apoptosis assay by nuclei fragmentation. HeLa/G5‐Luc, HeLa/G5‐SVS‐1 and HeLa/G5‐vWDm cells were infected with Ad/G4VP2 at 16 moi or mock‐infected. At 24 h after infection, cells were stained with Hoechst33258 for detecting nuclear fragmentation and with anti‐FLAG antibody for immunofluorescence staining for SVS‐1 expression. The number of nuclei with fragmentation per each microscopic field was counted. An average of six fields was calculated.
DNA ladder formation analysis. Two days after infection with Ad/G4VP2 at a moi of 16, 3 × 106 cells were washed once with PBS, and were processed as described previously for DNA laddering assay.( 15 )
Flow cytometric analysis of cell cycle distribution. HeLa/G5‐LUC, HeLa/G5‐SVS‐1 and HeLa/G5‐vWDm cells were seeded in 60‐mm dishes at an initial density of 5 × 105 cells/dish in growth medium. Twelve hours after plating, the cells were infected with Ad/G4VP2 at a moi indicated. Forty‐eight hours after infection, cells were processed as described previously for flow cytometric analysis of DNA content.( 15 )
Western blotting analysis. Western blotting analysis was performed essentially as described previously.( 14 , 15 ) In brief, proteins were separated using SDS‐PAGE and transferred to nitrocellulose membrane followed by probing with antibodies against cleaved caspases and procaspase. Transfection for HA‐Akt into HeLa/G5‐LUC, HeLa/G5‐SVS‐1 and HeLa/G5‐vWDm cells was carried out using Lipofectamine 2000 reagent (Life Technologies). At 24 h after transfection cells were infected with Ad/G4VP2 at a moi of 8. At 48 h after infection cells were processed for western blotting analysis using antiphospho(Ser‐473)‐Akt and anti‐Akt antibodies.
MTT assay. MTT assay was performed as described separately.( 14 )
Anchorage‐independent growth. Anchorage‐independent growth of cells was measured as described separately.( 14 )
Results
In order to examine the roles of the functional domains located on the SVS‐1 protein, we generated an SVS‐1 mutant of von Willebrand factor type D domain, vWDm, by substituting conserved GLLG at amino acid positions 591–594 in the vWD domain of SVS‐1 with AAAA (Fig. 1). We first examined subcellular localization of vWDm in HeLa cells. Cells were transiently transfected with pcDNA3‐FSVS‐1V5his or pcDNA3‐vWDm, stained with anti‐FLAG or anti‐V5 antibodies with or without Triton X‐100 treatment. As shown in Fig. 2 and separately,( 14 ) the outer surface of the plasma membrane of wild‐type SVS‐1‐expressing and Triton X‐100‐untreated cells was stained positive with anti‐FLAG antibody. In contrast, vWDm‐expressing cells were not stained without permeabilization, but stained positive only after permeabilization (Fig. 2), clearly showing that vWDm protein was not exposed to the outer surface but retained in the interior of the cells. Additionally, both SVS‐1‐ and vWDm‐transfected cells were stained positive with V5 antibody only after Triton X‐100 treatment, showing that the C‐terminus of both wild‐type and vWDm is located in the inner surface of the plasma membrane. Observation with the confocal microscope suggested that both the N‐terminus and the C‐terminus of the mutant vWDm is localized in the inner surface of the plasma membrane, while the N ‐terminus of wild‐type SVS‐1 is localized in the outer surface of the plasma membrane (Fig. 2).
Figure 1.

Alignment of amino acid sequences of vWD of SVS‐1 and several vWD‐containing proteins. Amino acid sequences of the predicted vWD of SVS‐1 protein and several vWD‐containing proteins are shown using the NCBI ‘Conserved Domains’ database (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtm). Only the latter half of the domains is aligned. The conserved consensus sequence GXXG motif is underlined and highlighted.
Figure 2.
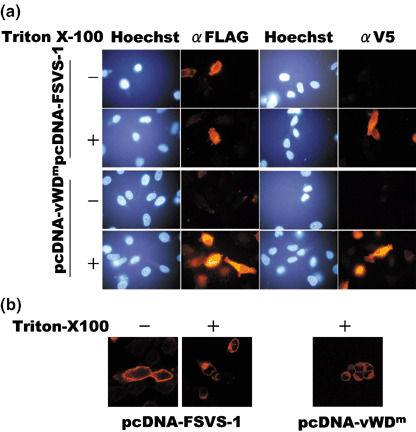
Subcellular localization of wild‐type and von Willebrand factor Type D domain mutant (vWDm) SVS‐1 protein. (a) HeLa cells were transfected with pcDNA3‐FSVS‐1 or pcDNA3‐vWDm. At 48 h after transfection cells were fixed and permeabilized with Triton X‐100 or not and stained with anti‐FLAG or anti‐V5 antibodies staining the N‐terminus and the C‐terminus, respectively, of SVS‐1. (b) Fixed cells were washed with PBS with or without 0.2% Triton X‐100, stained with anti‐FLAG antibody staining the N‐terminus of SVS‐1, and observed using a laser confocal microscope.
Infection of cells with Ad/G4VP2 induces overexpression of vWDm. HeLa/G5‐FSVS‐1, HeLa/G5‐vWDm and HeLa/G5‐Luc cells were infected with Ad/G4VP2, and the expression of FSVS‐1, vWDm and luciferase was detected using immunofluorescence staining and western blot analysis with anti‐FLAG and anti‐Luc antibodies (2, 3 and data not shown). Overexpression of wild‐type FSVS‐1 and vWDm in HeLa cells induced detachment of cells from the substratum and cell aggregation or compaction of rounded‐up cells (Fig. 3). In addition overexpression of vWDm induced nuclear fragmentation (Fig. 4). Western blotting analysis confirmed the expression of the SVS‐1 protein and vWDm at 48 h after infection (Fig. 5). Additionally, of great interest is the finding that intact vWDm protein with 110 kDa was processed much less efficiently and in a slightly different manner, as compared with the wild‐type protein.
Figure 3.
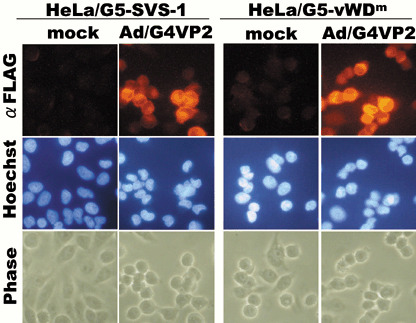
Induced expression of FSVS‐1 and von Willebrand factor Type D domain mutant (vWDm) by Ad/G4VP2 infection, as assayed by immunofluorescence. At 48 h after infection at 4 moi or mock infection, HeLa/G5‐FSVS‐1 and HeLa/G5‐vWDm cells were fixed, permeabilized and stained with anti‐FLAG antibody.
Figure 4.
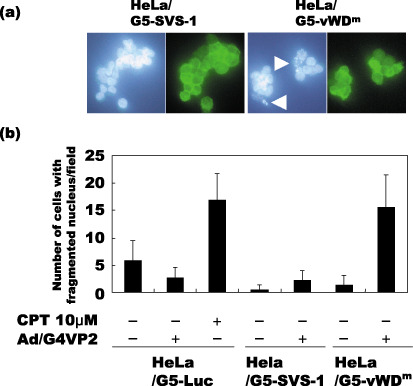
Apoptosis of HeLa/G5‐vWDm cells induced by expression of von Willebrand factor type D domain mutant (vWDm). HeLa/G5‐LUC, HeLa/G5‐SVS‐1 and HeLa/G5‐vWDm cells were infected with Ad/G4VP2 at 16 moi, harvested 1 day after infection, then monitored for nuclei fragmentation. (a) Open arrowheads indicate nuclei fragmentation. (b) The number of cells with fragmented nuclei per each microscopic field was counted.
Figure 5.
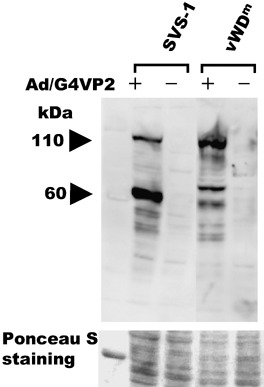
Induced expression of FSVS‐1 and von Willebrand factor Type D domain mutant (vWDm) by Ad/G4VP2 infection, as assayed using western blotting. At 48 h after infection at 2 moi, HeLa/G5‐FSVS‐1 and HeLa/G5‐vWDm cells were harvested, lyzed and analyzed using western blotting with anti‐FLAG antibody. As a loading control the membrane was stained with Ponceau S.
Then we tested using MTT assay whether overexpression of vWDm inhibits cell growth as well as SVS‐1. The growth rate of HeLa/G5‐FSVS‐1 and HeLa/G5‐vWDm cells infected with an indicated moi of Ad/G4VP2 was significantly reduced as compared with that of HeLa/G5‐Luc or mock infected cells (Fig. 6). Thus adenovirus‐mediated vWDm gene expression suppressed growth of HeLa cells as well as or to a greater extent than the wild‐type SVS‐1 in a dose‐dependent manner.
Figure 6.
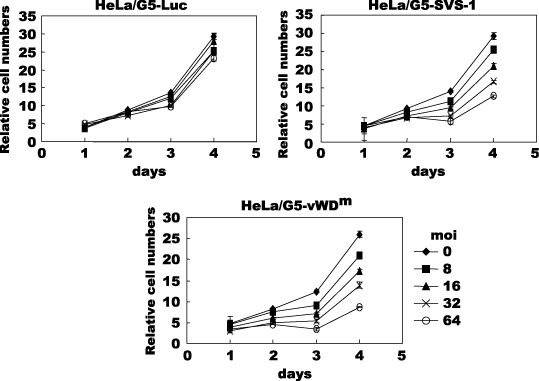
Overexpression of FSVS‐1 and von Willebrand factor Type D domain mutant (vWDm) inhibits cell proliferation. Cells, HeLa/G5‐Luc, HeLa/G5‐FSVS‐1 and HeLa/G5‐vWDm, were infected with various moi of Ad/G4VP2 and the growth of cells was followed by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay at the indicated number of days after infection. Relative cell numbers were expressed relative to the number of cells on day 0.
Suppression of anchorage‐independent growth by vWDm. HeLa/G5‐Luc, HeLa/G5‐FSVS‐1 and HeLa/G5‐vWDm cells were infected with Ad‐G4VP2, and at 48 h post infection the cells were assayed for colony formation in suspension in methylcellulose medium on polyHEMA coated dishes (Fig. 7). The vWDm significantly suppressed anchorage‐independent growth of HeLa cells as well as wild‐type SVS‐1. A reduction of 98% in colony numbers was observed with HeLa/G5‐vWDm compared with mock infection, while an 80% reduction was observed with HeLa/G5‐FSVS‐1 (Fig. 7b). These results clearly show that the vWDm suppresses anchorage‐independent growth, one of the prominent characteristics of tumor cells.
Figure 7.
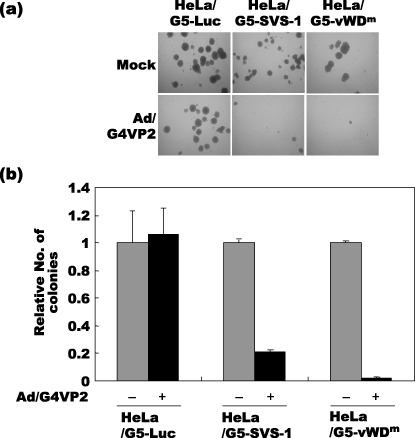
Inhibition of anchorage‐independent growth by SVS‐1 and von Willebrand factor Type D domain mutant (vWDm). (a) At 48 h after Ad/G4VP2 infection of HeLa/G5‐Luc, HeLa/G5‐FSVS‐1 and HeLa/G5‐vWDm cells at 16 moi, 1 × 104 cells were seeded per dish in methylcellulose medium on polyHEMA coated plates. After 12 days of incubation the numbers of colonies were counted. (b) The average number of colonies from three dishes of mock and Ad/G4VP2‐infected cells was calculated.
Induction of apoptosis by vWDm. To test whether expression of the vWDm in tumor cells induces apoptosis, we examined the induction of fragmentation of nuclei and DNA, and the DNA content of the cells using flow cytometry. Two days after infection, HeLa/G5‐vWDm cells appeared to undergo apoptosis by induction of apoptotic bodies (Fig. 4) and nucleosome‐level DNA degradation (Fig. 8), as much as those induced by a chemotherapeutic agent, camptothecin, used as a positive control, while no degradation of nuclei or DNA was observed in HeLa/G5‐Luc and HeLa/G5‐FSVS‐1 cells. Analysis of the cell cycle distribution of Luc‐, SVS‐1‐, and vWDm‐expressing cells using flow cytometry showed that, as compared with Luc and SVS‐1‐induced cells, a significant fraction of HeLa/G5‐vWDm cells appeared to be accumulated in the subG1 phase in a moi‐dependent manner (Fig. 9).
Figure 8.

DNA breakdown in HeLa/G5‐vWDm cells induced by expression of von Willebrand factor type D domain mutant (vWDm). HeLa/G5‐LUC, HeLa/G5‐SVS‐1 and HeLa/G5‐vWDm cells were infected with Ad/G4VP2 at 16 moi, harvested at the indicated number of hours after infection, and DNA was extracted from the cells and electrophoresed on agorose gels for DNA degradation.
Figure 9.
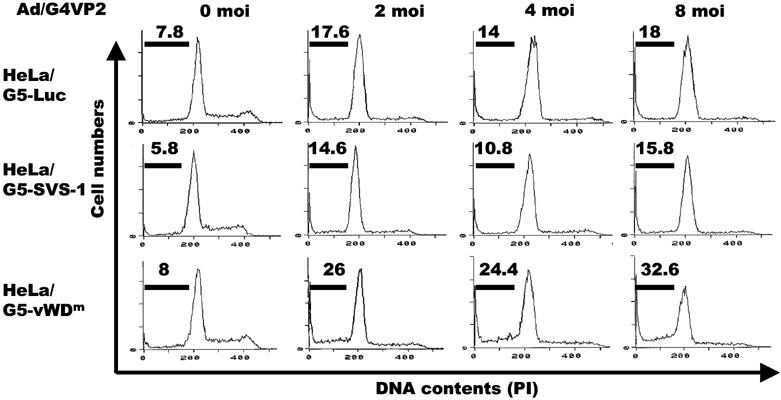
Cell cycle distribution of cells induced by expression of FSVS‐1‐vWDm. HeLa/G5‐LUC, HeLa/G5‐SVS‐1 and HeLa/G5‐vWDm cells were infected with Ad/G4VP2 at indicated moi, harvested 2 days after infection and subjected to flow cytometric analysis.
We measured activation of caspases using anti‐activated‐caspase‐3, ‐8 and ‐9 antibodies and anti‐procaspase 12 antibody. As shown in Fig. 10a, caspase‐3 and ‐9 activity in vWDm‐induced cells increased after 48 h of infection at 8 moi, whereas caspase‐8 and ‐12 activities were not affected, demonstrating that the intrinsic mitochondrial apoptotic pathway is activated by vWDm, whereas apoptotic pathways through death receptors or ER stress are not involved. It is noted that camptothecin, an antitumor agent used as a positive control, induced a dramatic increase in activity of all caspases examined.
Figure 10.
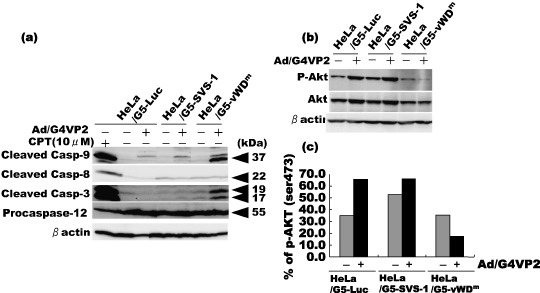
Activation of caspases‐3 and ‐9, and inactivation of Akt in cells induced by expression of FSVS‐1‐vWDm. HeLa/G5‐LUC, HeLa/G5‐SVS‐1 and HeLa/G5‐vWDm cells were infected with Ad/G4VP2 at 8 moi, and harvested 2 days after infection. (a) Western blotting analysis was performed using antibodies for cleaved caspase‐3, ‐8, ‐9 and pro‐caspase‐12. (b) Cells were transfected with pcDNA‐HA‐Akt. At 24 h after transfection, cells were infected with Ad/G4VP2 at a moi of 8. At 48 h after infection, cell lysates were subjected to western blotting analysis with anti‐phospho‐Akt (Ser473) and anti‐Akt antibodies. (c) Band intensities of Akt and phospho‐Akt were quantified by NIH image. The values of phospho‐Akt are indicated relative to those of Akt.
Given that in apoptosis of cells the protein kinase, Akt/PKB, in the survival pathway is often inactivated, we examined the activation (phosphorylation) status of Akt. As the activation of Akt is normally mediated by phosphorylation at Thr308 and Ser473 residues, we examined the amount of phosphorylated Akt at Ser473 after infection with Ad/G4VP2 using an anti‐phospho‐Akt (Ser473) antibody. As shown in Fig. 10b, vWDm expression induced a significant decrease in the amount of phospho‐Akt relative to that in Luc‐ or SVS‐1‐induced cells. Of additional interest is a small but significant increase in the relative amount of phospho‐Akt in Ad/G4VP2‐infected HeLa/G5‐Luc or HeLa/G5‐SVS‐1 cells (Fig. 10b), a result that is reminiscent of the finding that increased migratory and invasive activity in Ad/G4VP2‐infected HeLa/G5‐Luc and HeLa/G5‐SVS‐1 cells at low moi.( 14 ) These results indicate that overexpression of vWDm suppressed Akt kinase activity and induced apoptosis.
Discussion
The von Willebrand factor (vWF) domain is found in various plasma proteins such as complement factors B, C2, CR3, CR4, integrins, collagen types VI, VII, XII and XIV and other extracellular proteins. Proteins that incorporate vWF domains participate in numerous biologic events, for example, cell adhesion, migration, homing, pattern formation, and signal transduction, involving interaction with a large array of ligands.( 16 , 17 ) In some types of von Willebrand factor disease, missense mutations have been found in A or D domains, which affect binding capacity to factor VIII, multimerization of vWF protein or proteolysis.( 18 )
In the present report we showed that, in sharp contrast with the wild‐type SVS‐1 protein, the artificially constructed mutant of SVS‐1 in von Willebrand factor type D domain which had the conserved sequence GXXG replaced by AAAA led tumor cells to apoptosis, thus the mutation being a ‘gain‐of‐function’ mutation. Laser confocal microscopic observation clearly showed that the mutant protein is not exposed to the outer surface of the plasma membrane inaccessible by antibodies, but appears to be localized on the inner surface of the plasma membrane (Fig. 2). Therefore, the mutant protein is assumed to undergo conformational change distinct from that of the wild‐type protein and is unable to pass through the plasma membrane. The protein may interact with some other proteins there and in as yet unknown fashion to transmit signal to activate proapoptotic machinery, leading to activation of the mitochondrial apoptotic pathway including activation of caspase‐9 and ‐3 and finally to degradation of DNA.
Gain‐of‐function mutations are often observed in a variety of systems, including tumor suppressor protein p53.( 19 , 20 ) Of relevance is to refer to a p53 mutant, p53‐46F, in which Ser‐46 is replaced by phenylalanine, which induces apoptosis more efficiently than wild‐type p53 in a number of tumor cells.( 9 ) An increased level of p53 Ser‐15 phosphorylation was observed in all cell lines infected with Ad‐p53‐46F. This has previously been suggested to induce stabilization of the p53 protein.( 21 ) Several other mutant forms of p53 have been shown to possess a more potent ability than wild‐type protein in inducing apoptosis in vitro, including S121F, S121C, S121A, A121Y, R290G, K291E, K291Q, K291T, K292T and K292I.( 20 , 22 )
Using western blotting analysis, we showed that the vWDm protein appeared to be less efficiently processed than the wild‐type protein (Fig. 5). In concert with the localization of the mutant protein in the inner surface of the plasma membrane, it was suggested that the mutant vWDm protein undergoes conformational change such that it is not transported to the outer surface of the cell (Fig. 2) and is less efficient in proteolytic processing than the wild‐type protein (Fig. 5).
Apoptosis, a form of programmed cell death, plays a crucial role in the regulation of the development and maintenance of tissues in metazoans.( 23 , 24 ) In apoptosis, a genetic program is activated in which the balance is shifted such that proapoptotic gene functions dominate those of antiapoptotic genes, resulting in cell death.( 24 ) Deregulation of apoptosis has been implicated in the pathogenesis of many diseases, including cancer.( 25 , 26 )
Akt/PKB kinase is a well‐characterized effector of phosphatidylinositol 3‐kinase (PI3K), and its deregulation plays important roles in the pathogenesis of human cancer. Akt/PKB activation requires phosphorylation of Thr‐308 in the activation loop by the phosphatidylinositol‐dependent kinase 1 (PDK1) and Ser473 within the carboxyl‐terminal hydrophobic motif by an unknown kinase.( 27 , 28 , 29 ) At least 10 kinases have been proposed to function as the hydrophobic motif domain protein kinase (HMD‐PK) that phosphorylates Akt on Ser‐473, including integrin‐linked kinase (ILK), protein kinase C (PKC), DNA‐dependent protein kinase (DNA‐PK), ataxia telangiectasia mutated (ATM) gene product, and the mammalian target of rapamycin (mTOR).( 30 ) Recently, in Drosophila and human cells, the mTOR kinase and its associated protein, rictor, have been shown to be necessary for Ser473 phosphorylation. A reduction in rictor or mTOR expression inhibited the Akt/PKB phosphorylation.( 31 , 32 ) Phosphorylation of Ser‐473 results in a ‘disorder to order’ transition, allowing interaction between the hydrophobic motif domain and the N‐terminal lobe, leading to the activation of the kinase.( 33 ) The rictor–mTOR complex directly phosphorylated Akt/PKB on Ser‐473 in vitro and facilitated Thr‐308 phosphorylation by PDK1.( 31 , 32 )
We showed that overexpression of vWDm induced apoptosis and dephosphorylation of Akt at Ser‐473, and in turn the activation of caspases‐3 and ‐9, suggesting apoptosis involving the intrinsic mitochondrial pathway.
The available data suggest that, on encountering ER stress, proapoptotic Bcl2‐related proteins, Bak and Bax, undergo conformational alteration in the ER membrane to permit Ca2+ efflux into the cytoplasm.( 34 , 35 ) In vitro experiments support the idea that the increase in Ca2+ concentration (from micromolar to millimolar) in the cytoplasm activates the calcium‐dependent protease, m‐calpain, which subsequently cleaves and activates the ER‐resident procaspase‐12.( 36 ) Activated caspase‐12 cleaves and activates procaspase‐9 and consequently leads to activation of the caspase cascade.( 37 ) Overexpression of vWDm did not cause an increase in caspase‐8 and a decrease in procaspase‐12, demonstrating that the vWDm protein does not have an impact on ER stress‐induced apoptosis or the death receptor pathway.
Evidence has been accumulated showing that various cellular damaging stimuli which lead to apoptosis, such as radiation and chemical agents, inactivate the survival pathway, PI‐3K/Akt, by dephosphorylaton of Akt.( 38 , 39 ) Thus Akt plays an important role in tumorigenesis. Downregulation of Akt activity, for example by PTEN phosphatase, suppresses tumorigenesis. Overexpression of the vWDm gene using adenovirus vector caused dephosphorylation of Akt that may play a major role in apoptosis induction by vWDm.
In brief, we have shown that the vWD domain mutant, vWDm, unlike the wild‐type counterpart expressing on the outer surface of the plasma membrane, is expressed underneath the plasma membrane and possesses apoptosis‐inducing activity, as shown by inhibition of growth in both liquid and semisolid medium, induction of nuclei‐ and DNA‐fragmentation and activation of caspases‐3 and ‐9. The vWDm mutant protein and/or its cDNA may serve as a novel and promising candidate antitumor agent.
References
- 1. Fang B, Roth JA. Tumor‐suppressing gene therapy. Cancer Biol Ther 2003; 2 (4 Suppl. 1): S115–21. [PubMed] [Google Scholar]
- 2. Hernandez‐Alcoceba R, Sngro B, Prieto J. Gene therapy of liver cancer. World Gastroenterol 2006; 14: 6085–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hollstein M, Sidransky D, Vogelstein B et al. p53 mutations in human cancers. Science 1991; 253: 49–53. [DOI] [PubMed] [Google Scholar]
- 4. Greenblatt MS, Bennett WP, Hollstein M et al. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54: 4855–78. [PubMed] [Google Scholar]
- 5. Soussi T, Beroud C. Assessing TP53 status in human tumors to evaluate clinical outcome. Nat Rev Cancer 2001; 1: 233–40. [DOI] [PubMed] [Google Scholar]
- 6. Kato S, Han SY, Liu W et al. Understanding the function‐structure and function‐mutation relationship of p53 tumor suppressor protein by high‐resolution missense mutations analysis. Proc Natl Acad Sci USA 2003; 100: 8424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Resnick MA, Inga A. Functional mutants of the sequence‐specific transcription factor p53 and implications for master genes of diversity. Proc Natl Acad Sci USA 2003; 100: 9934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soussi T, Kato S, Levy PP et al. Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations. Hum Mutat 2005; 25: 6–17. [DOI] [PubMed] [Google Scholar]
- 9. Nakamura Y, Futamura M, Kamino H, Yoshida K, Nakamura Y, Arakawa H. Identification of p53‐46F as a super p53 with an enhanced ability to induce p53‐dependent apoptosis. Cancer Sci 2006; 97: 633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ji L, Fang B, Yen N et al. Induction of apoptosis and inhibition of tumorigenicity and tumor growth by adenovirus vector‐mediated fragile histidine triad (FHIT) gene overexpression. Cancer Res 1999; 59: 3333–9. [PubMed] [Google Scholar]
- 11. Saeki T, Mhashilkar A, Swanson X et al. Inhibition of human lung cancer growth following adenovirus‐mediated mda‐7 gene expression in vivo . Oncogene 2002; 21: 4558–66. [DOI] [PubMed] [Google Scholar]
- 12. Roz L, Gramegna M, Ishii H et al. Restoration of fragile histidine triad (FHIT) expression induces apoptosis and suppresses tumorigenicity in lung and cervical cancer cell lines. Proc Natl Acad Sci USA 2002; 99: 3615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tambe Y, Isono T, Haraguchi S et al. A novel apoptotic pathway induced by the drs tumor suppressor gene. Oncogene 2004; 23: 2977–87. [DOI] [PubMed] [Google Scholar]
- 14. Sugahara T, Yamashita Y, Shinomi M et al. Isolation of a novel mouse gene, mSVS‐1/SUSD2, reversing tumorigenic phenotypes of cancer cells in vitro . Cancer Sci 2007; doi: 10.1111/j.1349‐7006.2007.00466.x. [DOI] [PMC free article] [PubMed]
- 15. Yanagihara M, Sasaki‐Takahashi N, Sugahara T et al. Leptosins isolated from marine fungus Leptoshaeria species inhibit DNA topoisomerases I and/or II and induce apoptosis by inactivation of Akt/protein kinase B. Cancer Sci 2005; 96: 816–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colombatti A, Bonaldo P, Doliana R. Type A modules: interacting domains found in several non‐fibrillar collagens and in other extracellular matrix proteins. Matrix 1993; 13: 297–306. [DOI] [PubMed] [Google Scholar]
- 17. Bork P. Shuffled domains in extracellular proteins. FEBS Lett 1991; 286: 47–54. [DOI] [PubMed] [Google Scholar]
- 18. Hilbert L, Jorieux S, Proulle V et al. Two novel mutations, Q1053H and C1060R, located in the D3 domain of von Willebrand factor, are responsible for decreased FVIII‐binding capacity. Br J Haematol 2003; 120: 627–32. [DOI] [PubMed] [Google Scholar]
- 19. Payne SR, Kemp CJ. Tumor suppressor genetics. Carcinogenesis 2005; 26: 2031–45. [DOI] [PubMed] [Google Scholar]
- 20. Van Oijen MGCT, Slootweg PJ. Gain‐of‐function mutations in the tumor suppressor gene p53 . Clin Cancer Res 2000; 6: 2138–45. [PubMed] [Google Scholar]
- 21. Shieh SY, Ikeda M, Taya Y et al. DNA damage‐induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91: 325–34. [DOI] [PubMed] [Google Scholar]
- 22. Kakudo Y, Shibata H, Otsuka K et al. Lack of correlation between p53‐dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s. Cancer Res 2005; 65: 2108–14. [DOI] [PubMed] [Google Scholar]
- 23. Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 2004; 29: 233–42. [DOI] [PubMed] [Google Scholar]
- 24. Steller H. Mechanisms and genes of cellular suicide. Science 1995; 267: 1445–9. [DOI] [PubMed] [Google Scholar]
- 25. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A 2001; 98: 10 983–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vivanco I, Sawyers CL. The phosphatidylinositol 3‐Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2: 489–501. [DOI] [PubMed] [Google Scholar]
- 27. Alessi DR, James SR, Downes CP et al. Characterization of a 3‐phosphoinositide‐dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997; 7: 261–9. [DOI] [PubMed] [Google Scholar]
- 28. Stephens L, Anderson K, Stokoe D et al. Protein kinase B kinases that mediate phosphatidylinositol 3–5‐trisphosphate‐dependent activation of protein kinase B. Science 1998; 279: 710–14. [DOI] [PubMed] [Google Scholar]
- 29. Alessi DR, Deak M, Casamayor A et al. 3‐Phosphoinositide‐dependent protein kinase‐1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 1997; 7: 776–89. [DOI] [PubMed] [Google Scholar]
- 30. Dong LQ, Liu F. PDK2: the missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am J Physiol Endocrinol Metab 2005; 289: E187–96. [DOI] [PubMed] [Google Scholar]
- 31. Sarbassov DD, Guertin DA, Ali SM et al. Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 2005; 307: 1098–101. [DOI] [PubMed] [Google Scholar]
- 32. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3‐L1 adipocytes. J Biol Chem 2005; 280: 40406–16. [DOI] [PubMed] [Google Scholar]
- 33. Yang J, Cron P, Good VM et al. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3‐peptide and AMP‐PNP. Nat Struct Biol 2002; 9: 940–4. [DOI] [PubMed] [Google Scholar]
- 34. Zong WX, Li C, Hatzivassiliou G et al. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 2003; 162: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scorrano L, Oakes SA, Opferman JT et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003; 300: 135–9. [DOI] [PubMed] [Google Scholar]
- 36. Nakagawa T, Yuan J. Cross‐talk between two cysteine protease families. Activation of caspase‐12 by calpain in apoptosis. J Cell Biol 2000; 150: 887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rao RV, Castro‐Obregon S, Frankowski H et al. Coupling endoplasmic reticulum stress to the cell death program. An Apaf‐1‐independent intrinsic pathway. J Biol Chem 2002; 277: 21 836–42. [DOI] [PubMed] [Google Scholar]
- 38. Fujiwara H, Yamakuni T, Ueno M et al. IC101 induces apoptosis by Akt dephosphorylation via an inhibition of heat shock protein 90‐ATP binding activity accompanied by preventing the interaction with Akt in L1210 cells. J Pharmacol Exp Ther 2004; 310: 1288–95. [DOI] [PubMed] [Google Scholar]
- 39. Kulp SK, Yang YT, Huang CC et al. 3‐Phosphoinositide‐dependent protein kinase‐1/Akt signaling represents a a major cyclooxygenase‐2‐independent target for celecoxib in prostate cancer cells. Cancer Res 2004; 64: 1444–51. [DOI] [PubMed] [Google Scholar]