

Abstract
Interleukin‐17 (IL‐17), a potent pro‐inflammatory cytokine, plays an active role in inflammation and cancer. Recently, we found that increased IL‐17‐producing cells correlate with poor survival and increased lymphangiogenesis in non‐small‐cell lung cancer (NSCLC), but the mechanism is unknown. Here, we show that IL‐17 promotes lymphangiogenesis via inducing vascular endothelial growth factor‐C (VEGF‐C) production by lung cancer cells. We found that IL‐17 receptor (IL‐17R) is expressed on the surface of Lewis lung carcinoma (LLC) cells but not on lymphatic endothelial cells (LEC). Moreover, LEC chemotaxis and tube formation (measures of net lymphangiogenic potential) were increased by conditioned medium from recombinant mouse IL‐17 (rmIL‐17)‐stimulated LLC but not by rmIL‐17. Interleukin‐17 increased production of VEGF‐C in lung cancer cell lines. The enhanced chemotaxis and endothelial cord formation in the presence of LLC/rmIL‐17 was inhibited by addition of recombinant mouse VEGF R3/Fc chimera. Treatment of the A549 cells with rIL‐17 significantly increased VEGF‐C expression, which was extracellular signal‐regulated protein kinase 1/2 (ERK 1/2) dependent. Importantly, we found significant correlations between IL‐17 expression, VEGF‐C expression and lymphatic vascular density (LVD) in NSCLC. We conclude that IL‐17 is involved in lymphangiogenesis in NSCLC by enhancing production of VEGF‐C, and IL‐17 may be an important target for the treatment of NSCLC. (Cancer Sci 2010; 101: 2384–2390)
Non‐small‐cell lung cancer (NSCLC) is one of the most common malignancies worldwide.( 1 ) The outcome of patients with NSCLC remains poor and mediastinal lymph node metastasis is one of the most important prognostic variables.( 2 ) Lymphangiogenesis, which is thought to play an important role in cancer lymphatic spread to the regional lymph nodes, is dependent on lymphangiogenic factors. Among these factors, vascular endothelial growth factor‐C (VEGF‐C), which is a ligand for VEGF receptor 3 (VEGF R3), has been reported to act as an important lymphatic‐specific growth factor.( 3 ) The VEGF R3 is a tyrosine kinase receptor that is expressed predominantly in lymphatic endothelial cells (LEC).( 4 )
Interleukin‐17 (IL‐17) is a pro‐inflammatory cytokine mainly produced by activated T cells.( 5 ) Accumulating evidence has shown that IL‐17‐positive cells are frequently present in multiple inflammation‐associated cancers and that IL‐17 promotes angiogenesis in tumor models.( 6 ) Expression of IL‐17 correlates well with the numbers of blood vessels in human ovarian cancer,( 7 ) hepatocellular carcinoma( 8 ) and NSCLC,( 9 ) and correlates with poor survival in hepatocellular carcinoma( 8 ) Recently, we found that increased IL‐17‐producing cells correlate with poor survival and increased lymphangiogenesis in NSCLC patients.( 10 ) However, the mechanism underlying the role of IL‐17 in lymphangiogenesis still remains to be explored.
The IL‐17 receptor (IL‐17R) is a type I transmembrane protein with no homology to other known cytokine receptors. Interleukin‐17 has several biological activities including induction of IL‐6, IL‐8, tumor necrosis factor (TNF)‐α, IL‐1β and VEGF.( 11 , 12 ) The proinflammatory function and intracellular signaling pathway of IL‐17 is similar to those of IL‐1 and Toll‐like receptors (TLR). Interleukin‐1 is known to promote tumor‐associated lymphatic vessel growth through enhancing the expression of VEGF‐C.( 13 ) Therefore, we hypothesized that, like IL‐1, IL‐17 might play a deleterious role in patients with NSCLC by promoting lymphangiogenesis via enhancing the expression of VEGF‐C.
In the present study, we sought to elucidate the mechanisms by which IL‐17 enhances lymphangiogenesis in NSCLC. Our results show that IL‐17 is involved in lymphangiogenesis in NSCLC by enhancing production of VEGF‐C.
Materials and Methods
Reagents and antibodies. Recombinant human IL‐17 (rhIL‐17) protein, recombinant mouse IL‐17 (rmIL‐17), goat anti‐human IL‐17 mAb and recombinant mouse VEGF R3/Fc (rVEGF R3/Fc) chimera were from R&D Systems (Minneapolis, MN, USA). Rabbit anti‐human VEGF‐C polyclonal Ab and mouse anti‐human D2‐40 mAb were from Zhongshan Company (Beijing, China). Extracellular signal‐regulated protein kinase 1/2 (ERK1/2) antibody, p‐ERK1/2 antibody and ERK1/2 inhibitor PD98059 were purchased from Biyuntian Biotech (Jiangsu, China).
Tissue samples. Formalin‐fixed, paraffin‐embedded tissue samples from 36 primary human NSCLC were randomly obtained at surgery from the department of Pathology, Xinqiao Hospital. None of the patients received any anticancer therapy prior to sample collection. This study was approved by the local ethics committee and written informed consent was obtained from each patient.
Cell culture. LLC, A549 and SPC‐A‐1 cell lines were obtained from American Type culture Collection (ATCC) (Manassas, VA, USA). Cells were maintained in RPMI 1640 with 10% FCS (Invitrogen, Shanghai, China). Isolation and culture of LEC were performed as described previously.( 14 ) The LEC were maintained in endothelial cell basal medium‐2 (EBM‐2; Cambrex BioScience, Wokingham, UK) supplemented with 20% FBS and 50 ng/mL endothelial cell growth supplement (Cambrex BioScience) at 37°C in a humidified atmosphere of 5% CO2.
Flow cytometry. Cells (1 × 106) were washed twice with PBS containing 2% FCS and 0.1% NaN3 and then stained as follows. The LLC cells and LEC were incubated with phycoerythrin (PE)‐conjugated rabbit anti‐mouse IL‐17R mAb or with PE‐labeled rabbit IgG2a isotype (eBioscience, San Diego, CA, USA) at 4°C for 30 min. A549 cells were incubated with PE‐labeled mouse anti‐human IL‐17R mAb or PE‐conjugated mouse IgG1 isotype control (eBioscience) at 4°C for 30 min. The cells were analyzed with FACScan flow cytometer and CellQuest software (Coulter, Fullerton, CA, USA).
Real‐time RT‐PCR. The LLC, SPC‐A‐1 and A549 cells (1 × 105/mL) were stimulated with 0–500 ng/mL rmIL‐17 for 6 h, then the total RNA was isolated and subjected to quantitative RT‐PCR analysis in an ABI 7300 Prism Sequence Detection System (Applied Biosystems, Foster City, CA, USA) as described previously.( 14 ) Primers (Invitrogen) used for RT‐PCR were as following: mouse VEGF‐C, 5′‐GAGGAGCAGTT‐GCGGTCTG‐3′ (sense) and 5′‐CCTGGTATTGAGGGTGGG‐3′ (antisense); mouse β‐actin, 5′‐CCCTGTATGCCTCTGGTC‐3′ (sense) and 5′‐GTCTTTACGGATGTCAACG‐3′ (antisense); human VEGF‐C, 5′‐GAGGAGCAGTTGCGGTCTG‐3′ (sense) and 5′‐CCTGGTATTGAGGGTGGG‐3′ (antisense); human β‐actin, 5′‐CCCTGTATGCCTCTGGTC‐3′ (sense) and 5′‐GTCT‐TTACGGATGTCAACG‐3′ (antisense). The relative gene expression levels were calculated using the comparative Ct (△△Ct) method (according to Applied Biosystems), where the relative expression is calculated as 2−ΔΔCt, and Ct represents the threshold cycle.
Cytokine ELISA. The LLC cells (1 × 105/mL) were cultured with or without 10 ng/mL rmIL‐17 for 48 h. Cell‐free supernatants were collected and stored at −70°C. Concentrations of VEGF‐C were measured using commercially available ELISA kits (R&D Systems). Absorbance at 450 nm was determined using a microplate reader (Bio‐Rad, Hercules, CA, USA).
Western blotting. Protein extraction and immunoblotting have been described previously.( 15 ) ERK 1/2 expression was analyzed as an internal control. All experiments were repeated twice and blots were analyzed using quantity one 1‐D analysis software (Bio‐Rad).
MTT assay. Cells were seeded into 96‐well flat‐bottomed plates and cultured in RPMI 1640 containing 3% FCS with 0–500 ng/mL rmIL‐17. On day 5, cells were washed twice with medium and 100 μL MTT (Sigma, St Louis, MO, USA) solution (2.5 mg/mL in RPMI 1640 with 10% FCS) was added to each well. Plates were incubated for 90 min. Next, the MTT solution was removed and 50 μL dimethylsulfoxide (DMSO) (Sigma) was added to each well to solubilize the formazan crystals formed in the viable cells. The absorbance was read at a wavelength of 590 nm on an enzyme‐linked immunosorbent assay (ELISA) plate reader.
Migration assays. For migration assays, 24‐well culture inserts with a porous polycarbonate membrane (8.0 μm pore size, Millipore, Billerica, MA, USA) were coated with fibronectin (BD Biosciences Discovery Labware, Bedford, MA, USA). The LLC cells were incubated in serum‐free DMEM for 24 h with or without rmIL‐17 (10 ng/mL), after which culture supernatants were collected and termed conditioned medium (CM). The LEC (5 × 104cells/insert) were placed in the upper chamber then cultured with rmIL‐17 (10 ng/mL) in DMEM and EBM‐2 (1:1) or CM (LLC media and EBM‐2 [1:1], or LLC/rmIL‐17 medium and EBM‐2 [1:1], or LLC/rmIL‐17 medium and EBM‐2 [1:1] and rVEGF R3/Fc chimera [1 μg/mL]). The control medium was DMEM and EBM‐2 (1:1). After 48 h of incubation, non‐migratory cells in the upper chamber were scraped off and the migratory cells on the bottom part of the filter were identified by fixing in 70% methanol for 10 min and staining with crystal violet. In each individual experiment, cells that migrated through the filter were counted under the microscope at ×200 magnification in 12 randomly selected fields. The data were expressed as a percentage of the control. Each experiment was performed in triplicate.
Tube‐like structure formation assay. The LEC (6 × 104 cells/well) were seeded into a 24‐well plate pre‐coated with 100 μL Matrigel (10 mg/mL; Clontech, Palo Alto, CA, USA) and cultured with rmIL‐17 (10 ng/mL) in DMEM and EBM‐2 (1:1) or CM (LLC media and EBM‐2 [1:1], LLC/rmIL‐17 medium and EBM‐2 [1:1], or LLC/rmIL‐17 medium and EBM‐2 [1:1] and rVEGF R3/Fc chimera [1 μg/mL]). The control medium was DMEM and EBM‐2 (1:1). After 3 days of incubation, formation of tube‐like structures was monitored by microscopic observation at ×100 magnification and over 12 different fields of each well were photographed to measure the length of the tube‐like structures, as described previously.( 16 )
Assessment of tissue staining. All of the hematoxylin‐and‐eosin‐stained and the immunohistochemical sections were independently and blindly reviewed by two pathologists and the kappa values were tested. The expression of IL‐17 was assessed as previously described.( 10 ) The labeling intensity of VEGF‐C was assessed according to a published paper.( 17 ) Lymphatics were highlighted by D2‐40 immunostaining. Lymphatic vessel density was analyzed using a method previously described by Weidner( 18 ) for assessing microvascular density.
Statistical analysis. Statistical analyses were performed using the SPSS‐PC package (version 12.0; SPSS, Chicago, IL, USA). The correlation between IL‐17, VEGF‐C and lymphatic vascular density (LVD) was assessed by the chi square test and by the Student’s t test. All data from quantitative assays were expressed as the mean ± standard deviation and statistically analyzed using one‐way anova analysis and independent‐samples t‐test. P < 0.05 was considered to be statistically significant.
Results
Effects of rmIL‐17 and CM from LLC/rmIL‐17 on lymphatic vascular endothelial cells. Previously, we found that increased IL‐17‐producing cells correlate with increased lymphangiogenesis in NSCLC patients. There are two possibilities regarding the underlying mechanism of IL‐17 in promotion of lymphangiogenesis. One is that IL‐17 promotes lymphangiogenesis via a direct effect on LEC; inducing proliferation and migration of, and tube formation by, LEC. Another possibility is that IL‐17 induces the expression of lymphangiogenic factor(s) by cancer cells that then act on the LEC. The IL‐17 activity, either on cancer cells or LEC, depends on binding to IL‐17R. Therefore, we examined IL‐17R expression on lung cancer cell lines and LEC using FACS. Interestingly, we found that IL‐17R is expressed on the surface of LLC and A549 cells but not on LEC (Fig. 1). Next, we examined the effects of IL‐17 on in vitro lymphangiogenesis‐related functions of lymphatic vascular endothelial cells. The rmIL‐17 had no direct effect on the proliferation or chemotactic responses of, or endothelial cord formation by, LEC (Fig. 2). The LLC‐derived supernatant caused an increase in chemotaxis and endothelial cord formation by LEC compared with the rmIL‐17‐treated group (P < 0.05, Fig. 2). Moreover, significantly increased chemotaxis and endothelial cord formation by LEC was observed in response to CM from LLC/rmIL‐17 (P < 0.05, Fig. 2).
Figure 1.
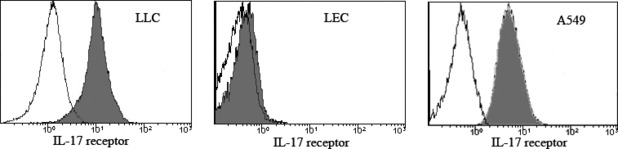
Interleukin‐17R receptor (IL‐17R) expression on LLC cells, lymphatic endothelial cells (LEC) and A549 cells. The LLC cells or LEC were incubated with phycoerythrin (PE)‐labeled rabbit anti‐mouse IL‐17R mAb or with PE conjugated rabbit IgG2a isotype control. The A549 cells were incubated with PE conjugated mouse anti‐human IL‐17R mAb or with PE‐labeled mouse IgG1 isotype control. The expression of IL‐17R on the surface of the LLC cells, LEC and A549 cells was analyzed by flow cytometry (control, unfilled; IL‐17R, filled histogram).
Figure 2.
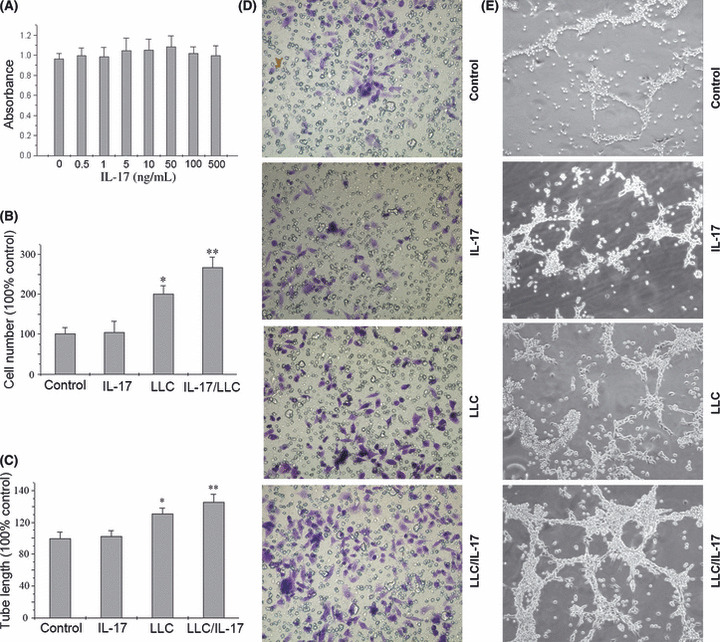
Effect of recombinant mouse interleukin‐17 (rmIL‐17) and conditioned medium on the proliferation and migration of, and tube‐like structure formation by, lymphatic endothelial cells (LEC). (A) After 48 h of culture of LEC with rmIL‐17 (0–500 ng/mL), the number of viable LEC was determined by MTT assay. (B,D) The LEC were seeded in the upper well of a 24‐well trans‐well chamber and the rmIL‐17, LLC medium and LLC/rmIL‐17‐conditioned medium was placed in the lower chamber. After 48 h of incubation the invasive LEC on the outside surface of the upper chambers were visualized by a crystal violet stain (magnification, ×200). (C,E) The LEC were seeded in a 96‐well plate pre‐coated with Matrigel in the presence or absence of rmIL‐17, LLC medium and LLC/rmIL‐17‐conditioned medium, and the formation of capillary‐like structures was assessed. The presence or absence of tube‐like structures was determined after 3 days of incubation (magnification, ×200). In B and C, the results shown are the average of three independent experiments. Data are displayed as the mean ± SD. *P < 0.05, LLC versus rmIL‐17; **P < 0.05, LLC/rmIL‐17 versus LLC.
Interleukin‐17 enhanced chemotaxis and endothelial cord formation by increasing the expression levels of VEGF‐C. Among the known pro‐lymphangiogenic factors, vascular endothelial growth factor‐C (VEGF‐C) has been reported to act as an important lymphatic‐specific growth factor. Therefore, we hypothesized that IL‐17 might promote lymphangiogenesis by inducing the production of VEGF‐C by lung cancer cells. To examine whether IL‐17 induces expression of VEGF‐C in lung cancer cell lines in vitro, we used real‐time RT‐PCR and ELISA to analyze VEGF‐C mRNA and protein expression in mouse Lewis lung cancer cells treated with rmIL‐17. We also used real‐time RT‐PCR to analyze VEGF‐C mRNA expression in human lung cancer cell lines, including A549 and SPC‐A‐1. We found that rmIL‐17 significantly increased the expression of VEGF‐C mRNA and protein in LLC cells (Fig. 3). As shown in Figure 3, stimulation of the LLC cells with 0.1, 1, 10, 100 or 500 ng/mL rmIL‐17 for 6 h resulted in increased VEGF‐C mRNA expression (Fig. 3A). The VEGF‐C mRNA expression peaked in response to the 10 ng/mL rmIL‐17 treatment. The VEGF‐C protein production by unstimulated and 10 ng/mL rmIL‐17‐stimulated LLC cells was 203.0 ± 13.1 and 277.3 ± 14.0 ng/mL, respectively (Fig. 3B). rhIL‐17‐induced expression of VEGF‐C mRNA was also observed in A549 and SPC‐A‐1 (Fig. 3C). The enhanced chemotaxis and endothelial cord formation in the presence of LLC/rmIL‐17 was inhibited by addition of rVEGF R3/Fc chimera (1 μg/mL) (P ≤ 0.05, Fig. 4). These results indicate that IL‐17 has lymphangiogenic activity by inducing VEGF‐C production.
Figure 3.
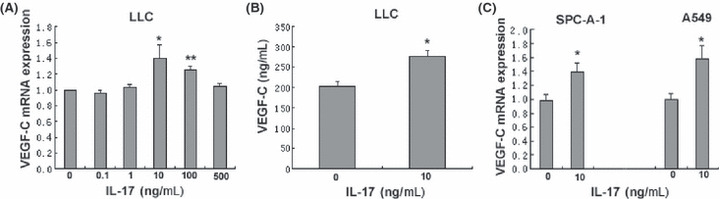
Vascular endothelial growth factor‐C (VEGF‐C) expression in LLC cells. (A) recombinant mouse interleukin‐17 (rmIL‐17) increases VEGF‐C mRNA expression in LLC cells. Cells were stimulated with rmIL‐17 (0.1–500 ng/mL) for 6 h. The VEGF‐C mRNA expression was determined by real‐time RT‐PCR, normalized to β‐actin and expressed relative to untreated LLC cells. *P < 0.05, rmIL‐17 10 ng/mL versus untreated; **P < 0.05, rmIL‐17 100 ng/mL versus untreated. (B) rmIL‐17 induces VEGF‐C protein production by LLC cells. Cells were cultured in the presence or absence of rmIL‐17 (10 ng/mL) for 48 h. At the end of the incubation period, cell culture supernatants were collected and analyzed for VEGF‐C content by ELISA. *P < 0.05, rmIL‐17 10 ng/mL versus untreated. (C) Recombinant human IL‐17 (rhIL‐17) increases VEGF‐C mRNA expression in SPC‐A‐1 and A549 cells. Cells were stimulated with or without rhIL‐17 (10 ng/mL) for 6 h. The VEGF‐C mRNA expression was determined by real‐time RT‐PCR, normalized to ß‐actin and expressed relative to untreated SPC‐A‐1 or A549 cells. *P ≤ 0.05, rhIL‐17 10 ng/mL versus untreated. Data shown are the mean ± SD of three representative experiments.
Figure 4.
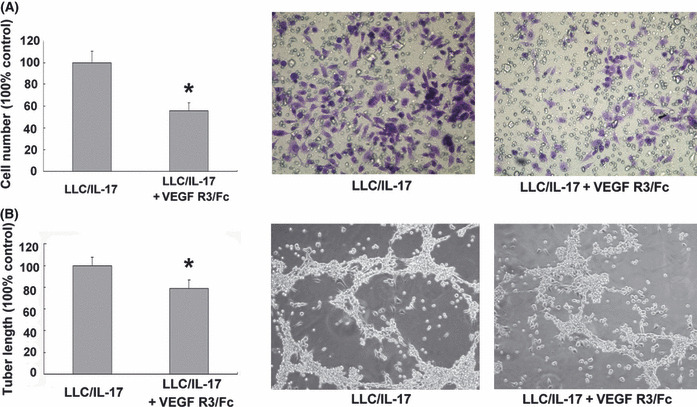
Effect of recombinant mouse vascular endothelial growth factor (rVEGF) R3/Fc chimera and conditioned medium on the migration of, and tube‐like structure formation by, lymphatic endothelial cells (LEC). (A) The LEC were seeded in the upper well of a 24‐well trans‐well chamber and LLC/recombinant mouse interleukin‐17 (rmIL‐17)‐conditioned medium or LLC/rmIL‐17‐conditioned medium ± rVEGF R3/Fc chimera (1 μg/mL) was placed in the lower chamber. After 48 h of incubation, the invasive LEC on the outside surface of the upper chambers were visualized by a crystal violet stain (magnification, ×200). (B) The LEC were seeded in a 96‐well plate pre‐coated with Matrigel in the presence of LLC/rmIL‐17‐conditioned medium or LLC/rmIL‐17‐conditioned medium ± rVEGF R3/Fc chimera (1 μg/mL) and the formation of capillary‐like structures was assessed. The presence or absence of tube‐like structures was determined after 3 days of incubation (magnification, ×200). In A and B, the results shown are the average of three independent experiments. Data is displayed as the mean ± SD. *P ≤ 0.05, LLC/rmIL‐17 versus LLC/rmIL‐17 ± rVEGF R3/Fc chimera.
Correlations between LVD, IL‐17 and VEGF‐C in vivo. We have found that IL‐17 induces lung cancer cells to produce VEGF‐C in vitro. To investigate the role of IL‐17 in the induction of VEGF‐C production by cancer cells in vivo, we performed IL‐17 and VEGF‐C immunohistochemistry on NSCLC tissues from 36 primary human NSCLC cases. We found that specific IL‐17/VEGF‐C staining (shown by immunochemistry as a brown color) was observed mainly within the cytoplasm. The IL‐17‐ and VEGF‐C‐positive cells were localized in the tumor and peritumoral regions, but not in normal mucosa. The VEGF‐C staining was mainly confined to the carcinoma cells and macrophages, while the IL‐17 staining was mainly found in the inflammatory cells among the tumor stroma (Fig. 5). In a previous study, we have shown a correlation between LVD and the frequency of intratumoral IL‐17‐positive cells using immunohistochemical staining of 52 NSCLC patients. In this study of 36 NSCLC patients, the α2 test showed a significant correlation between expression of IL‐17 and VEGF‐C (P ≤ 0.01, Table 1). A similar association was observed between VEGF‐C expression and LVD (P ≤ 0.01, Table 2).
Figure 5.
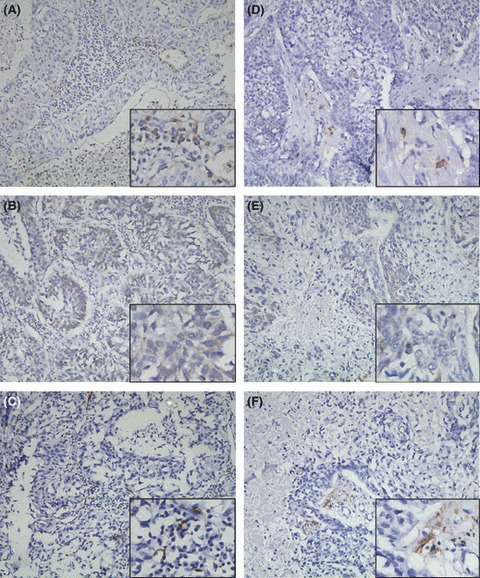
Expression of interleukin‐17 (IL‐17), vascular endothelial growth factor‐C (VEGF‐C) and D2‐40 in tumors from non‐small‐cell lung cancer (NSCLC) patients in situ. High expression of IL‐17 (A), VEGF‐C (B) and D2‐40 (C) on sections of case 18. Low expression of IL‐17 (D), VEGF‐C (E) and D2‐40 (F) on sections of case 10 (magnification, ×200).
Table 1.
. Association of IL‐17 expression with VEGF‐C expression in NSCLC
| VEGF‐C (high) | VEGF‐C (low) | Total | |
|---|---|---|---|
| IL‐17 (high) | 17 | 2 | 19 |
| IL‐17 (low) | 6 | 11 | 17 |
| Total | 23 | 13 | 36 |
P < 0.01 high group versus low group (Fisher’s exact test). IL‐17, interleukin‐17; NSCLC, non‐small‐cell lung cancer; VEGF‐C, vascular endothelial growth factor‐C.
Table 2.
Association of LVD with expression of IL‐17 and VEGF‐C in NSCLC
| n | LVD | P | |
|---|---|---|---|
| IL‐17 (high) | 19 | 11.1 ± 5.3 | 0.002 |
| IL‐17 (low) | 17 | 5.8 ± 4.2 | |
| VEGF‐C (high) | 23 | 10.4 ± 5.6 | 0.006 |
| VEGF‐C (low) | 13 | 5.4 ± 3.3 |
P < 0.01 high group versus low group (Student’s T test). IL‐17, interleukin‐17; NSCLC, non‐small‐cell lung cancer; VEGF‐C, vascular endothelial growth factor‐C.
Treatment of A549 cells with rhIL‐17 causes activation of ERK 1/2 and VEGF‐C synthesis. As described above, stimulation of human lung cancer cell line A549 cells with IL‐17 significantly increased the production of VEGF‐C. We explored the possibility that IL‐17‐induced VEGF‐C production relies on mitogen‐activated protein kinase (MAPK) activation. To address this issue, A549 cells were treated with rhIL‐17 and both phosphorylated and total MAPK were examined. As reported by Ning et al.,( 15 ) the addition of rhIL‐17 to A549 cultures activated ERK 1/2 but not other MAP kinases (Fig. 6A and data not shown). To assess the role of ERK 1/2 activation in IL‐17‐induced VEGF‐C production, A549 cells were stimulated with rhIL‐17 in the presence or absence of PD98059, a specific inhibitor of ERK 1/2. Inhibition of ERK 1/2 activity by PD98059 significantly inhibited IL‐17‐induced VEGF‐C expression at the RNA level (Fig. 6B).
Figure 6.
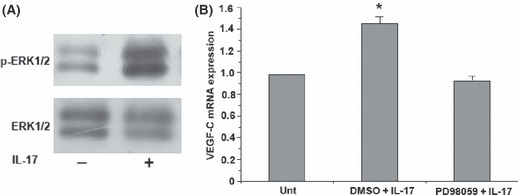
Recombinant human IL‐17 (rhIL‐17) induces vascular endothelial growth factor‐C (VEGF‐C) production by A549 cells via extracellular signal‐regulated protein kinase 1/2 (ERK 1/2) activation. (A) Western blot analysis of ERK1/2 activation in response to rhIL‐17. The A549 cells were incubated in the presence or absence of rhIL‐17 for 20 min and ERK activation was assessed by western blot using specific antibodies recognizing only the phosphorylated form of the protein. Stimulation of A549 with rhIL‐17 resulted in the phosphorylation of ERK1/2. (B) The A549 cells were pre‐incubated with vehicle control (DMSO; 0.1%) or ERK 1/2 inhibitor (PD98059, 50 μM) for 30 min, followed by stimulation with rhIL‐17 (10 ng/mL) for 3 h. The expression of VEGF‐C transcripts was determined by real‐time RT‐PCR. Data are the mean ± SD from three representative experiments.*P < 0.05, IL‐17 + DMSO versus IL‐17 + PD98059.
Discussion
Chronic inflammation has been implicated in the pathogenesis of many different forms of cancer including NSCLC.( 19 ) Interleukin‐17 has been recently described as a pro‐inflammatory cytokine mainly secreted by activated T lymphocytes,( 20 ) which is capable of promoting angiogenesis.( 21 , 22 ) Recently, we found that increased IL‐17‐producing cells correlate with poor survival and increased lymphangiogenesis in NSCLC, but the mechanism is unknown.
To investigate the action of IL‐17 in enhancing lymphangiogenesis in NSCLC, we first tested IL‐17R expression on lung cancer cells and LEC and found that LLC cells, but not LEC, expressed IL‐17R. These findings indicate that IL‐17‐induced lymphangiogenesis might be mediated by lung cancer cells, IL‐17R signaling selectively up‐regulating lymphangiogenic factor expression by lung cancer cells rather than IL‐17 acting directly on LEC. To examine this possibility, we performed LEC migration and tube formation assays using CM from LLC/rmIL‐17. We found that CM from LLC/rmIL‐17 promoted LEC migration and tube formation, while IL‐17 had no effect on LEC proliferation in vitro. These findings again indicate that IL‐17 might promote lymphangiogenesis via selectively up‐regulating lymphangiogenic factor expression by lung cancer cells.
Interleukin‐17 up‐regulates VEGF production by fibroblasts and promotes fibroblast‐induced new vessel formation in inflammation and tumors.( 23 ) Previous studies have shown that the expression of VEGF‐C, a key lymphatic‐specific growth factor, is often induced by inflammatory factors, such as tumor necrosis factor‐α and IL‐1β, as well as by several growth factors expressed by human lung fibroblasts and rheumatoid synoviocytes.( 24 ) Therefore, we investigated whether IL‐17 might promote lymphangiogenesis via up‐regulation of VEGF‐C expression by lung cancer cells. Indeed, treatment of human and mouse lung cancer cells with IL‐17 potently up‐regulated VEGF‐C mRNA and protein expression. Moreover, by addition of rVEGF R3/Fc chimera, the enhanced chemotaxis and endothelial cord formation in the presence of LLC/rmIL‐17 was inhibited. In accordance with our previous findings, high IL‐17 expression correlated with high LVD as well as high tumor grade and lymphatic node metastasis in NSCLC.( 10 ) We also found a significant correlation between IL‐17, VEGF‐C and LVD in 36 cases of NSCLC. These data strongly suggest enhanced expression of IL‐17 in the cancer microenvironment is important for VEGF‐C expression by lung cancer cells and that IL‐17 stimulates VEGF‐C protein synthesis through paracrine actions on lung cancer cells. Further studies using tumor models genetically engineered to knockdown IL‐17R in LLC cells are needed to provide additional understanding of the role of IL‐17 in lymphangiogenesis in NSCLC.
Furthermore, the increase in production of VEGF‐C elicited by IL‐17 was mediated via the p‐ERK1/2 stress kinase in A549 cells. Interleukin‐17 signaling has been shown to involve MAPK. Inoue et al. recently showed that IL‐17A‐mediated promotion of primary bronchial epithelial cell growth relies on activation of ERK 1/2 but not p38 or JNK.( 25 ) Treatment of MKN28 cells with IL‐17 caused activation of ERK 1/2 but not other MAPK.( 26 ) Consistent with the results reported by Ning et al.,( 15 ) p‐ERK1/2, but not other MAP kinases, was activated by IL‐17 treatment of A549 cells. Furthermore, we also found that treatment of A549 cells with the p‐ERK inhibitor PD98059 inhibited IL‐17‐stimulated VEGF‐C synthesis. Therefore, the ERK MAP kinase pathway plays a dominant role in IL‐17A‐elicited VEGF‐C production by A549 cells. Interleukin‐17‐induced lymphangiogenesis thus might be, in part, mediated by VEGF‐C proteins induced through activation of p‐ERK.
In summary, our results provide strong evidence that IL‐17 promotes lymphangiogenesis via up‐regulation of VEGF‐C expression by NSCLC cells. Thus, targeting IL‐17 is likely to have beneficial clinical effects not only by inhibiting inflammation and tumor blood vessel formation but also by inhibiting the tumor‐promoting lymphangiogenesis pathways.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
The authors thank Dr A.L.J Symonds (Institute of Cellular and Molecular Science, University of London, UK) for revision and correction of this manuscript. This work was supported by the National Nature Science Foundation of China (No. 30870516), the outstanding Youth Scientist Foundation of Chongqing (No. CSTC, 2008BA5035) and the National Key Basic Research Program of China (973 program, No. 2010CB529404).
References
- 1. Machtay M, Jeremic B. Complex and controversial issues in locally advanced non‐small cell lung carcinoma. Semin Surg Oncol 2003; 21: 128–37. [DOI] [PubMed] [Google Scholar]
- 2. Renyi‐Vamos F, Tovari J, Fillinger J et al. Lymphangiogenesis correlates with lymph node metastasis, prognosis, and angiogenic phenotype in human non‐small cell lung cancer. Clin Cancer Res 2005; 11: 7344–53. [DOI] [PubMed] [Google Scholar]
- 3. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF‐C‐induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood 2007; 109: 1010–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kukk E, Lymboussaki A, Taira S et al. VEGF‐C receptor binding and pattern of expression with VEGFR‐3 suggests a role in lymphatic vascular development. Development 1996; 122: 3829–37. [DOI] [PubMed] [Google Scholar]
- 5. Yao Z, Fanslow WC, Seldin MF et al. Herpesvirus Saimiri encodes a new cytokine, IL‐17, which binds to a novel cytokine receptor. Immunity 1995; 3: 811–21. [DOI] [PubMed] [Google Scholar]
- 6. Murugaiyan G, Saha B. Protumor vs antitumor functions of IL‐17. J Immunol 2009; 183: 4169–75. [DOI] [PubMed] [Google Scholar]
- 7. Miyahara Y, Odunsi K, Chen W, Peng G, Matsuzaki J, Wang RF. Generation and regulation of human CD4+IL‐17‐producing T cells in ovarian cancer. Proc Natl Acad Sci U S A 2008; 105: 15505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang JP, Yan J Xu J et al. Increased intratumoral IL‐17‐producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol 2009; 50: 980–9. [DOI] [PubMed] [Google Scholar]
- 9. Numasaki M, Watanabe M, Suzuki T et al. IL‐17 enhances the net angiogenic activity and in vivo growth of human non‐small cell lung cancer in SCID mice through promoting CXCR‐2‐dependent angiogenesis. J Immunol 2005; 175: 6177–89. [DOI] [PubMed] [Google Scholar]
- 10. Chen X, Wan J, Liu J et al. Increased IL‐17‐producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer 2010; 69: 348–354. [DOI] [PubMed] [Google Scholar]
- 11. Kolls JK, Linden A. Interleukin‐17 family members and inflammation. Immunity 2004; 21: 467–76. [DOI] [PubMed] [Google Scholar]
- 12. Honorati MC, Cattini L, Facchini A. IL‐17, IL‐1beta and TNF‐alpha stimulate VEGF production by dedifferentiated chondrocytes. Osteoarthritis Cartilage 2004; 12: 683–91. [DOI] [PubMed] [Google Scholar]
- 13. Watari K, Nakao S, Fotovati A et al. Role of macrophages in inflammatory lymphangiogenesis: Enhanced production of vascular endothelial growth factor C and D through NF‐kappaB activation. Biochem Biophys Res Commun 2008; 377: 826–31. [DOI] [PubMed] [Google Scholar]
- 14. Wang J, Zhang B, Guo Y et al. Artemisinin inhibits tumor lymphangiogenesis by suppression of vascular endothelial growth factor C. Pharmacology 2008; 82: 148–55. [DOI] [PubMed] [Google Scholar]
- 15. Ning W, Choi AM, Li C. Carbon monoxide inhibits IL‐17‐induced IL‐6 production through the MAPK pathway in human pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol 2005; 289: L268–73. [DOI] [PubMed] [Google Scholar]
- 16. Wang J, Guo Y, Zhang BC, Chen ZT, Gao JF. Induction of apoptosis and inhibition of cell migration and tube‐like formation by dihydroartemisinin in murine lymphatic endothelial cells. Pharmacology 2007; 80: 207–18. [DOI] [PubMed] [Google Scholar]
- 17. Saintigny P, Kambouchner M, Ly M et al. Vascular endothelial growth factor‐C and its receptor VEGFR‐3 in non‐small‐cell lung cancer: concurrent expression in cancer cells from primary tumour and metastatic lymph node. Lung Cancer 2007; 58: 205–13. [DOI] [PubMed] [Google Scholar]
- 18. Weidner N. Tumor angiogenesis: review of current applications in tumor prognostication. Semin Diagn Pathol 1993; 10: 302–13. [PubMed] [Google Scholar]
- 19. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 20. Park H, Li Z, Yang XO et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6: 1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Numasaki M, Fukushi J, Ono M et al. Interleukin‐17 promotes angiogenesis and tumor growth. Blood 2003; 101: 2620–7. [DOI] [PubMed] [Google Scholar]
- 22. Kato T, Furumoto H, Ogura T et al. Expression of IL‐17 mRNA in ovarian cancer. Biochem Biophys Res Commun 2001; 282: 735–8. [DOI] [PubMed] [Google Scholar]
- 23. Honorati MC, Neri S, Cattini L, Facchini A. Interleukin‐17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage 2006; 14: 345–52. [DOI] [PubMed] [Google Scholar]
- 24. Ristimaki A, Narko K, Enholm B, Joukov V, Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor‐C. J Biol Chem 1998; 273: 8413–8. [DOI] [PubMed] [Google Scholar]
- 25. Inoue D, Numasaki M, Watanabe M et al. IL‐17A promotes the growth of airway epithelial cells through ERK‐dependent signaling pathway. Biochem Biophys Res Commun 2006; 347: 852–8. [DOI] [PubMed] [Google Scholar]
- 26. Sebkova L, Pellicano A, Monteleone G et al. Extracellular signal‐regulated protein kinase mediates interleukin 17 (IL‐17)‐induced IL‐8 secretion in Helicobacter pylori‐infected human gastric epithelial cells. Infect Immun 2004; 72: 5019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]