

Abstract
The promoter region of estrogen receptor 1 (ESR1) has been shown to be methylated in normal colorectal mucosa in an age‐dependent manner. However, the methylation of this region in colorectal tumors has not sufficiently been investigated. The methylation status of ESR1 in 105 colorectal adenoma tissues was examined by MethyLight and presented as the percentage of methylated references (PMR). Factors that affect the PMR of ESR1 in adenomas were determined using parameters including patient age, sex, past history of malignancy, family history of colorectal cancer, smoking and drinking habits, clinical characteristics of adenomas (location, size, macroscopic appearance, and histology), and K‐ras mutation. Multiple linear regression revealed that the PMR was not correlated with patient age. K‐ras mutation was significantly correlated with the higher methylation status of ESR1 in adenoma (t‐value = 3.21, P = 0.0018), whereas alcohol exposure was significantly correlated with lower methylation status (t‐value = –2.37, P = 0.02). Because methylation of O6‐methylguanine DNA methyltransferase (MGMT) has been reported to be correlated with K‐ras G‐to‐A transition, methylation of ESR1 was compared with that of MGMT with regard to K‐ras mutation. Contrary to expectations, methylation of MGMT was not significantly correlated with K‐ras G‐to‐A transition, but that of ESR1 was strongly correlated with K‐ras G‐to‐A transition. Thus, the methylation status of ESR1 in adenomas was not correlated with patient age, but was associated with K‐ras mutation, suggesting that methylation of ESR1 in tumors functions differently from that in normal colon mucosa. (Cancer Sci 2009; 100: 1005–1011)
Abbreviations:
- ACTB
β‐actin gene
- CIMP
CpG island methylator phenotype
- CRC
colorectal cancer
- ER
estrogen receptor
- ESR1
estrogen receptor 1
- MGMT
O6‐methylguanine DNA methyltransferase
- PCR
polymerase chain reaction
- PMR
percentage of methylated reference
- RASSF
Ras association domain families
Recently, epigenetic changes, including DNA methylation, have been found to play an important role in carcinogenesis in addition to genetic changes. DNA methylation is a covalent chemical modification resulting in the addition of a methyl (CH3) group at the C5‐position of cytosine in the sequence context 5′‐CG‐3′. This epigenetic effect is particularly striking when DNA methylation affects promoter CpG islands. Methylation of cytosines within CpG islands is associated with the loss of protein expression by the repression of transcription. Therefore, gene hypermethylation is associated with the silencing of tumor‐suppressor genes in cancer.( 1 ) In colon carcinogenesis, methylation of various genes such as MGMT,( 2 , 3 , 4 ) human mutL homolog (hMLH1),( 5 , 6 , 7 ) secreted frizzled‐related protein,( 8 , 9 , 10 ) and RASSF2 ( 11 , 12 ) has been reported to be closely related in cancer development. According to the frequency of CpG island methylation, the idea of a CIMP in carcinogenesis of the colon has also been proposed.( 13 )
Meanwhile, methylation of some CpG islands occurs in normal colon mucosa, and some such methylations increase with age.( 13 , 14 , 15 , 16 , 17 ) This has led to a proposal by Toyota et al. that methylation of some genes is age related (type A genes), whereas for other genes, methylation is cancer specific (type C genes).( 13 ) Methylation of the promoter region of ESR1 in the normal colon mucosa was initially reported in 1994.( 14 ) This locus was shown to be methylated in normal colon mucosa in an age‐dependent manner; thus, this gene is regarded as a representative type A gene. Age‐related methylation in normal mucosa like the ESR1 locus has been suggested as a predisposing factor for the increased risk of cancer with age.( 14 , 18 )
On the other hand, ESR1 is densely methylated in colorectal neoplasia.( 19 ) However, the correlation of methylation of ESR1 in the normal colon and neoplasias has not been investigated, maybe because the initial report by Issa et al. indicated that methylation of ESR1 was equally observed in all examined colon neoplasias.( 14 ) Because the methodology used in that report was qualitative, and not very sophisticated, the methylation status of ESR1 in neoplasia was not accurately quantified. Recently, however, quantitative methods of methylation analysis such as MethyLight have become available. Several reports have quantified the methylation of ESR1 in colon neoplasias,( 20 , 21 ) and have revealed that methylation levels range to some extent.( 21 ) If the methylation of ESR1 in aging is linked with that in cancer, there should be some correlation of methylation levels of ESR1 in normal mucosa and neoplasias in individual patients.
Accordingly, in the present study, we investigated the methylation status of ESR1 in colorectal adenomas, and determined whether the methylation of ESR1 in adenomas is also age dependent, as is the case in normal mucosa. In this context, the correlation of the methylation levels of ESR1 between each patient's normal mucosa and adenomas was examined. Moreover, factors that affect the methylation status of ESR1 in colon adenomas were identified among various parameters including clinical factors and K‐ras mutation.
Materials and Methods
Patients and tissue samples. Colorectal adenoma tissues were consecutively collected from individuals who provided written informed consent and underwent endoscopic resection of colorectal polyps at Okayama University Hospital from June 2003 to March 2005. In addition, normal mucosal samples of these patients were collected if the patient's consent was obtained. A total of 105 adenomas from 90 patients, and normal mucosal samples of 47 patients were collected and examined. There were no cases with inflammatory bowel disease or a known history of hereditary colorectal cancer (familial adenomatous polyposis or hereditary non‐polyposis colorectal cancer) in the analyzed patients.
Clinical information about patients was obtained, including age, sex, history of any kind of malignancy, family history of CRC, smoking habits, and alcohol consumption. Family history was defined as having a first‐degree relative with CRC. Smokers were defined as consuming 20 cigarettes a day for 10 years or more. Alcohol drinkers were defined as those who drank more than approximately 20 g per day.
At the time of resection, the location, size, and macroscopic appearance of the adenomas were determined. Adenoma locations were classified into two groups: ‘proximal’, defined as the cecum and ascending and transverse colon; and ‘distal’, defined as the descending and sigmoid colon and rectum. Adenoma size was recorded as the maximum diameter of the extirpated specimen. Histological studies were carried out on all removed adenomas. The resected adenomas were fixed and embedded in paraffin. Serial sections were obtained and stained with hematoxylin–eosin. All cases were reviewed by two board‐certificated pathologists, and were classified as low‐grade or high‐grade dysplasia according to World Health Organization criteria.( 22 ) Hyperplastic polyps and serrated adenomas were not included in this analysis. We classified the macroscopic appearance of adenomas, based on colonoscopy observations, into flat‐type or protruded‐type according to the definition described previously.( 23 )
A small tissue fragment was excised from resected adenomas for DNA extraction, and the remaining portion was submitted for histological diagnosis. Adenoma and normal mucosa samples were stored at –80°C until the analysis began.
This study protocol was approved by the institutional review board of Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences. Informed consent was obtained from each patient.
Methylation analysis of adenomas and normal colon mucosa. Genomic DNA was extracted from adenomas and normal colorectal tissues using a QIAmp DNA mini kit (Qiagen, Hilden, Germany). Bisulfite modification of DNA was carried out using an EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA), according to the manufacturer's instructions. The methylation status of the CpG locus of the promoter region of ESR1 and MGMT was determined by the fluorescence‐based, real‐time PCR assay MethyLight as described previously.( 20 , 24 ) For the MethyLight assay, primers and probes designed specifically for bisulfite‐converted DNA of the gene of interest were used. Those for ACTB were also used to normalize for input DNA. The primer and probe sequences used were as follows (forward PCR primer, TaqMan probe, and reverse PCR primer, respectively): ESR1 (GGCGTTCGTTTTGGGATTG, 6FAM5′‐CGATAAAACCGAACGACCCGACGA‐3′TAMRA, GCCGACACGCGAACTCTAA); MGMT (GCGTTTCGACGTTC GTAGGT, 6FAM5′‐CGCAAACGATACGCACCGCGA‐3′TAMRA, CACTCTTCCGAAAACGAAACG); and ACTB (TGGTGATGG AGGAGGTTTAGTAAGT, 6FAM5′‐ACCACCACCCAACACAC AATAACAAACACA‐3′TAMRA, AACCAATAAAACCTACTCC TCCCTTAA).( 20 , 25 , 26 )
The specificity of the reactions for methylated DNA was confirmed separately using the RKO (for ESR1) or SW48 (for MGMT) CRC cell line (American Type Culture Collection, Manassas, VA, USA) as the methylated control. For data presentation, the PMR was indicated. PMR at a specific locus was calculated by dividing the gene : ACTB ratio of a sample by that of RKO or SW48 and multiplying by 100.( 24 , 25 )
Bisulfite sequencing. To verify the ESR1 methylation status determined by MethyLight, bisulfite sequencing was carried out as described previously.( 27 ) Briefly, amplification of the bisulfite‐treated DNA was carried out using the following primers: 5′‐GGTTTTTGAGTTTTTTGTTTTG‐3′ (forward) and 5′‐AACTTAC TACTATCCAAATACACCTC‐3′ (reverse). Temperature conditions for PCR were as follows: initial denaturation of 5 min at 95°C, followed by 35 cycles of 95°C for 30 s, 55°C for 15 s, and 72°C for 30 s and then a final extension for 4 min at 72°C. PCR products were cloned into the pCRII vector of the TA Cloning Kit (Invitrogen, Purchase, NY, USA), and extracted DNA for each adenoma or normal mucosa sample were sequenced using M13 primers with a Big Dye Terminator v3.1 kit and an ABI Genetic Analyzer 3100 (ABI, Foster City, CA, USA).
K‐ras mutation in adenomas. K‐ras codon‐12 and codon‐13 point mutations were detected by direct sequencing. The following primers were used for the sequencing reactions: 5′‐GCCTGCTGAAAAT GACTGAAT‐3′ (forward) and 5′‐GACCATTCTTTGATACAGA TAA‐3′ (reverse). DNA sequencing was carried out on an ABI Genetic Analyzer 3100, using the BigDye 3.1 cycle sequencing technology (ABI).
Statistical analysis. The associations between age and methylation, and between methylation in adenomas and methylation in normal mucosa were evaluated using linear regression. The relationship between the PMR of adenoma ESR1 and clinical factors was evaluated using univariate and multivariate linear regression. The primary independent variables were clinical factors of patients or adenomas, including age, sex, history of any kind of malignancy, family history of CRC, smoking habits, alcohol consumption, location, size, macroscopic appearance, histological grade, and K‐ras mutation. The primary outcome variable was the PMR of the ESR1 locus. Multivariate linear regression analyses were then carried out on all clinical variables, which we evaluated in univariate analyses. The differences in PMR among more than three groups were analyzed using the Kruskal–Wallis rank test and Mann–Whitney U‐test. These analyses were carried out using SAS version 9.1 (SAS Institute, Cary, NC, USA). All P‐values were two sided and considered significant when less than 0.05.
Results
Clinicopathological characteristics of patients and adenomas. We evaluated 105 colorectal adenomas derived from 90 patients. The clinicopathological characteristics of patients and adenomas are depicted in Table 1. Patients included 26 (29%) women and 64 (71%) men, with a median age of 62 years (range, 31–83 years). Of these patients, 53 (59%) were alcohol drinkers and 48 (53%) were smokers. In 11 (12%) patients, two or more polyps were simultaneously resected and analyzed.
Table 1.
Clinical characteristics of patients and adenomas
| Characteristic | No. (%) patients (n = 90) | No. (%) adenomas (n = 105) |
|---|---|---|
| Age | 62 (31–83) | 62 (31–83) |
| Sex | ||
| Male | 64 (71) | 77 (73) |
| Female | 26 (29) | 28 (27) |
| History of malignancy | ||
| Yes | 18 (20) | 19 (18) |
| No | 72 (80) | 86 (82) |
| Family history of colorectal cancer | ||
| Yes | 8 (9) | 9 (9) |
| No | 82 (91) | 96 (91) |
| Alcohol exposure | ||
| Drinker | 53 (59) | 68 (65) |
| Occasionally or none | 37 (41) | 37 (35) |
| Tobacco exposure | ||
| Smoker | 48 (53) | 59 (56) |
| None | 42 (47) | 46 (44) |
| Location of polyps | ||
| Proximal colon | 31 (34) | 39 (37) |
| Distal colon | 59 (66) | 66 (63) |
| Size of polyps | 12 (4–40) mm | 12 (4–40) mm |
| Macroscopic appearance | ||
| Flat | 13 (14) | 16 (15) |
| Protruded | 77 (86) | 89 (85) |
| Histology | ||
| High‐grade | 29 (32) | 32 (30) |
| Low‐grade | 61 (68) | 73 (70) |
| K‐ras mutation | ||
| Yes | 27 (30) | 28 (27) |
| No | 63 (70) | 77 (73) |
Percentages for patients are based on the largest polyp.
Of these 105 adenomas, 39 (37%) adenomas were located in the proximal colon, whereas 66 (63%) were in the distal colon. Almost three‐quarters (77; 73%) of the adenomas were larger than 1 cm in diameter, including 28 (27%) adenomas with a size in excess of 2 cm. Regarding macroscopic appearance, 89 (85%) were categorized as protruded‐type and 16 (15%) as flat‐type adenomas. Histological examinations revealed that 32 (30%) were high‐grade dysplasia. K‐ras mutation was identified in 28 (27%) adenomas.
Methylation status of ESR1 in adenomas and normal mucosa. The methylation status of ESR1 in 105 adenomas was determined using the MethyLight assay, which is capable of quantifying methylation. The PMR of ESR1 in adenomas ranged from 2 to 286 (median 74). No significant correlation was observed between the PMR and patient age (Fig. 1a). PMR of the normal mucosa of 47 patients were also examined, and ranged from 1 to 39 (median 16). As in previous reports,( 14 , 16 , 28 ) the PMR of the normal colon was significantly correlated with patient age (P = 0.0004; Fig. 1b). Then the PMR values were compared between adenoma and normal colon mucosa in each patient who could provide both adenoma and normal colon samples; however, no correlation was observed between the two (P = 0.82; Fig. 1c). In addition, we carried out bisulfite sequencing of the ESR1 locus in representative adenoma and matched normal colon samples (Fig. 2). In normal mucosal samples, the PMR were relatively low (6.2 and 22.6) and methylation status shown with bisulfite sequencing was sparse, whereas in adenoma samples, the PMR were high (153.1 and 159.2) and the sequencing revealed that the locus was densely methylated. Thus, the ESR1 locus is methylated in an age‐dependent manner in normal mucosa, whereas the methylation in adenomas seemed to occur independently of patient age. There seems to be no link between the methylation of ESR1 in normal mucosa and that in adenomas.
Figure 1.
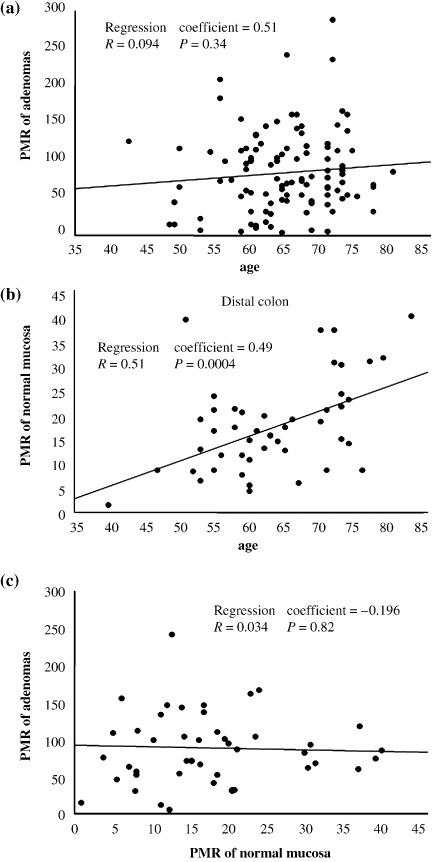
Methylation status of estrogen receptor 1 (ESR1) in adenomas and normal mucosa. (a) The percentage of methylated references (PMR) of ESR1 in 105 adenomas are shown and stratified by patient age. No significant correlation was observed between PMR and patient age. (b) The PMR of ESR1 in normal colon mucosa of 47 patients who provided normal samples are shown. PMR of normal mucosa were significantly correlated with patient age (P = 0.0004). (c) The PMR of ESR1 in adenoma and those in normal mucosa were compared in individual patients. No correlation was observed.
Figure 2.
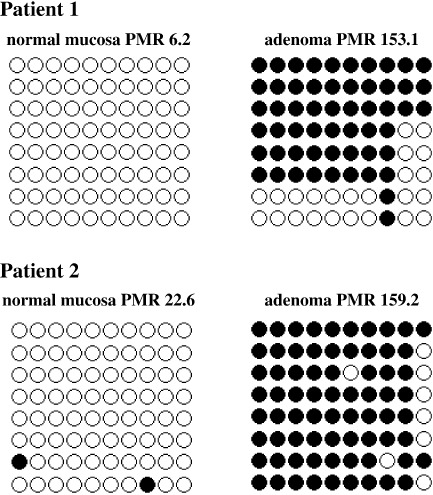
Bisulfite sequencing of the estrogen receptor 1 (ESR1) locus in representative patients. In normal mucosal samples, the percentage of methylated references (PMR) were relatively low (6.2 and 22.6) and methylation status shown with bisulfite sequencing was sparse, whereas in adenoma samples, the PMR were high (153.1 and 159.2) and the sequencing revealed that the locus was densely methylated.
Factors that affect methylation of ESR1 in adenomas. Because methylation in adenomas was not correlated with aging, the methylation may occur through different mechanisms from that in normal mucosa. In order to identify factors contributing to methylation of ESR1 in adenomas, multivariate analysis was carried out using age, sex, history of any kind of malignancy, family history of CRC, smoking habits, alcohol consumption, adenoma location, size, macroscopic appearance, histological grade, and K‐ras mutation. Analysis revealed that K‐ras mutation was the most significant factor for higher methylation of ESR1 (t‐value = 3.21, P = 0.0018). On the other hand, alcohol use was a significant factor for lower methylation of ESR1 (t‐value = –2.37, P = 0.02). Other factors were not correlated with methylation of ESR1 in adenomas (Table 2).
Table 2.
Factors associated with methylation of estrogen receptor 1 in adenomas
| Characteristic | Percentage of methylated reference | Univariate | Multivariate | |||
|---|---|---|---|---|---|---|
| t | P | Adjusted r2 | t | P | ||
| Age | 0.96 | 0.34 | 0.009 | 1.76 | 0.08 | |
| 62 years (31–83 years) | – | |||||
| Sex | –2.28 | 0.02 | 0.039 | 0.49 | 0.62 | |
| Male | 67 (2–286) | |||||
| Female | 113 (30–206) | |||||
| History of malignancy | 1.08 | 0.28 | 0.002 | 0.42 | 0.68 | |
| Yes | 70 (13–143) | |||||
| No | 78 (2–286) | |||||
| Family history of colorectal cancer | –1.48 | 0.14 | 0.011 | –1.27 | 0.21 | |
| Yes | 39 (4–143) | |||||
| No | 74 (2–286) | |||||
| Alcohol exposure | –2.93 | 0.004 | 0.068 | –2.37 | 0.02 | |
| Drinker | 63 (2–239) | |||||
| Occasionally or none | 112 (13–286) | |||||
| Tobacco exposure | –1.30 | 0.20 | 0.006 | 0.29 | 0.77 | |
| Smoker | 66 (2–286) | |||||
| None | 78 (9–206) | |||||
| Location of polyps | 0.04 | 0.97 | <0.001 | –0.48 | 0.64 | |
| Proximal | 74 (3–239) | |||||
| Distal | 75 (2–286) | |||||
| Size of polyp | 2.06 | 0.04 | 0.030 | 0.06 | 0.95 | |
| 11 cm (4–40 cm) | – | |||||
| Macroscopic appearance | –2.03 | 0.04 | 0.029 | –1.64 | 0.10 | |
| Flat | 47 (4–153) | |||||
| Protruded | 84 (2–286) | |||||
| Histology | 1.08 | 0.28 | 0.002 | 0.21 | 0.83 | |
| High grade | 98 (2–239) | |||||
| Low grade | 71 (3–286) | |||||
| K‐ras mutation | 3.82 | 0.0002 | 0.116 | 3.21 | 0.0018 | |
| Yes | 110 (46–239) | |||||
| No | 65 (2–286) | |||||
Adjusted r 2 for multiple linear regression = 0.165.
Methylation of ESR1 in adenomas with K‐ras mutation or alcohol exposure. Figure 3 shows the correlation between methylation of ESR1 and K‐ras mutation or alcohol exposure in adenomas. Samples were classified into four groups: adenomas with wild‐type K‐ras derived from drinkers (median, 56; range, 2–233); those with wild‐type K‐ras derived from non‐drinkers (median, 97; range, 13–286); those with mutant K‐ras derived from drinkers (median, 103; range, 47–239); and those with mutant K‐ras derived from non‐drinkers (median, 120; range, 62–206). The Kruskal–Wallis rank test revealed that the difference in the PMR of ESR1 among the four groups was statistically significant (P = 0.0002). Adenomas derived from drinkers are likely to exhibit low methylation of ESR1; in contrast, adenomas with K‐ras mutations are likely to exhibit high methylation of ESR1. In particular, adenomas with PMR below 47 did not carry K‐ras mutations. These results suggest that methylation of ESR1 is strongly correlated with K‐ras mutation in colorectal neoplasias, and that alcohol negatively affects the methylation of ESR1, resulting in suppression of the occurrence of K‐ras mutation in those tumors.
Figure 3.
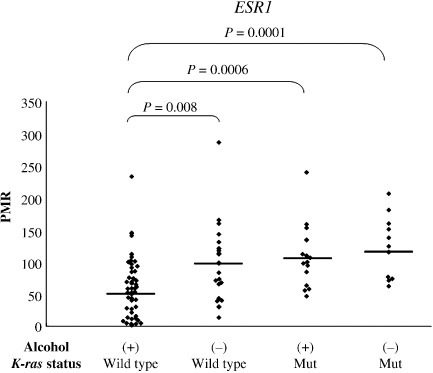
Methylation of estrogen receptor 1 (ESR1) in adenomas stratified by K‐ras status and alcohol exposure. Adenomas were stratified into four groups according to K‐ras status (wild‐type or mutant) and alcohol exposure (+ or –). The Kruskal–Wallis rank test revealed that the differences in the percentage of methylated references (PMR) of ESR1 among the four groups were statistically significant (P < 0.0001). Adenomas derived from drinkers were likely to exhibit lower methylation of ESR1; in contrast, adenomas with K‐ras mutations were likely to exhibit higher methylation of ESR1. In particular, no adenomas with PMR below 47 carried a K‐ras mutation. Horizontal lines represent median methylation levels for each group.
K‐ras G‐to‐A transition and methylation of ESR1 and MGMT. We have shown here that methylation of ESR1 is closely correlated with K‐ras mutation in colorectal adenomas. On the other hand, previous reports have indicated that methylation of MGMT, a DNA repair gene involved in avoiding G‐to‐A mutation, is closely correlated with the K‐ras G‐to‐A transition.( 3 , 4 ) We compared methylation of MGMT and the methylation of ESR1 in our samples with regard to K‐ras status (Fig. 4). Samples were classified into three groups according to K‐ras status: adenomas with a G‐to‐A transition; those with other mutations; and wild‐type adenomas. There was a significant difference in the PMR of ESR1 among the three groups (P = 0.0002, Kruskal–Wallis rank test). In contrast, no significant difference was observed in the PMR of MGMT among the three groups (P = 0.35). Furthermore, the PMR of ESR1 was significantly higher in adenomas with the K‐ras G‐to‐A transition than in the wild‐type adenomas (P = 0.0003). Meanwhile, for the PMR of MGMT, no correlation was observed between the two. Thus, methylation of ESR1 is more specifically correlated with K‐ras mutation than methylation of MGMT.
Figure 4.
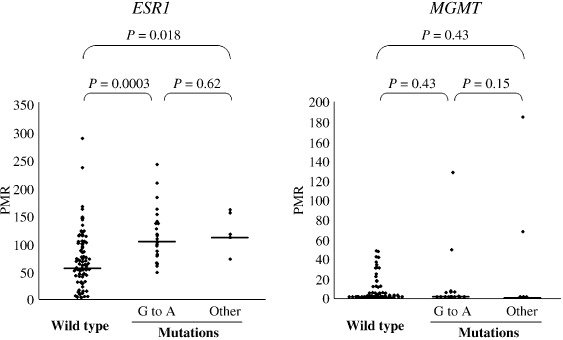
Prevalence of the percentage of methylated reference (PMR) of estrogen receptor 1 (ESR1) and O6‐methylguanine DNA methyltransferase (MGMT) in adenomas according to K‐ras status. Adenoma samples were classified into three groups according to K‐ras status: adenomas with the G‐to‐A transition, those with other mutations, and wild‐type adenomas. The Kruskal–Wallis rank test revealed that the PMR of ESR1 was significantly correlated with K‐ras status (P = 0.0002). On the other hand, the PMR of MGMT was not significantly correlated with K‐ras status (P = 0.35). In addition, the PMR of ESR1 was significantly higher in adenomas with the K‐ras G‐to‐A transition than in wild‐type adenomas (P = 0.0003). Meanwhile, the PMR of MGMT was not significantly different between adenomas with the K‐ras G‐to‐A transition and wild‐type adenomas (P = 0.43). Horizontal lines represent median methylation levels for each group.
Discussion
Methylation of ESR1 in colon mucosa and neoplasia was initially reported in 1994.( 14 ) Although the methylation has been confirmed to occur in an age‐dependent manner in the normal colon mucosa, it has not been examined whether the methylation in neoplasias is age dependent, due to the insensitivity of analyzed methods. If the methylation of ESR1 in normal mucosa is truly linked to the predisposition to cancer due to aging, the methylation in neoplasia should be expected to be age dependent to some extent. Moreover, we hypothesized that methylation of ESR1 may correlate with some of the markers of adenoma malignancy, such as adenoma size and high histological grade, because ESR1 is considered to function as a tumor‐suppressor gene.( 14 , 15 )
In our analysis, contrary to expectations, the methylation of ESR1 in neoplasias was not age dependent. However, intriguingly, there were correlations between methylation and both alcohol use and K‐ras mutation. These results suggest that methylation of ESR1 in neoplasia may have a distinct meaning from that in normal mucosa.
Although methylation of ESR1 in normal mucosa has been discussed extensively, the reports regarding methylation in neoplasias have been relatively few. It is believed, however, that age‐related methylation is linked to cancer‐specific methylation, without there being any definite evidence of such a link. In the present study, we demonstrated that methylation of ESR1, the representative locus of age‐related methylation, occurs in a different manner in normal mucosa and neoplasias. This suggests that there are major differences in the biological mechanism and meanings between methylation in the normal mucosa and methylation in neoplasias. The hypothesis that progression of age‐related methylation in the normal mucosa is directly linked with cancer susceptibility is not necessarily correct. In fact, many other genes that are methylated in neoplasias are not methylated in normal mucosa, even in elderly patients. Thus, methylation of age‐related loci observed in neoplasias would not be an age‐related but neoplasia‐specific event.
As shown herein, methylation of ESR1 in colorectal neoplasia would be a neoplasia‐specific event. If so, there should be a biological role of this methylation in neoplasias. Issa et al. showed that introduction of ERα into colon cancer cells inhibited cell growth, suggesting that methylation and loss of the function of the ER gene may promote cell growth.( 14 ) Although relatively few reports have referred to the function of estrogen receptors, the function of estrogen itself with regard to tumor growth or suppression has frequently been reported. Some reports have suggested a tumor‐promoting effect of estrogen,( 29 , 30 , 31 ) but others have indicated a tumor‐suppressing effect.( 32 , 33 , 34 ) Therefore, the biological meanings of the methylation of ESR1 with regard to the function of estrogen would be controversial. However, epidemiological studies regarding hormone replacement therapy have suggested that estrogen has a protective effect against colon cancer development.( 21 , 35 , 36 , 37 ) In this context, the methylation of ESR1 would reduce the estrogen signal, resulting in progression of colorectal neoplasia.
Correlations between the methylation status of colorectal neoplasia and K‐ras mutation have been reported. In particular, the methylation of MGMT has been shown to be closely correlated with G‐to‐A transition of the K‐ras gene, because MGMT is believed to function to avoid such mutations.( 38 , 39 , 40 ) In the present study, however, methylation of MGMT was less specific for G‐to‐A transition of the K‐ras gene than that of ESR1. Therefore, ESR1 may function more importantly in inducing K‐ras mutations during colon carcinogenesis.
Alternatively, a recent report indicated that K‐ras mutation itself can cause methylation of various genes.( 41 ) Based on this hypothesis, if K‐ras mutation initially occurs in colorectal neoplasia, it then might induce methylation of genes including MGMT, ESR1, and RASSF2. In this context, the idea of a CIMP should be considered, although ESR1 is not a marker of CIMP. In general, CIMP is strongly correlated with BRAF mutation.( 42 ) In our study, however, none of the adenoma samples showed BRAF mutation (data not shown). Meanwhile, recently, Shen et al. reported that one category of CIMP (CIMP2) is closely correlated with K‐ras mutation.( 43 ) Thus, further analysis is required to elucidate the correlation between K‐ras, BRAF mutations, and methylation.
In the present study, alcohol drinkers were more likely to exhibit lower methylation of ESR1. Alcohol consumption can cause DNA hypomethylation through a deficiency of S‐adenocylmethionine, which is a methyl donor that is maintained by folate supplementation, because alcohol has been shown to cleave folate,( 44 ) impair folate absorption,( 45 ) increase folate excretion,( 46 ) and interfere with methionine synthase activity.( 47 , 48 ) Thus, these effects of alcohol on folate status may easily work in the colon cells as well as in intensely methylated gene loci. In addition, epidemiological data indicated that high alcohol intake was associated with K‐ras mutation in colon cancer.( 49 ) Thus, alcohol may work against the susceptibility to methylation and K‐ras mutation in colon carcinogenesis cases.
There are limitations to the present study. Although accurate comparison of the PMR between normal mucosa and adenoma needs elimination of normal cells from adenoma samples, we could not do so due to the small sizes of the adenoma samples. However, the bias due to this drawback may be small, because our data indicated that the PMR of adenomas was one order higher than that of normal mucosa. Next, although we have demonstrated a correlation between methylation of ESR1 and K‐ras mutation in colorectal adenomas, the precise mechanism of inducing K‐ras mutation by methylation of ESR1 could not be proved. Further investigations for the function of estrogen or estrogen receptors may elucidate a more definite explanation for our results.
In conclusion, methylation of ESR1 in colorectal adenomas is not age dependent, and is not correlated with that in normal mucosa. Alcohol intake is correlated with lower methylation, and K‐ras mutation, especially a G‐to‐A transition, is correlated with higher methylation of ESR1 of adenomas. The correlation between methylation of ESR1 and K‐ras G‐to‐A transition was more significant than that between methylation of MGMT and the K‐ras G‐to‐A transition. These results suggest that methylation of ESR1 may work distinctly and independently during colon carcinogenesis.
References
- 1. Baylin SB, Herman JG, Graff JR et al . Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 1998; 72: 141–96. [PubMed] [Google Scholar]
- 2. Esteller M, Risques RA, Toyota M et al . Promoter hypermethylation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase is associated with the presence of G : C to A : T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res 2001; 61: 4689–92. [PubMed] [Google Scholar]
- 3. Esteller M, Toyota M, Sanchez‐Cespedes M et al . Inactivation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K‐ras in colorectal tumorigenesis. Cancer Res 2000; 60: 2368–71. [PubMed] [Google Scholar]
- 4. Shen L, Kondo Y, Rosner GL et al . MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 2005; 97: 1330–8. [DOI] [PubMed] [Google Scholar]
- 5. Haydon AM, Jass JR. Emerging pathways in colorectal‐cancer development. Lancet Oncol 2002; 3: 83–8. [DOI] [PubMed] [Google Scholar]
- 6. Herman JG, Umar A, Polyak K et al . Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998; 95: 6870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hawkins N, Norrie M, Cheong K et al . CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology 2002; 122: 1376–87. [DOI] [PubMed] [Google Scholar]
- 8. Caldwell GM, Jones C, Gensberg K et al . The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res 2004; 64: 883–8. [DOI] [PubMed] [Google Scholar]
- 9. Caldwell GM, Jones CE, Taniere P et al . The Wnt antagonist sFRP1 is downregulated in premalignant large bowel adenomas. Br J Cancer 2006; 94: 922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki H, Watkins DN, Jair KW et al . Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 2004; 36: 417–22. [DOI] [PubMed] [Google Scholar]
- 11. Harada K, Hiraoka S, Kato J et al . Genetic and epigenetic alterations of Ras signalling pathway in colorectal neoplasia: analysis based on tumour clinicopathological features. Br J Cancer 2007; 97: 1425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akino K, Toyota M, Suzuki H et al . The Ras effector RASSF2 is a novel tumor‐suppressor gene in human colorectal cancer. Gastroenterology 2005; 129: 156–69. [DOI] [PubMed] [Google Scholar]
- 13. Toyota M, Ahuja N, Ohe‐Toyota M et al . CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Issa JP, Ottaviano YL, Celano P et al . Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994; 7: 536–40. [DOI] [PubMed] [Google Scholar]
- 15. Toyota M, Issa JP. CpG island methylator phenotypes in aging and cancer. Semin Cancer Biol 1999; 9: 349–57. [DOI] [PubMed] [Google Scholar]
- 16. Kawakami K, Ruszkiewicz A, Bennett G et al . DNA hypermethylation in the normal colonic mucosa of patients with colorectal cancer. Br J Cancer 2006; 94: 593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakagawa H, Nuovo GJ, Zervos EE et al . Age‐related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite‐unstable colorectal cancer development. Cancer Res 2001; 61: 6991–5. [PubMed] [Google Scholar]
- 18. Issa JP. Aging, DNA methylation and cancer. Crit Rev Oncol Hematol 1999; 32: 31–43. [DOI] [PubMed] [Google Scholar]
- 19. Ahuja N, Li Q, Mohan AL et al . Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58: 5489–94. [PubMed] [Google Scholar]
- 20. Eads CA, Danenberg KD, Kawakami K et al . MethyLight: a high‐throughput assay to measure DNA methylation. Nucleic Acids Res 2000; 28: E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woodson K, Weisenberger DJ, Campan M et al . Gene‐specific methylation and subsequent risk of colorectal adenomas among participants of the polyp prevention trial. Cancer Epidemiol Biomarkers Prev 2005; 14: 1219–23. [DOI] [PubMed] [Google Scholar]
- 22. Hamilton SRAL. Pathology and genetics of tumours of the digestive system. WHO Classification of Tumours, Vol. 2. Lyon: IARC, 2000. [Google Scholar]
- 23. Hiraoka S, Kato J, Tatsukawa M et al . Laterally spreading type of colorectal adenoma exhibits a unique methylation phenotype and K‐ras mutations. Gastroenterology 2006; 131: 379–89. [DOI] [PubMed] [Google Scholar]
- 24. Eads CA, Lord RV, Wickramasinghe K et al . Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res 2001; 61: 3410–18. [PubMed] [Google Scholar]
- 25. Widschwendter M, Siegmund KD, Muller HM et al . Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res 2004; 64: 3807–13. [DOI] [PubMed] [Google Scholar]
- 26. Virmani AK, Tsou JA, Siegmund KD et al . Hierarchical clustering of lung cancer cell lines using DNA methylation markers. Cancer Epidemiol Biomarkers Prev 2002; 11: 291–7. [PubMed] [Google Scholar]
- 27. Hesson LB, Wilson R, Morton D et al . CpG island promoter hypermethylation of a novel Ras‐effector gene RASSF2A is an early event in colon carcinogenesis and correlates inversely with K‐ras mutations. Oncogene 2005; 24: 3987–94. [DOI] [PubMed] [Google Scholar]
- 28. Shannon B, Kay P, House A et al . Hypermethylation of the MYF‐3 gene in colorectal cancers: associations with pathological features and with microsatellite instability. Int J Cancer 1999; 84: 109–13. [DOI] [PubMed] [Google Scholar]
- 29. Xu X, Thomas ML. Estrogen receptor‐mediated direct stimulation of colon cancer cell growth in vitro . Mol Cell Endocrinol 1994; 105: 197–201. [DOI] [PubMed] [Google Scholar]
- 30. Di Domenico M, Castoria G, Bilancio A et al . Estradiol activation of human colon carcinoma‐derived Caco‐2 cell growth. Cancer Res 1996; 56: 4516–21. [PubMed] [Google Scholar]
- 31. English MA, Stewart PM, Hewison M. Estrogen metabolism and malignancy: analysis of the expression and function of 17β‐hydroxysteroid dehydrogenases in colonic cancer. Mol Cell Endocrinol 2001; 171: 53–60. [DOI] [PubMed] [Google Scholar]
- 32. Schwartz B, Smirnoff P, Shany S et al . Estrogen controls expression and bioresponse of 1,25‐dihydroxyvitamin D receptors in the rat colon. Mol Cell Biochem 2000; 203: 87–93. [DOI] [PubMed] [Google Scholar]
- 33. Smirnoff P, Liel Y, Gnainsky J et al . The protective effect of estrogen against chemically induced murine colon carcinogenesis is associated with decreased CpG island methylation and increased mRNA and protein expression of the colonic vitamin D receptor. Oncol Res 1999; 11: 255–64. [PubMed] [Google Scholar]
- 34. Kennelly R, Kavanagh DO, Hogan AM et al . Oestrogen and the colon: potential mechanisms for cancer prevention. Lancet Oncol 2008; 9: 385–91. [DOI] [PubMed] [Google Scholar]
- 35. Fernandez E, La Vecchia C, Braga C et al . Hormone replacement therapy and risk of colon and rectal cancer. Cancer Epidemiol Biomarkers Prev 1998; 7: 329–33. [PubMed] [Google Scholar]
- 36. Calle EE, Miracle‐McMahill HL, Thun MJ et al . Estrogen replacement therapy and risk of fatal colon cancer in a prospective cohort of postmenopausal women. J Natl Cancer Inst 1995; 87: 517–23. [DOI] [PubMed] [Google Scholar]
- 37. Calle EE. Hormone replacement therapy and colorectal cancer: interpreting the evidence. Cancer Causes Control 1997; 8: 127–9. [DOI] [PubMed] [Google Scholar]
- 38. Povey AC, Badawi AF, Cooper DP et al . DNA alkylation and repair in the large bowel: animal and human studies. J Nutr 2002; 132: 3518S–21S. [DOI] [PubMed] [Google Scholar]
- 39. Pegg AE. Repair of O6‐alkylguanine by alkyltransferases. Mutat Res 2000; 462: 83–100. [DOI] [PubMed] [Google Scholar]
- 40. Kaina B, Ochs K, Grosch S et al . BER, MGMT, and MMR in defense against alkylation‐induced genotoxicity and apoptosis. Prog Nucleic Acid Res Mol Biol 2001; 68: 41–54. [DOI] [PubMed] [Google Scholar]
- 41. Gazin C, Wajapeyee N, Gobeil S et al . An elaborate pathway required for Ras‐mediated epigenetic silencing. Nature 2007; 449: 1073–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weisenberger DJ, Siegmund KD, Campan M et al . CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38: 787–93. [DOI] [PubMed] [Google Scholar]
- 43. Shen L, Toyota M, Kondo Y et al . Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci USA 2007; 104: 18654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shaw S, Jayatilleke E, Herbert V et al . Cleavage of folates during ethanol metabolism: Role of acetaldehyde/xanthine oxidase‐generated superoxide. Biochem J 1989; 257: 277–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Romero JJ, Tamura T, Halsted CH. Intestinal absorption of [3H]folic acid in the chronic alcoholic monkey. Gastroenterology 1981; 80: 99–102. [PubMed] [Google Scholar]
- 46. Eichner ER, Hillman RS. The evolution of anemia in alcoholic patients. Am J Med 1971; 50: 218–32. [DOI] [PubMed] [Google Scholar]
- 47. Barak AJ, Beckenhauer HC, Tuma DJ. Hepatic transmethylation and blood alcohol levels. Alcohol 1991; 26: 125–8. [DOI] [PubMed] [Google Scholar]
- 48. Kenyon SH, Nicolaou A, Gibbons WA. The effect of ethanol and its metabolites upon methionine synthase activity in vitro . Alcohol 1998; 15: 305–9. [DOI] [PubMed] [Google Scholar]
- 49. Slattery ML, Curtin K, Anderson K et al . Associations between dietary intake and Ki‐ras mutations in colon tumors: a population‐based study. Cancer Res 2000; 60: 6935–41. [PubMed] [Google Scholar]