

Abstract
Id‐1 (inhibitor of differentiation or DNA binding), a member of the basic helix‐loop‐helix transcription factor family, is up‐regulated in many types of human cancer and its expression levels are correlated with poor treatment outcome and shorter survival. In this study, we provided evidence to suggest that Id‐1 is a universal survival factor that plays a key role in protection against anticancer drug‐induced apoptosis. Using nine anticancer drugs and five cancer cell lines derived from nasopharyngeal carcinoma (CNE1), cervical carcinoma (HeLa), breast cancer (MCF7), hepatocarcinoma (Huh7) and prostate cancer (PC3), we found that down‐regulation of Id‐1 expression at both transcriptional and protein levels was associated with increased apoptosis rates and increased cleaved PARP after exposure to all anticancer agents. Treatment with a caspase 9 inhibitor, Z‐LEHD‐FMK, protected cancer cells from drug‐induced PARP cleavage. However, overexpression of Id‐1 in a p53 mutated cell line, CNE1, was able to suppress PARP cleavage in response to all anticancer drugs examined. In contrast, down‐regulation of Id‐1 through small RNA technology in CNE1 cells led to increased sensitivity to all six types of chemotherapeutic drugs. Our results demonstrate that Id‐1 may be a general negative regulator of anticancer drug‐induced apoptosis and suggest a novel therapeutic target in inducing chemosensitization in cancer cells. Our evidence also provides a possible underlying mechanism responsible for the positive role of Id‐1 in the progression of human cancer. (Cancer Sci 2007; 98: 308–314)
Id‐1(inhibitor of differentiation or DNA binding), a member of the basic helix‐loop‐helix (HLH) transcription factor family, has been suggested as a potential oncogene. This is because overexpression of Id‐1 in normal cells is able to promote cell proliferation through the inactivation of tumor suppressor pathways such as p16INK4a/RB.( 1 , 2 ) In addition, up‐regulation of Id‐1 has been reported in more than 20 types of human cancer, most of which are of epithelial origin, including breast,( 3 ) pancreas,( 4 ) cervical,( 5 ) head and neck,( 6 ) and prostate cancers.( 7 ) Id‐1 expression levels are also positively correlated with advanced tumor stage as well as poor prognosis in several types of human cancer.( 4 , 5 , 7 ) Furthermore, recently it has been reported that patients with higher levels of Id‐1 expression have much shorter overall survival in ovarian cancer,( 8 ) melanoma,( 9 ) and breast cancer.( 10 ) Up‐regulation of Id‐1 in early stage cervical cancer has been suggested as a marker for poor prognosis.( 5 ) These results indicate that overexpression of Id‐1 may play an important part not only in tumorigenesis but also in the progression of human cancer. This hypothesis is also confirmed by several cDNA array studies demonstrating that up‐regulation of Id‐1 is one of the most frequent events during the development of tumorigenesis in animal models.( 11 , 12 , 13 ) Recently, inactivation of Id‐1 is reported to have a therapeutic potential. For example, in cultured prostate cancer cells, down‐regulation of Id‐1 by siRNA technology leads to chemosensitization to a commonly used anticancer drug, taxol.( 14 ) Furthermore, in a breast cancer animal model, inactivation of Id‐1 is able to suppress the metastatic spread of cancer cells to the lung.( 12 ) These lines of evidence strongly indicate Id‐1 as a promising therapeutic target for the treatment of human cancer.
Although the precise molecular mechanisms responsible for its cancer promoting action are not well understood, recent evidence suggests that high levels of Id‐1 expression in cancer cells might lead to suppression of the apoptosis pathway, resulting in promotion of cell survival and cancer progression. Several lines of evidence support this hypothesis. First, overexpression of Id‐1 confers resistance to TNF‐α‐induced apoptosis in prostate cancer cells through activation of the nuclear factor (NF)‐κB pathway.( 15 ) In addition, ectopic expression of Id‐1 leads to resistance to an anticancer drug, taxol, through suppression of the apoptosis in nasopharyngeal carcinoma (NPC) cells.( 16 ) Furthermore, overexpression of Id‐1 results in acquired drug resistance to doxorubicin in androgen independent prostate cancer cells.( 17 ) In contrast, suppression of Id‐1 gene expression through small RNA technology leads to hypersensitivity to taxol‐induced apoptosis in prostate cancer cells.( 14 ) In addition, in gastric cancer cells, down‐regulation of Id‐1 is associated with the nonsteroidal anti‐inflammatory drug sulindac sulfide induced cell death.( 18 ) In melanoma cells, Id‐1 was greatly reduced when cells were undergoing All‐trans‐retinoid acid‐induced apoptosis.( 19 ) Based on these lines of evidence we hypothesized that Id‐1 might be a common antiapoptotic factor and high levels of Id‐1 in cancer cells could act as protection against a variety of anticancer drug‐induced cell death. To test this hypothesis, using six types of anticancer agents with different mechanisms of action, we treated five cancer cell lines derived from five types of human cancer, all of which showed an up‐regulation of Id‐1 expression in clinical specimens,( 3 , 5 , 7 , 20 , 21 ) and studied the role of Id‐1 in anticancer drug‐induced apoptosis. Our results suggest that Id‐1 is a novel universal antiapoptotic factor that may play a key role in the protection of anticancer drug‐induced apoptosis.
Materials and Methods
Anticancer agents. Two DNA damaging agents (cisplatin, mechlorethamine), one antibiotic (mitomycin C), two topoisomerase II inhibitors (doxorubicin, etoposide), one antimetabolite (methotrexate), two microtubule disrupting agents (taxol, vincristine), and a green tea extract, epigallocatechin gallate (EGCG) were purchased from Calbiochem, CA, USA except for mechlorethamine (Sigma, MO, USA). Taxol and etoposide were diluted in dimethylsulfoxide, while mitomycin C was diluted with hot phosphate‐buffered saline (PBS), and the rest of the drugs were all diluted in PBS. The stock solutions of each drug were further diluted again in culture medium to obtain the desired concentrations. Table S1 lists the summary of drug concentrations used in this study.
Cell lines and cell culture conditions. CNE1 (nasopharyngeal carcinoma cell line),( 22 ) HeLa (cervical cancer cell line), MCF7 (breast cancer cell line), Huh7 (hepatocarcinoma cell line) and PC3 (androgen‐independent prostate cancer cell line) obtained from American Type Culture Collection (Rockville, MD, USA), were maintained in RPMI 1640 medium (Sigma) supplemented with 5% (v/v) fetal bovine serum (FBS), penicillin (100 units/mL) and streptomycin (100 µg/mL) at 37°C, 5% CO2, except for Huh7 which was cultured in Dulbecco's modified Eagle's medium containing 10% FBS. A stable Id‐1 transfectant cell line and the vector control generated from CNE1 cells (CNE1 pBabe and CNE1 Id‐1) in a previous study( 21 ) were also included.
Terminal deoxynucleotidyl transferase fluorescence‐dUTP nick end labeling (TUNEL) assay. Using an in situ cell death detection kit, fluorescein was used to detect apoptotic cells according to the recommended protocol (Roche Dignostics). Briefly, cells were seeded on 4‐mm well diameter Chamber slides (ICN, Biomedicals, Aurora, OH, USA) and 24 h later the cells were treated with drugs with or without Caspase 9 inhibitor Z‐LEHD‐FMK (Calbiochem). They were fixed with 4% paraformaldehyde and incubated with reagents supplied in the kit after the permeabilization process. Then propidium iodide (10 µg/mL in PBS) was added to visualize cell nucleus. The percentage of TUNEL positive cells was calculated under a fluorescent microscope. At least 500 cells were counted in each experiment and the percentage of apoptotic cells was calculated as the number of apoptotic cells over the total number of cells counted × 100.
MTT assay. Cell viability was measured using 3‐(4, 5‐dimethyl thiazol‐2‐yl)‐2, 5‐diphenyl tetrazolium bromide (MTT) proliferation assay kit (Boeringher, MO, USA), and the experimental procedures were described previously.( 14 ) Briefly, 3000–6000 cells were seeded in 96‐well plates and cultured for 24 h, drugs (dose 3) were added, respectively. Cell viability was examined 24 h after treatment.
Western blotting. Detailed experimental procedures were described previously.( 23 ) Briefly, whole‐cell lysate was prepared by resuspending cell pellets in lysis buffer, and protein concentrations were measured using the protein assay kit (Bio‐Rad, Hercules, CA, USA). Protein suspension (15–30 µg) was then loaded onto sodium dodecylsulfate–polyacrylamide gel electrophoresis for electrophoresis and then transferred to a polyvinylidene difluoride membrane (Amersham, Piscataway, NJ, USA). The membrane was then incubated with primary antibodies for 1–2 h at room temperature against Id‐1(Santa Cruz Biotechnology, CA, USA), Bax, PARP (Cell Signaling Technology, MA, USA) and β‐actin (Santa Cruz Biotechnology), respectively. After washing with Tris‐buffered saline Tween‐20 (TBS‐T), the membrane was incubated with a secondary antibody against rabbit or goat IgG and the signals were visualized using the enhanced chemiluminescence plus Western blotting system (Amersham).
Reverse transcription‐polymerase chain reaction. Total RNA was isolated from drug‐treated and control cells using a Trizol reagent according to the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA). For reverse transcription‐polymerase chain reaction (RT‐PCR), cDNAs were synthesized using the SuperScript First Strand Synthesis System (Invitrogen). The cDNA was then amplified by PCR with Id‐1 specific primers (sense: 5′‐CCG GCA AGA CAG CGA GCG GTG CG‐3′; antisense: 5′‐GGC GCT GAT CTC GCC GTT GAG GG‐3′) as described.( 24 ) The PCR conditions were as follows: an initial denaturation at 95°C was followed by 28 cycles of PCR (94°C for 1 min, 58°C for 1 min and 72°C for 1 min), and a final extension at 72°C for 5 min. The GAPDH was amplified as an internal loading control. PCR products were electrophoresed on 1.5% agarose gels and analyzed using a gel documentation system (Ultra‐Violet Product Limited, CA, USA).
Transient si‐RNA transfection. siGENOME siRNA targeted the Id‐1 gene. It was purchased commercially (Dharmacon, CO, USA) and dissolved in RNase‐free distilled water. Cells were seeded and cultured overnight. Then the si‐genome duplex RNA (60 nM) (Si‐Id‐1: Sense: UAAACGUGCUGCUCUACGA, Antisense: UCGUAGAGCACGUUUA; Si‐Id‐1‐2: Sense: GGACGAGCAGCAGGUAAAC; Antisense: GUUUACCUGCUGCUCGUCC) and si‐CON (60 nM) (Sense: UAGCGACUAAACACAUCAA, Antisense: UUGAUGUGUUUAGUCGCUA) was mixed, respectively, with lipofectamine 2000 (Invitrogen), then added into the culture medium. The cells were collected after 48 h, or treated with drugs after 24 h and then collected after 48 h.
Results
Down‐regulation of Id‐1 is correlated with increased anticancer drug‐induced apoptosis and decreased cell viability. To test the hypothesis that Id‐1 might play a positive role in protecting cancer cells from programmed cell death, we studied the role of Id‐1 on five cancer cell lines derived from nasopharyngeal carcinoma (CNE1), cervical carcinoma (HeLa), breast cancer (MCF7), hepatocarcinoma (Huh7) and prostate cancer (PC3), all of which have been shown to overexpress Id‐1 in clinical specimens.( 3 , 5 , 7 , 20 , 21 ) Nine commonly used anticancer drugs including two DNA damaging agents (cisplatin, mechlorethamine), one antibiotic (mitomycin C), two topoisomerase II inhibitors (doxorubicin, etoposide), one antimetabolite (methotrexate) and two microtubule disrupting agents (taxol, vincristine), were used to examine the association between Id‐1 expression and the anticancer drug‐induced apoptosis. In addition, a green tea extract, epigallocatechin gallate (EGCG), which has been shown to induce apoptosis in cancer cells through multiple pathways,( 25 ) was also included as a representative for alternative anticancer agents. As shown in Fig. 1, after exposure to the anticancer agents (summary of drug doses is shown in Table S1), all of the cell lines showed a dose‐dependent down‐regulation of the Id‐1 protein. In addition, RT‐PCR experiments showed that the decreased Id‐1 protein expression was associated with decreased Id‐1 mRNA level in response to the anticancer drugs (Fig. S1). However, the expression of Bax, a proapoptotic factor, was not significantly altered by any of the treatments, indicating that down‐regulation of Id‐1 may not be a result of overall transcriptional suppression of the drug treatment. In contrast, the expression of cleaved PARP, an indicator of activation of the apoptosis pathway, was increased (see arrows). The increased PARP cleavage was associated with increased TUNEL positive cells (Fig. 2) and decreased cell viability (Fig. 1b). These results suggest that decreased Id‐1 expression is correlated with the activation of apoptosis pathway and apoptotic cell death in human cancer cells in response to all nine anticancer drugs.
Figure 1.

Effect of anticancer drugs on Id‐1, Bax and PARP expression and cell viability. (a) Western blotting analysis of Id‐1, Bax and PARP expression in response to six types of anticancer drugs. Five cancer cell lines, CNE1 (NPC), HeLa (cervical cancer), MCF7 (breast cancer), Huh7 (hepatocarcinoma) and PC3 (prostate cancer), were exposed with three doses of each anticancer drug for 24 h, respectively, and protein lysates were analyzed. Note that decreased Id‐1 is associated with increased cleaved PARP expression. (b) Cell viability of five cancer cell lines, after exposure to six types of anticancer drugs (dose 3) for 24 h, examined by 3‐(4, 5‐dimethyl thiazol‐2‐yl)‐2, 5‐diphenyl tetrazolium bromide (MTT) assay. CNE1, Hela, MCF7, Huh7 and PC3 cells were seeded in 96‐well plates and drugs (dose 3) were added 24 h later. Cell viability was examined 24 h after treatment. Results represented the optical density (OD) ratio between the treated and untreated cells. Each data point represented the mean and standard deviation. Note that cell viability is decreased after drug treatment. CP, cisplatin; EGCG, epigallocatechin gallate; MCH, mechlorethamine; MTX, methotrexate.
Figure 2.
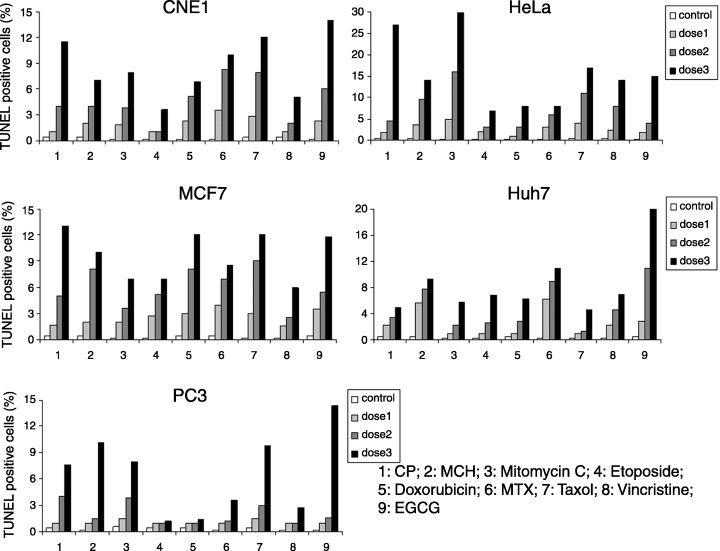
Evidence of anticancer drug‐induced apoptosis in cancer cells. Cells were treated with three doses of each anticancer drug for 24 h, respectively, and the percentage of TUNEL positive cells was determined. 1, cisplatin (CP); 2, mechlorethamine (MCH); 3, Mitomycin; 4, Etopside; 5, Doxorubicin; 6, methotrexate (MTX); 7, Taxol; 8, Vincristine; 9, epigallocatechin gallate (EGCG). Less than 1% TUNEL positive cells was observed in the untreated samples. Note that increased percentage of TUNEL positive cells is associated with increased drug concentration.
To further confirm these results, we then treated the cells with Z‐LEHD‐FMK, a caspase 9 inhibitor, which has been shown to suppress the apoptosis pathway.( 26 ) As shown in Fig. 3, the expression of cleaved PARP was decreased in the cells treated with both anticancer drugs and Z‐LEHD‐FMK compared to the cells treated with drugs alone (Fig. 3a). The decreased PARP cleavage was also associated with a decreased percentage of TUNEL positive cells in the Z‐LEHD‐FMK treated cells (Fig. 3b). Treatment with a higher dose of Z‐LEHD‐FMK led to suppression of cleaved PARP levels but not the expression of Id‐1 (Fig. 3c). Our data suggest that apoptosis might be the result of Id‐1 downregulation by anticancer drug treatment, because in the absence of Z‐LEHD‐FMK, increased cleaved PARP was associated with decreased Id‐1 expression after drug treatment. However, in the presence of Z‐LEHD‐FMK, an inhibitor of caspase 9, though the expression of Id‐1 was decreased after drug treatment, cleavage of PARP was inhibited. These results suggest that Id‐1 could be one of the upstream regulators of apoptosis. These results further support the suggestion that the down‐regulation of Id‐1 mediated chemosensitivity is through the apoptosis pathway and indicate that Id‐1 could be an upstream regulator of the anticancer drug‐induced apoptosis.
Figure 3.
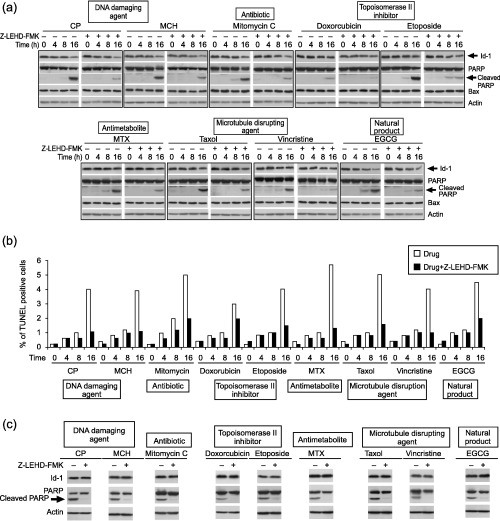
Effect of Caspase 9 inhibitor on anticancer drug‐induced apoptosis. (a) Western blotting analysis of Id‐1, PARP and Bax expression of CNE1 with or without treatment with a caspase 9 inhibitor, Z‐LEHD‐FMK, 20 µM, and with or without drugs (dose 3). (b) Anticancer drug‐induced apoptosis in response to Z‐LEHD‐FMK examined by TUNEL assay. (c) Effect of a higher dose of Z‐LEHD‐FMK (40 µM) on the expression of PARP and Id‐1.
Overexpression of Id‐1 suppresses anticancer drug‐induced apoptosis. To investigate if overexpression of Id‐1 could protect cancer cells from apoptosis against all nine anticancer drugs, we then treated two previously generated CNE1 sublines (pBabe and Id‐1) with relatively high (CNE1 Id‐1) and low (CNE1 pBabe)( 21 ) Id‐1 expression (Fig. 4a). As shown in Fig. 4(b), after treatment with the same doses of anticancer drugs, the pBabe cells showed much lower Id‐1 protein levels (arrows) compared to the Id‐1 transfectants in a dose dependent manner, although decreased Id‐1 expression was observed in both cell lines. In addition, the relatively higher Id‐1 protein levels in the Id‐1 transfectants were associated with lower levels of cleaved PARP (see arrows) and decreased percentages of TUNEL positive cells (data not shown) and increased cell viability (Fig. 4c) after exposure to all nine anticancer agents. These results indicate that high levels of Id‐1 are able to protect cancer cells from apoptosis induced by a variety of anticancer drugs.
Figure 4.
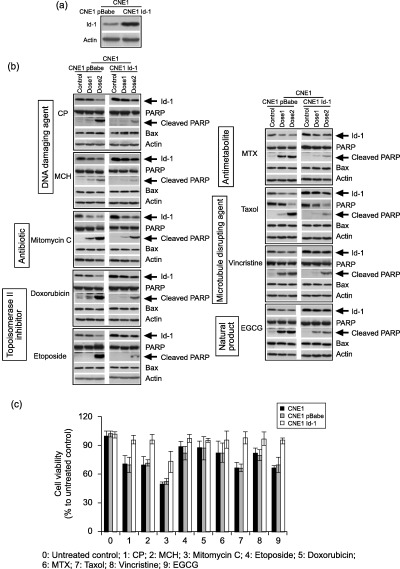
Effect of Id‐1 overexpression on anticancer drug‐induced apoptosis. (a) Differential Id‐1 expression between CNE1 pBabe and CNE1 Id‐1 transfectants cultured in serum free medium. Stable transfectants were generated using an Id‐1 expression vector as described. (b) Western blotting analysis of Id‐1, PARP and Bax expression in response to anticancer drugs after 24 h treatment in the cells with high and low levels of Id‐1 protein. (c) Cell viability of CNE1, CNE1 pBabe and CNE1 Id‐1, after exposure to six types of anticancer drugs (dose3) for 24 h, examined by 3‐(4, 5‐dimethyl thiazol‐2‐yl)‐2, 5‐diphenyl tetrazolium bromide (MTT) assay. CP, cisplatin; EGCG, epigallocatechin gallate; MCH, mechlorethamine; MTX, methotrexate.
Down‐regulation of Id‐1 leads to increased sensitivity to anticancer drug‐induced apoptosis. To further investigate the possibility of using Id‐1 as a target to increase chemosensitivity in human cancer cells, we then suppressed Id‐1 expression in CNE1 cells through small RNA interference and studied the effect of Id‐1 suppression on anticancer drug‐induced apoptosis. As shown in Fig. 5(a), Id‐1 expression was much lower in the si‐Id‐1 transfectants (up to an 80% decrease) compared to the control cells, indicating a successful inhibition of Id‐1 expression. Western blotting analysis showed that after exposure to nine anticancer drugs, respectively, the differential Id‐1 expression between the si‐Id‐1 and the control cells was even greater in a dose‐dependent manner (Fig. 5b). The decreased Id‐1 protein expression was associated with higher levels of cleaved PARP in the si‐Id‐1‐1 cells (Fig. 5b; arrows) as well as an increased percentage of TUNEL positive cells (data not shown). These results were confirmed in the cells transfected with another Si‐Id‐1 sequence (Si‐Id‐1‐2, Fig. 5c,d). These results further support the negative role of Id‐1 in anticancer drug‐induced apoptosis and suggest inactivation of Id‐1 might be a target to induce chemosensitization to a variety of anticancer drugs.
Figure 5.
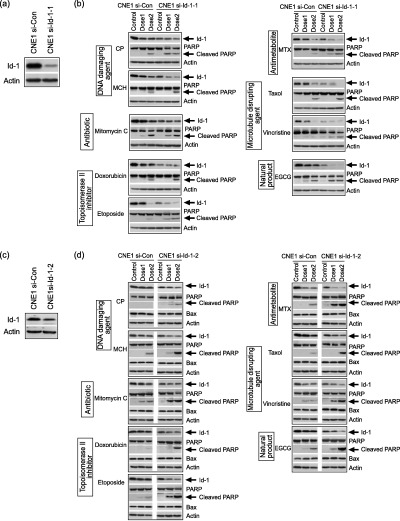
Effect of Id‐1 inactivation on anticancer drug‐induced apoptosis. (a), (c) Differential Id‐1 expression between CNE1 cells expressing high and low levels of Id‐1. Two Si‐RNA sequences targeted to Id‐1 (Si‐Id‐1; Si‐Id‐1‐2) and control sequence were transfected with LipofectamineTM 2000 in CNE1 cells, respectively. (b), (d) Western blotting analysis of Id‐1, PARP and Bax expression in response to anticancer drugs for 24 h.
Discussion
Overexpression of Id‐1 has been reported in over 20 types of human cancer,( 27 ) and its expression levels have been associated with tumor progression, poor prognosis and shorter survival.( 5 , 6 , 7 , 8 , 9 , 10 ) In this study, we demonstrated that the presence of high levels of Id‐1 protein in cancer cells is able to provide protection against activation of the apoptosis pathway by nine anticancer drugs. Since these agents induce cancer cell death through at least six different pathways, it is possible that Id‐1 might be a common mediator of anticancer drug‐induced apoptosis. Increased Id‐1 expression in cancer cells might serve as a universal antiapoptotic factor leading to promotion of cell survival. In addition, our results that down‐regulation of Id‐1 leads to increased sensitivity to chemodrug‐induced apoptosis implicate a novel molecular therapeutic target for inducing chemosensitization in human cancer cells.
As discussed, overexpression of Id‐1 has been reported to confer resistance to certain anticancer drugs such as taxol,( 14 ) TNF‐α,( 15 ) and doxorubicin.( 17 ) However, the present study has provided the first evidence to demonstrate that the antiapoptotic effect of Id‐1 was observed not only in a variety of cancer types, but also against six types of anticancer drugs. More importantly, our results could provide an explanation for the previous clinical studies on nasopharyngeal carcinoma, cervical carcinoma, breast cancer, hepatocarcinoma and prostate cancer as to why overexpression of Id‐1 is correlated with disease progression in cancer patients.( 3 , 5 , 7 , 20 , 21 ) In addition, our results also suggest a possible mechanism underlying the association of Id‐1 with poor prognosis and shorter survival reported in several types of human cancer patients.( 5 , 6 , 7 , 8 , 9 , 10 ) Targeting genes that are essential for cell survival has always been a key interest in the field of drug development. In this study, our results that suppression of Id‐1 by small RNA interference led to increased sensitivity to nine anticancer drug‐induced apoptosis (Fig. 5), suggest that down‐regulation of Id‐1 could be a therapeutic strategy to induce chemosensitization of conventional anticancer drugs in advanced cancers.
It has been well established that the p53‐dependent pathways play a key role in mediating anticancer drug‐induced apoptotic cell death.( 28 ) Although a majority of studies suggest that the oncogenic role of Id‐1 is mainly mediated through p16INK4a/RB pathway,( 1 , 2 ) recently it was reported that mutational inactivation of p53 in a prostate cancer cell line, LNCaP, was able to induce Id‐1 expression that could contribute to its increased proliferative potential.( 29 ) However, in this study, although the five cancer cell lines examined contained differential p53 status from a functional wild type p53 (MCF7, HeLa),( 30 , 31 ) a mutated p53 gene (CNE1, Huh7),( 32 , 33 ) to a deleted p53 (PC3),( 34 ) a consistent negative correlation between Id‐1 expression and drug‐induced apoptosis was observed. This suggests that the Id‐1 mediated antiapoptotic effect could be independent of a p53 pathway. Interestingly however, the basal levels of Id‐1 protein seemed to be higher in the p53 deleted PC3 cell line compared to the lines with a wild type p53 (Fig. 1). Although it is far from clear how Id‐1 interacts with the apoptosis pathway in response to multiple extracellular stresses, or the molecular mechanisms responsible for its antiapoptotic effect, it is possible that it could be regulated through several signaling pathways. This hypothesis is strongly supported by the results demonstrated in this study that down‐regulation of Id‐1 was universally observed in response to a broad spectrum of anticancer drugs, which induce cancer cell death through diverse pathways from directly binding to DNA to microtubule disruption. In additional support to this argument, previous reports also showed that the Id‐1‐induced protection against apoptosis was mediated through various pathways including the NF‐κB, c‐Jun N‐terminal kinase (JNK) and Raf/mitogen‐activated protein kinase kinase (MEK) pathways.( 14 , 15 , 16 ) With these lines of evidence in mind and taking into account our new results, it is possible that like Bcl‐2, Id‐1 might be a novel universal survival factor whose main function is to protect cancer cells from extracellular stress‐induced apoptosis. Overexpression of Id‐1 in cancer cells could provide a survival advantage in response to chemodrug‐induced apoptosis, therefore promoting drug resistance leading to cancer progression. Currently, we are investigating how Id‐1 regulates the apoptosis cascade and what role it plays in mediating cellular responses to a variety of anticancer drugs. In conclusion, the current study demonstrates that Id‐1 is a novel universal antiapoptotic factor, and provides the first evidence to suggest a therapeutic target for the management of advanced cancers.
Supporting information
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
This work was supported by Research Grant Council (Hong Kong) grants to X. H. Wang (HKU7478/03M) and Y. C. Wong (HKU 7314/01M, HKU7490/03M and 7470/04M).
References
- 1. Alani RM, Hasskarl J, Grace M, Hernandez MC, Israel MA, Munger K. Immortalization of primary human keratinocytes by the helix‐loop‐helix protein, Id‐1. Proc Natl Acad Sci USA 1999; 96: 9637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ohtani N, Zebedee Z, Huot TJ et al. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001; 409: 1067–70. [DOI] [PubMed] [Google Scholar]
- 3. Lin CQ, Singh J, Murata K et al. A role for Id‐1 in the aggressive phenotype and steroid hormone response of human breast cancer cells. Cancer Res 2000; 60: 1332–40. [PubMed] [Google Scholar]
- 4. Maruyama H, Kleeff J, Wildi S et al. Id‐1 and Id‐2 are overexpressed in pancreatic cancer and in dysplastic lesions in chronic pancreatitis. Am J Pathol 1999; 155: 815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schindl M, Oberhuber G, Obermair A, Schoppmann SF, Karner B, Birner P. Overexpression of Id‐1 protein is a marker for unfavorable prognosis in early‐stage cervical cancer. Cancer Res 2001; 61: 5703–6. [PubMed] [Google Scholar]
- 6. Langlands K, Down GA, Kealey T. Id proteins are dynamically expressed in normal epidermis and dysregulated in squamous cell carcinoma. Cancer Res 2000; 60: 5929–33. [PubMed] [Google Scholar]
- 7. Ouyang XS, Wang X, Lee DT, Tsao SW, Wong YC. Over expression of ID‐1 in prostate cancer. J Urol 2002; 167: 2598–602. [PubMed] [Google Scholar]
- 8. Schindl M, Schoppmann SF, Strobel T et al. Level of Id‐1 protein expression correlates with poor differentiation, enhanced malignant potential, and more aggressive clinical behavior of epithelial ovarian tumors. Clin Cancer Res 2003; 9: 779–85. [PubMed] [Google Scholar]
- 9. Straume O, Akslen LA. Strong expression of ID1 protein is associated with decreased survival, increased expression of ephrin‐A1/EPHA2, and reduced thrombospondin‐1 in malignant melanoma. Br J Cancer 2005; 93: 933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schoppmann SF, Schindl M, Bayer G et al. Overexpression of Id‐1 is associated with poor clinical outcome in node negative breast cancer. Int J Cancer 2003; 104: 677–82. [DOI] [PubMed] [Google Scholar]
- 11. Borlak J, Meier T, Halter R, Spanel R, Spanel‐Borowski K. Epidermal growth factor‐induced hepatocellular carcinoma: gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene 2005; 24: 1809–19. [DOI] [PubMed] [Google Scholar]
- 12. Fong S, Itahana Y, Sumida T et al. Id‐1 as a molecular target in therapy for breast cancer cell invasion and metastasis. Proc Natl Acad Sci USA 2003; 100: 13 543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ouyang XS, Wang X, Lee DT, Tsao SW, Wong YC. Up‐regulation of TRPM‐2, MMP‐7 and ID‐1 during sex hormone‐induced prostate carcinogenesis in the Noble rat. Carcinogenesis 2001; 22: 965–73. [DOI] [PubMed] [Google Scholar]
- 14. Zhang X, Ling MT, Wang X, Wong YC. Inactivation of Id‐1 in prostate cancer cells: a potential therapeutic target in inducing chemosensitization to taxol through activation of JNK pathway. Int J Cancer 2006; 118: 2072–81. [DOI] [PubMed] [Google Scholar]
- 15. Ling MT, Wang X, Ouyang XS, Xu K, Tsao SW, Wong YC. Id‐1 expression promotes cell survival through activation of NF‐κB signalling pathway in prostate cancer cells. Oncogene 2003; 22: 4498–508. [DOI] [PubMed] [Google Scholar]
- 16. Cheung HW, Ling MT, Tsao SW, Wong YC, Wang X. Id‐1‐induced Raf/MEK pathway activation is essential for its protective role against taxol‐induced apoptosis in nasopharyngeal carcinoma cells. Carcinogenesis 2004; 25: 881–7. [DOI] [PubMed] [Google Scholar]
- 17. Lin JC, Chang SY, Hsieh DS, Lee CF, Yu DS. Modulation of mitogen‐activated protein kinase cascades by differentiation‐1 protein: acquired drug resistance of hormone independent prostate cancer cells. J Urol 2005; 174: 2022–6. [DOI] [PubMed] [Google Scholar]
- 18. Jang TJ, Jung KH, Choi EA. Id‐1 gene downregulation by sulindac sulfide and its upregulation during tumor development in gastric cancer. Int J Cancer 2006; 118: 1356–63. [DOI] [PubMed] [Google Scholar]
- 19. Zhang H, Rosdahl I. Expression profiles of Id1 and p16 proteins in all‐trans‐retinoic acid‐induced apoptosis and cell cycle re‐distribution in melanoma. Cancer Lett 2005; 217: 33–41. [DOI] [PubMed] [Google Scholar]
- 20. Matsuda Y, Yamagiwa S, Takamura M et al. Overexpressed Id‐1 is associated with a high risk of hepatocellular carcinoma development in patients with cirrhosis without transcriptional repression of p16. Cancer 2005; 104: 1037–44. [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Xu K, Ling MT et al. Evidence of increased Id‐1 expression and its role in cell proliferation in nasopharyngeal carcinoma cells. Mol Carcinog 2002; 35: 42–9. [DOI] [PubMed] [Google Scholar]
- 22. Zhang S, Wu Y, Zeng Y, Zech L, Klein G. Cytogenetic studies on an epithelioid cell line derived from nasopharyngeal carcinoma. Hereditas 1982; 97: 23–8. [DOI] [PubMed] [Google Scholar]
- 23. Ouyang XS, Wang X, Ling MT, Wong HL, Tsao SW, Wong YC. Id‐1 stimulates serum independent prostate cancer cell proliferation through inactivation of p16 (INK4a)/pRB pathway. Carcinogenesis 2002; 23: 721–5. [DOI] [PubMed] [Google Scholar]
- 24. Matejka GL, Thornemo M, Kernholt A, Lindahl A. Expression of Id‐1 mRNA and protein in the post‐ischemic regenerating rat kidney. Exp Nephrol 1998; 6: 253–64. [DOI] [PubMed] [Google Scholar]
- 25. Shimizu M, Deguchi A, Lim JT, Moriwaki H, Kopelovich L, Weinstein IB. (‐)‐Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor‐2 signaling pathways in human colon cancer cells. Clin Cancer Res 2005; 11: 2735–46. [DOI] [PubMed] [Google Scholar]
- 26. Chou AH, Tsai HF, Wu YY et al. Hepatitis C virus core protein modulates TRAIL‐mediated apoptosis by enhancing Bid cleavage and activation of mitochondria apoptosis signaling pathway. J Immunol 2005; 174: 2160–6. [DOI] [PubMed] [Google Scholar]
- 27. Wong YC, Wang X, Ling MT. Id‐1 expression and cell survival. Apoptosis 2004; 9: 279–89. [DOI] [PubMed] [Google Scholar]
- 28. Neubauer A, Thiede C, Huhn D, Wittig B. P53 and induction of apoptosis as a target for anticancer therapy. Leukemia 1996; 10 (Suppl 3): S2–4. [PubMed] [Google Scholar]
- 29. Tepper CG, Gregg JP, Shi XB et al. Profiling of gene expression changes caused by p53 gain‐of‐function mutant alleles in prostate cancer cells. Prostate 2005; 65: 375–89. [DOI] [PubMed] [Google Scholar]
- 30. Vayssade M, Haddada H, Faridoni‐Laurens L et al. P73 functionally replaces p53 in Adriamycin‐treated, p53‐deficient breast cancer cells. Int J Cancer 2005; 116: 860–9. [DOI] [PubMed] [Google Scholar]
- 31. Charlot JF, Nicolier M, Pretet JL, Mougin C. Modulation of p53 transcriptional activity by PRIMA‐1 and Pifithrin‐alpha on staurosporine‐induced apoptosis of wild‐type and mutated p53 epithelial cells. Apoptosis 2006; 11: 813–27. [DOI] [PubMed] [Google Scholar]
- 32. Qi V, Weinrib L, Ma N, Li JH, Klamut H, Liu FF. Adenoviral p53 gene therapy promotes heat‐induced apoptosis in a nasopharyngeal carcinoma cell line. Int J Hyperthermia 2001; 17: 38–47. [DOI] [PubMed] [Google Scholar]
- 33. Chi TY, Chen GG, Lai PB. Eicosapentaenoic acid induces Fas‐mediated apoptosis through a p53‐dependent pathway in hepatoma cells. Cancer J 2004; 10: 190–200. [DOI] [PubMed] [Google Scholar]
- 34. Zhao R, Xiang N, Domann FE, Zhong W. Expression of p53 enhances selenite‐induced superoxide production and apoptosis in human prostate cancer cells. Cancer Res 2006; 66: 2296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item