

Abstract
Denosumab, a fully human monoclonal antibody to receptor activator of nuclear factor–kappa B ligand (RANKL), suppresses bone resorption. This open‐label, multicenter, phase 1 study evaluated the safety, pharmacodynamics, and pharmacokinetics of denosumab in Japanese women with breast cancer–related bone metastases. Patients (n = 18; median age, 57 years) received a single subcutaneous injection of denosumab 60 mg or 180 mg or three doses of denosumab 180 mg on days 1, 29, and 57 (every 4 weeks) and were followed for ≥ 141 days. No major safety concerns related to denosumab were noted in any cohort. All patients experienced at least 1 adverse event (AE); most were mild (grade ≤ 2). One patient reported grade 4 myositis and grade 3 anemia, malaise, and dysphagia that the investigator deemed treatment‐related; other treatment‐related AE were grade ≤ 2. No antidenosumab antibodies or clinically significant changes in laboratory findings, vital signs, or electrocardiograms were observed. Pharmacokinetics were approximately dose‐linear. Denosumab caused rapid, substantial, and sustained suppression of urinary N‐telopeptide corrected for creatinine (uNTx/Cr) across all doses; at day 85, the median change from baseline uNTx/Cr ranged from –61.9% to –90.8%. No dose‐limiting toxicity was observed at any dosage. Coupled with pharmacokinetic and pharmacodynamic data, these results were consistent with those observed in non‐Japanese populations. (Cancer Sci 2008; 99: 1237–1242)
Bone is a common site of metastasis in breast cancer. An estimated 75% of women with advanced breast cancer will develop bone metastases,( 1 , 2 , 3 ) which are characterized by pain, fracturing, and spinal cord compression that cause morbidity for many patients. Receptor activator of nuclear factor–kappa B ligand (RANKL) is a key mediator of the ‘vicious cycle’ of bone destruction in metastatic cancer. RANKL is a critical mediator of osteoclast differentiation, function, and survival.( 4 , 5 , 6 ) Within the bone microenvironment, tumor cells secrete factors that stimulate stromal cells and osteoblasts to express and secrete RANKL, which binds to its cognate receptor RANK on the surface of precursor and mature osteoclasts. Osteoclast‐mediated bone resorption releases growth factors that further stimulate tumor growth, resulting in a propagation of bone destruction and tumor cell proliferation.( 7 ) RANKL has recently been shown to promote migration of RANK‐expressing tumor cells to bone.( 8 )
Patients with bone metastases often have increased bone turnover that can be measured using biochemical markers of bone resorption and formation, such as urinary N‐telopeptide (uNTx) and bone‐specific alkaline phosphatase (BSAP). Elevated levels of bone turnover markers are correlated with an increased risk of skeletal complications, disease progression, and death.( 9 , 10 , 11 , 12 ) A key objective in the management of bone metastases is to minimize skeletal morbidity by re‐establishing the homeostasis of bone metabolism. If excessive osteolysis is inhibited, skeletal complications caused by bone metastases may be prevented or delayed.
Denosumab is a fully human monoclonal antibody that binds and inhibits RANKL, thus inhibiting osteoclast‐mediated bone destruction. Results from clinical trials in non‐Japanese women with breast cancer‐related bone metastases showed that denosumab suppressed bone turnover, and the incidence of adverse events was similar in the denosumab and control groups.( 2 , 13 ) The objectives of this trial were to evaluate the safety, pharmacokinetics, and pharmacodynamics of denosumab in Japanese women with bone metastases associated with breast cancer and to compare the results of this trial with those from a similar study in an analogous population of non‐Japanese women (NCT00091832, Clinical Trials.gov).( 2 , 13 , 14 )
Materials and Methods
This study was conducted according to the principles of the Japanese Ministry of Health, Labour, and Welfare, and the International Conference on Harmonisation regulations and guidelines. Institutional Review Boards at each clinical site approved the protocol and all amendments. An Efficacy and Safety Evaluation Committee monitored patient safety during the study as needed. Patients provided appropriate written informed consent.
Study design. In this phase 1 open‐label, multicenter, dose‐ascending single, and multiple dose study, patients were sequentially enrolled in one of three cohorts. Patients in the first cohort received a single 60‐mg subcutaneous injection of denosumab. If no safety signals were observed in the first cohort after 8–10 days, patients were enrolled in the second cohort and received a single 180‐mg subcutaneous injection of denosumab. After an 8‐ to 10‐day period for observation of safety of the second dose, patients were enrolled in the third cohort and received three 180‐mg subcutaneous injections of denosumab at 4‐week intervals (Q4W) on days 1, 29, and 57. Doses were chosen to be comparable with those administered in a study in non‐Japanese women with breast cancer and bone metastases.( 2 ) Although no formal stopping rules were specified in the protocol, safety signals that were considered when making dose escalation decisions included adverse events (AE), vital signs, and serum chemistry and hematology values.
Endpoints. The primary endpoint of the study was the subject incidence of AE, including physical findings, changes in laboratory values, vital signs, and 12‐lead electrocardiogram (ECG) data. Adverse events were classified using the Common Terminology Criteria for Adverse Events version 3.0. Secondary endpoints included serum denosumab levels (pharmacokinetics); the maximum observed serum concentration (Cmax), time at which Cmax was reached (Tmax); the area under the concentration‐time curve (AUC0‐t); the accumulation ratio (AR, for cohort 3 only), the beta‐phase half‐life (t1/2,β, for cohorts 1 and 2 only) after administration of the first dose (pharmacokinetics); the presence of serum antidenosumab antibodies; and the percent change in the bone turnover marker urinary N‐telopeptide corrected for creatinine (uNTx/Cr) from baseline to study day 85 (pharmacodynamics). Exploratory endpoints included pharmacokinetic parameter estimates following the last dose; the percent change in uNTx at study day 141 in cohort 3; the percent change in additional bone turnover markers (serum type I collagen cross‐link C telopeptide [sCTx], bone‐specific alkaline phosphatase, and osteocalcin) from baseline at study day 85 (all cohorts) and day 141 (cohort 3 only); and the proportion of patients experiencing skeletal‐related events (SRE), defined as bone fracture, surgery or radiation therapy to bone, and spinal cord compression.
Patient eligibility. Patients were non‐pregnant Japanese women with histologically or cytologically confirmed breast cancer and radiographic evidence of at least one bone metastasis, enrolled at three centers in Japan. Eligible patients were 20–74 years of age, with an Eastern Cooperative Oncology Group (ECOG) performance status (PS) ≤ 2 and adequate organ function. Concurrent chemotherapy or hormonal therapy was allowed as long as the regimen did not change within 13 days before or after administration of the denosumab dose. Patients with prior SRE were eligible to participate in the study except for those who had evidence of an impending fracture in weight‐bearing bones; major surgery to bone within 4 weeks before the first dose of denosumab; radiation therapy to bone within 2 weeks before the first dose of denosumab; or treatment with radioisotopes directed to bone within 8 weeks before the first dose of denosumab. Other exclusion criteria included cytotoxic chemotherapy within 13 days before denosumab administration; unresolved toxicities > grade 2 from previous chemotherapy regimens; central nervous system metastasis that was symptomatic or required treatment; or prior administration of osteoprotegerin or denosumab or administration of calcitonin, parathyroid hormone‐related peptides, mithramycin, gallium nitrate, or strontium ranelate within 6 months; or systemic corticosteroid treatment during the study. Patients were also excluded if they reported or had evidence of disorders that could affect bone metabolism; prior malignancies (excluding the targeted breast cancer, basal cell carcinoma, or cervical cancer in situ) within 3 years; uncontrolled systemic disease; major surgery or traumatic injury within 4 weeks; HIV infection; bisphosphonate use within 4 weeks of the first dose of denosumab; or an organic or psychiatric disorder that might prevent the patient from completing the study.
Study procedures. Patients in the 60 mg and 180 mg single‐dose cohorts received a single subcutaneous injection of denosumab in the anterior abdominal wall on study day 1. These patients were scheduled to have a total of nine study visits (study days 1, 2, 4, 8, 15, 22, 29, 57, and 85). Patients in the 180 mg Q4W cohort received a subcutaneous injection in the anterior abdominal wall on study days 1, 28, and 57. Patients in this cohort were scheduled to have a total of 12 study visits (study days 1, 8, 15, 22, 29, 57, 64, 71, 78, 85, 113, and 141). Screening assessments included medical and medication histories, physical examination including height and weight measurements, assessment of ECOG PS, hematology, serum chemistry, urinalysis, pregnancy tests, and spinal X‐ray imaging. Existing SRE were noted. Throughout the study, physical examinations, monitoring of vital signs, weight measurements, electrocardiograms, and collection of urine and blood were performed periodically. Adverse events, laboratory values, and concomitant medications were recorded and assessed at all study visits. Patients did not receive calcium or vitamin D supplementation in this study.
Statistics and data analysis. The study planned to enroll six patients in each of three treatment cohorts for a total sample size of 18 patients. The safety analysis subset included all patients who received at least 1 dose of denosumab. The pharmacokinetic (PK) analysis subset included all patients in the safety subset who had an evaluable serum denosumab concentration‐time profile. Pharmacodynamic evaluations were conducted among patients in the safety subset. Data were reviewed for safety before each dose escalation. Demographics and other baseline characteristics (values obtained 1 week before the first dose administration) were summarized using descriptive statistics. For continuous data, descriptive statistics included mean, median, standard deviation or standard error; and number of subjects, minimum, and maximum. Frequencies and percentages were presented for nominal categorical variables, including the number and percent of subjects.
Results
Patients. The first patient was enrolled on 22 November 2004 and the last patient visit occurred on 26 October 2005. A total of 19 patients were enrolled, including six in each cohort; a seventh patient in the 180 mg single‐dose cohort withdrew at the physician's discretion before receiving denosumab because of disease progression; this patient was not included in any results. All patients were Japanese women. The overall median age was 57 years (range, 28–67 years) (Table 1). Patients in the 60 mg and 180 mg Q4W cohorts were of similar ages; patients in the 180 mg single‐dose cohort had a median age of 47 years (range, 28–61 years). All women had an ECOG PS of 0 or 1. The median time since the original diagnosis was 6.2 years (range, 0.1–19.1), and the median time since the diagnosis of bone metastasis was 0.31 years (range, 0–5.6). Prior to the study, eight patients (44%) had never experienced an SRE, six patients (33%) had experienced only one SRE, and four patients (22%) had experienced two or more SRE (Table 1).
Table 1.
Baseline patient demographics and disease characteristics
| 60 mg SC (single dose) (n = 6) | 180 mg SC (single dose) (n = 6) | 180 mg Q4W (3 doses) (n = 6) | Total (n = 18) | |
|---|---|---|---|---|
| Sex – (%) | ||||
| Female | 6 (100) | 6 (100) | 6 (100) | 18 (100) |
| Race –n (%) | ||||
| Japanese | 6 (100) | 6 (100) | 6 (100) | 18 (100) |
| Age – years | ||||
| Median (min, max) | 58 (52, 66) | 47 (28, 61) | 60 (47, 67) | 57 (28, 67) |
| ECOG PS –n (%) | ||||
| 0 | 3 (50) | 3 (50) | 4 (67) | 10 (56) |
| 1 | 3 (50) | 3 (50) | 2 (33) | 8 (44) |
| Hormone receptor status –n (%) † | ||||
| Negative | 4 (67) | 0 (0) | 0 (0) | 4 (22) |
| Positive | 2 (33) | 5 (83) | 6 (100) | 13 (72) |
| Unknown | 0 (0) | 1 (17) | 0 (0) | 1 (6) |
| Time since original diagnosis – years | ||||
| Median (min, max) | 6.4 (0.9, 10.4) | 3.3 (0.1, 12.8) | 7.4 (1.6, 19.0) | 6.2 (0.1, 19.0) |
| Time since bone metastases – years | ||||
| Median (min, max) | 0.38 (0.0, 3.8) | 0.32 (0.1, 2.5) | 0.25 (0.1, 5.6) | 0.31 (0.0, 5.6) |
| Total number of previous SRE –n (%) | ||||
| 0 | 1 (17) | 4 (67) | 3 (50) | 8 (44) |
| 1 | 3 (50) | 1 (17) | 2 (33) | 6 (33) |
| 2 | 1 (17) | 1 (17) | 0 (0) | 2 (11) |
| 3 | 1 (17) | 0 (0) | 0 (0) | 1 (6) |
| >3 | 0 (0) | 0 (0) | 1 (17) | 1 (6) |
ECOG, Eastern Cooperative Oncology Group; PS, performance status; Q4W, every 4 weeks; SC, subcutaneous; SRE, skeletal‐related events.
Tumors were screened for expression of the estrogen receptor (ER) or progesterone receptor (PR).
Safety
Adverse events. No deaths occurred during the study, and no patients withdrew from the study because of AE. All 18 patients who received at least one dose of denosumab experienced at least one AE during the study, most of which were mild. The most common AE were fatigue, anorexia, headache, malaise, and nausea (Table 2).
Table 2.
Adverse events summary
| Event –n (%) (patients) | 60 mg SC (single dose) (n = 6) | 180 mg SC (single dose) (n = 6) | 180 mg Q4W (3 doses) (n = 6) | Total (n = 18) |
|---|---|---|---|---|
| All AE | 6 (100) | 6 (100) | 6 (100) | 6 (100) |
| Serious AE | 0 (0) | 2 (33) † | 0 (0) | 2 (11) |
| Treatment‐related AE | 3 (50) | 3 (50) | 3 (50) | 9 (50) |
| Serious treatment‐related AE | 0 (0) | 1 (16.7) ‡ | 0 (0) | 1 (5.6) |
| Deaths on study | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Adverse events occurring in 2 or more patients | ||||
| Fatigue | 2 (33) | 1 (17) | 2 (33) | 5 (28) |
| Anorexia | 2 (33) | 1 (17) | 1 (17) | 4 (22) |
| Headache | 2 (33) | 0 (0) | 2 (33) | 4 (22) |
| Malaise | 1 (17) | 3 (50) | 0 (0) | 4 (22) |
| Nausea | 1 (17) | 2 (33) | 1 (17) | 4 (22) |
| Arthralgia | 2 (33) | 0 (0) | 1 (17) | 3 (17) |
| Constipation | 1 (17) | 1 (17) | 1 (17) | 3 (17) |
| Diarrhea | 2 (33) | 0 (0) | 1 (17) | 3 (17) |
| Metastases to bone | 1 (17) | 1 (17) | 1 (17) | 3 (17) |
| Edema | 1 (17) | 1 (17) | 1 (17) | 3 (17) |
| Shoulder pain | 1 (17) | 1 (17) | 1 (17) | 3 (17) |
| Stomatitis | 2 (33) | 1 (17) | 0 (0) | 3 (17) |
| Alopecia | 2 (33) | 0 (0) | 0 (0) | 2 (11) |
| Chest pain | 1 (17) | 1 (17) | 0 (0) | 2 (11) |
| Hot flush | 0 (0) | 1 (17) | 1 (17) | 2 (11) |
| Hypoesthesia | 0 (0) | 0 (0) | 2 (33) | 2 (11) |
| Hypocalcemia | 1 (17) | 1 (17) | 0 (0) | 2 (11) |
| Insomnia | 0 (0) | 2 (33) | 0 (0) | 2 (11) |
| Metastases to liver | 0 (0) | 1 (17) | 1 (17) | 2 (11) |
| Nasopharyngitis | 1 (17) | 1 (17) | 0 (0) | 2 (11) |
| Neutrophil count decreased | 2 (33) | 0 (0) | 0 (0) | 2 (11) |
| Pain in extremity | 1 (17) | 1 (17) | 0 (0) | 2 (11) |
| White blood cell count decreased | 2 (33) | 0 (0) | 0 (0) | 2 (11) |
Includes 1 patient with grade 4 myositis and 1 patient with grade 3 febrile neutropenia.
Includes 1 patient with grade 4 myositis.
AE, adverse events; SC, subcutaneous.
Two patients, both in the 180 mg single‐dose group, reported serious AE (Table 2). One of these patients reported grade 4 myositis and grade 3 anemia, dysphagia, and malaise that were deemed by the investigator to be treatment‐related; she also experienced grade 4 metastatic brain cancer and depression and grade 3 herpes zoster infection and liver disorder that were not treatment‐related. The other patient experienced febrile neutropenia (absolute neutrophil count < 1.0 ¥ 109/L, temperature 38.5C), which was not deemed treatment‐related and was resolved with outpatient treatment. Two patients experienced mild, asymptomatic hypocalcemia (grades 1 and 2) that was deemed treatment‐related. Nine patients reported other grade 1 or 2 AE that were deemed by investigators to be treatment‐related (blurred vision, nausea, chest pain, fatigue, decreased white blood cell count, hyperkalemia, arthropathy, muscle spasms, pain in extremity, hypoesthesia, seborrheic dermatitis, and hot flush). No SRE occurred during the study.
Laboratory findings, vital signs, and ECG results. No clinically significant changes were observed in laboratory findings or vital signs except for the anemia, neutropenia, and liver disorder described above. Abnormal findings in ECGs were observed in four patients in the 60 mg cohort after dosing with denosumab. The findings included transient sinus tachycardia, supraventricular extrasystole, transient ST elevation; one patient with previously documented atrioventricular (AV) block experienced transient first‐degree AV block. None of these were considered as AE although the AV block was considered clinically significant by the investigator. No antidenosumab antibodies were observed.
Pharmacokinetics. Denosumab demonstrated approximately dose‐linear pharmacokinetics over the dose range investigated. Absorption of denosumab appeared to be rapid, with maximal exposures observed in median times of 8–10 days after a single dose and 14–18 days after Q4W dosing. In the 180 mg Q4W cohort, an approximate 2.2‐fold accumulation was observed from the first dose to the third dose. The serum denosumab concentration increased in an approximately dose‐proportional manner, with a four‐fold increase in mean Cmax and 3.8‐fold increase in AUC0‐t ‐values (Table 3). Serum concentration‐time profiles were biphasic, with an absorption phase and a beta phase that immediately followed peak concentrations. Mean half‐life values associated with the beta phase (t1/2,β) were comparable between the two single‐dose cohorts (25–29 days) (Fig. 1).
Table 3.
Pharmacokinetic parameters following denosumab administration
| Dose | Number of doses | Cmax mean (SD) (µg/L) | Tmax median (range) (day) | AUC0‐t mean (SD) (µgday/L) | t1/2, β mean (SD) (day) |
|---|---|---|---|---|---|
| 60 mg | Single | 7.73 (3.13) | 8.0 (7.0–28) | 351.0 (144.0) | 24.7 (2.44) |
| 180 mg | Single | 31.10 (14.9) | 10 (4.0–28) | 1320.0 (640.0) | 29.1 (7.15) |
| 180 mg Q4W | 1 | 24.10 (5.13) | 18 (7.0–28) | 545.0 (123.0) | NA |
| 3 | 48.0 (9.34) | 14 (7.0–21) | 1210.0 (240.0) | NA |
Cmax = Maximum observed serum concentration.
Tmax = Time at which Cmax was observed.
AUC0‐t = Area under the concentration‐time curve from time zero to the time of the last observation (which corresponds to AUC0‐84 for cohorts 1 and 2, and AUC0‐28 for cohort 3).
t1/2,β = Beta‐phase half‐life.
NA, not applicable.
Figure 1.
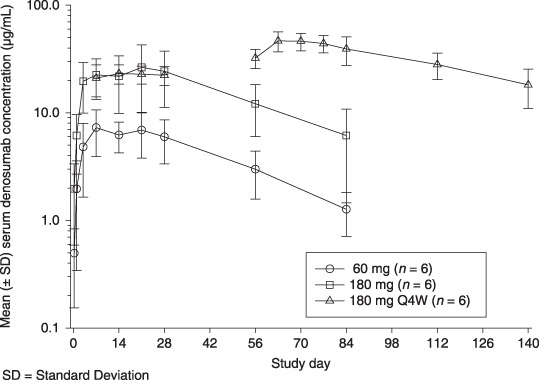
Denosumab demonstrated approximately dose‐linear pharmacokinetics. Serum concentration‐time profiles were biphasic, with an absorption phase and a beta phase that immediately followed peak concentrations. Mean half‐life values associated with the beta phase (t1/2,β) were comparable between the two single‐dose cohorts (25–29 days).
Pharmacodynamics. The suppression of uNTx/Cr was rapid, substantial, and sustained (Table 4). At day 85, the median percent changes from uNTx/Cr values at baseline were –91% (range, –23% to –93%) in the 60 mg single‐dose group, –62% (range, –74% to +54%) in the 180 mg single‐dose group, and –85% (range, –69% to –98%) in the 180 mg Q4W group. These values exclude five patients (two in the 180 mg single‐dose group, three in the 180 mg Q4W group) whose baseline values were below quantifiable limits (BQL). By day 2, the median change in uNTx/Cr was –70% (range, –85% to +10%) in the 60 mg group and –70% (range, –80% to –65%) in the 180 mg single‐dose group. In the 180 mg Q4W group, by week 2 (the first visit after administration of denosumab), uNTx/Cr changed a median of –64% (range, –10% to –96%). At day 141, the median percent change in uNTx/Cr in the 180 mg Q4W group (three patients) was –63% (range, –60% to –96%).
Table 4.
Denosumab effects: Changes from baseline in bone turnover markers
| Bone turnover marker | 60 mg SC (single dose) | 180 mg SC (single dose) | 180 mg Q4W (3 doses) | Total |
|---|---|---|---|---|
| n = 6 | n = 4 | n = 3 | n = 13 | |
| uNTx/Cr (nmol/mmol) – median percentage change (min, max) † | ||||
| Baseline | 109 (19, 233) | 55 (21, 60) | 29 (20, 389) | 60 (19, 389) |
| Day 2 | –70 (–85, 9) | –70 (–80, –65) | – | –69 (–85, 9) |
| Week 2 | –82 (–94, –40) | –75 (–90, –9) | –64 (–96, –10) | –77 (–96, –9) |
| Day 85 | –91 (–93, –23) | –62 (–74, 54) | –85 (–98, –69) | –85 (–98, 54) |
| Day 141 | – | – | –63 (–96, –60) | –63 (–96, –60) |
| sCTx (ng/mL) – median percentage change (min, max) † | n = 6 | n = 6 | n = 4 | n = 16 |
| Baseline | 0.5 (0.2, 1.8) | 0.3 (0.1, 0.9) | 0.2 (0.2, 1.7) | 0.3 (0.1, 1.8) |
| Day 2 | –69 (–83, –54) | –63 (–75, –8) | – | –65 (–83, –8.1) |
| Week 2 | –81 (–93, –69) | –77 (–85, –31) | –77 (–92, –50) | –80 (–93, –31) |
| Day 85 | –89 (–97, –68) | –76 (–84, 206) | –80 (–90, 53) | –82 (–97, 206) |
| Day 141 | – | – | –80 (–93, –62) | –80 (–93, –62) |
| BSAP (U/L) – median percentage change (min, max) | n = 6 | n = 6 | n = 6 | n = 18 |
| Baseline | 30.4 (25.0, 44.3) | 29.8 (17.9, 63.7) | 26.9 (15.3, 114.4) | 29.0 (15.3, 114.4) |
| Day 2 | –0.8 (–7.8, 14.7) | 1.9 (–11.8, 9.5) | – | –0.2 (–11.8, 14.7) |
| Week 2 | 0.4 (–25.4, 12.1) | 3.3 (–23.9, 10.3) | 0.7 (–18.4, 16.5) | 1.7 (–25.4, 16.5) |
| Day 85 | –48.3 (–60.0, 35.6) | –42.9 (–78.5, –9.5) | –34.3 (–63.2, 23.5) | –45.6 (–78.5, 35.6) |
| Day 141 | – | – | –53.4 (–68.5, –12.5) | –53.4 (–68.5, –12.5) |
| Osteocalcin (ug/L) – median percentage change (min, max) | n = 6 | n = 6 | n = 6 | n = 18 |
| Baseline | 13.8 (7.2, 17.6) | 9.6 (4.2, 13.3) | 10.9 (4.1, 30.5) | 12.2 (4.1, 30.5) |
| Day 2 | 13.6 (–5.3, 21.6) | –3.2 (–40.2, 50.0) | – | 1.4 (–40.2, 50.0) |
| Week 2 | 21.1 (9.5, 34.1) | 12.5 (–4.0, 84.4) | 22.9 (–15.9, 79.0) | 18.2 (–15.9, 84.4) |
| Day 85 | –28.0 (–50.3, –21.7) | –31.6 (–57.1, –18.8) | –7.7 (–57.9, 63.9) | –29.1 (–57.9, 63.9) |
| Day 141 | – | – | – 41.7 (–63.5, –1.5) | –41.7 (–63.5, –1.5) |
BSAP, bone‐specific alkaline phosphatase; SC, subcutaneous; sCTx, serum type I collagen cross‐link C‐telopeptide; uNTx/Cr, urinary N‐telopeptide corrected for creatinine.
Patients whose baseline values were below quantifiable limits were excluded from this analysis.
At day 85, the median percent change from baseline sCTx values was –89% (range, –68% to –97%) in the 60 mg group, –76% (range, –84% to +206%) in the 180 mg single‐dose group, and –80% (range, –90% to +53%) in the 180 mg Q4W group (Table 4). These results reflect the exclusion of two patients in the 180 mg Q4W group because of missing data and a baseline BQL value. At day 141, the median percent change in sCTx in the 180 mg Q4W group was –80% (range, –62% to –93%). Effects of denosumab therapy on bone turnover markers are summarized in Table 4.
Comparison with results in non‐Japanese women. In a randomised, phase 2, dose‐ranging study in women with breast cancer (study NCT00091832, Clinical Trials.gov),( 2 ) five denosumab dosing regimens were evaluated in 212 non‐Japanese women with breast cancer and bone metastases who were bisphosphonate‐naive. At comparable dosing levels after excluding patients with baseline BQL levels of uNTx/Cr, results in the current study were similar to those in the non‐Japanese study for the median percent change from baseline for uNTx/Cr (Fig. 2) and the median uNTx/Cr concentrations (Fig. 2b). No marked differences were seen in safety profiles or in serum concentrations or other PK parameters between Japanese and non‐Japanese patients.
Figure 2.
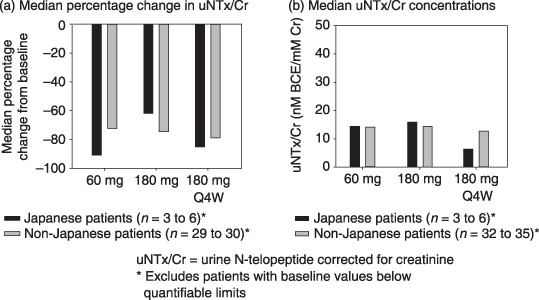
At comparable dosing levels, results of Japanese patients were similar to those of non‐Japanese patients for the median percent change from the baseline in (a) uNTx/Cr and (b) median uNTx/Cr concentrations. Q4W, every 4 weeks; uNTx/Cr, urine N‐telopeptide corrected for creatinine.
Discussion
As expected, the adverse event profile in this study was similar to that observed in advanced cancer patients undergoing systemic therapy, with no dose‐limiting toxicities observed. In a phase 2 study of non‐Japanese women with metastatic breast cancer, denosumab treatment was not associated with any severe or serious treatment‐related adverse events or with any dose‐dependent increase in adverse events.( 2 ) The only grade 4 AE that was deemed treatment‐related by the investigator was myositis, although the investigator considered paraneoplastic syndrome to be the primary possible etiology of the myositis. This patient, who had active metastatic disease, exhibited substantially elevated levels of creatine phosphokinase (CPK) at baseline. She developed a further elevation in CPK levels and proximal muscular weakness with myalgia in the extremities on day 29. The histological findings indicated non‐specific myositis with no apparent evidence of neurogenic change, collagen disorder, or viral infection. The patient was taking three concomitant medications (goserelin acetate, tamoxifen citrate, and loxoprofen sodium) that the investigator considered to be potentially suspect. Because of these factors, it is difficult to establish the role of denosumab in the development of myositis. This event was resolved after treatment with steroids.
The pharmacokinetics of denosumab with respect to dose and time were consistent in this population with results observed in analogous non‐Japanese populations. The range of baseline uNTx/Cr values observed in this study (19–389 nmol/mmol) was within the range observed in the phase 2 study of non‐Japanese patients with breast cancer (5 nmol/mmol to 942 nmol/mmol),( 2 ) and baseline variability among patients was similar in the two studies. Median percent changes in uNTx/Cr were also similar in both Japanese and non‐Japanese populations regardless of the median baseline uNTx/Cr levels. No relationship was observed between baseline uNTx/Cr levels and the median percent change in uNTx/Cr.( 2 ) In this study, the suppression of the bone turnover marker uNTx/Cr was rapid (occurring within 24 h), substantial (≥60%), and sustained (up to 12 weeks). These results are comparable to those seen in non‐Japanese patients, in which median reductions were ≥70%.( 2 , 13 , 14 ) The safety, pharmacokinetic, and pharmacodynamic profiles in denosumab‐treated Japanese women were not markedly different from those in non‐Japanese populations.( 2 , 13 , 14 ) These results demonstrate that the 120 mg Q4W regimen identified in a phase 2 study of non‐Japanese patients( 14 ) is appropriate for Japanese as well as non‐Japanese patients, a conclusion supported by the Japanese investigators in the current study. This study supports further investigation of denosumab for treatment of bone metastases and the prevention and treatment of SREs in Japanese patients with breast cancer. Multiple global phase 3 trials of denosumab are in progress for patients with advanced cancer and bone metastases; underlying malignancies include breast cancer (including Japanese patients), prostate cancer and other solid tumors, and multiple myeloma.
Acknowledgments
This study was sponsored by Amgen Limited. Amy Foreman‐Wykert of Kendle International and Sue Hudson of Medical Writing Associates provided medical writing assistance.
Funding: This study was sponsored by Amgen Limited.
References
- 1. Coleman RE. Skeletal complications of malignancy. Cancer 1997; 80: 1588–94. [DOI] [PubMed] [Google Scholar]
- 2. Lipton A, Steger G, Figueroa J et al . Randomized active‐controlled phase 2 study of denosumab efficacy and safety in patients with breast cancer‐related bone metastases. J Clin Oncol 2007; 25: 4431–7. [DOI] [PubMed] [Google Scholar]
- 3. Palma MA, Body JJ. Usefulness of bone formation markers in breast cancer. Int J Biol Markers 2005; 20: 146–55. [DOI] [PubMed] [Google Scholar]
- 4. Fuller K, Wong B, Fox S, Choi Y, Chambers TJ. TRANCE is necessary and sufficient for osteoblast‐mediated activation of bone resorption in osteoclasts. J Exp Med 1998; 188: 997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lacey DL, Tan HL, Lu J et al . Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol 2000; 157: 435–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lacey DL, Timms E, Tan HL et al . Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998; 93: 165–76. [DOI] [PubMed] [Google Scholar]
- 7. Roodman GD. Mechanisms of bone metastasis. N Engl J Med 2004; 350: 1655–64. [DOI] [PubMed] [Google Scholar]
- 8. Jones DH, Nakashima T, Sanchez OH et al . Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006; 440: 692–6. [DOI] [PubMed] [Google Scholar]
- 9. Brown JE, Cook RJ, Major P et al . Bone turnover markers as predictors of skeletal complications in prostate cancer, lung cancer, and other solid tumors. J Natl Cancer Inst 2005; 97: 59–69. [DOI] [PubMed] [Google Scholar]
- 10. Brown JE, Thomson CS, Ellis SP, Gutcher SA, Purohit OP, Coleman RE. Bone resorption predicts for skeletal complications in metastatic bone disease. Br J Cancer 2003; 89: 2031–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Costa L, Demers LM, Gouveia‐Oliveira A et al . Prospective evaluation of the peptide‐bound collagen type I cross‐links N‐telopeptide and C‐telopeptide in predicting bone metastases status. J Clin Oncol 2002; 20: 850–6. [DOI] [PubMed] [Google Scholar]
- 12. Coleman RE, Major P, Lipton A et al . Predictive value of bone resorption and formation markers in cancer patients with bone metastases receiving the bisphosphonate zoledronic acid. J Clin Oncol 2005; 23: 4925–35. [DOI] [PubMed] [Google Scholar]
- 13. Body JJ, Facon T, Coleman RE et al . study of the biological receptor activator of nuclear factor‐kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res 2006; 12: 1221–8. [DOI] [PubMed] [Google Scholar]
- 14. Peterson MC, Jang G, Kim W et al . Selection of a phase 3 dose regimen for denosumab based on pharmacokinetic (PK), pharmacodynamic (PD), and safety data from multiple subcutaneous (SC) dosing regimens in breast cancer patients (pts) with bone metastases (BM). ASCO Virtual Meeting, ASCO Virtual Meeting, 2006.