

Abstract
A total of 297 resected Japanese non‐small cell lung cancers (74 squamous cell carcinomas and 223 adenocarcinomas) were analyzed to evaluate the validity of the p53 mutation spectrum as a fingerprint for mutagenic substances as etiological factors. Frequencies of G→T transversions in smokers were significantly higher than in non‐smokers (P = 0.003) and the average incidence of G→T at hot spot codons of adduct formation was higher than that in other codons in smokers and in the hot spots in non‐smokers. Further, the mutation showed a marked strand bias. G→A transitions at CpG sites (CpG→CpA) were equally distributed in smokers and non‐smokers, and on both strands. A→G transitions did not show any variation with smoking status in terms of frequency, but exhibited a marked strand bias. Taken together, the G→T may be a fingerprint of direct mutagenic action of tobacco‐related compounds, the A→G being a new marker for other environmental chemicals, while the CpG→CpA may be attributable to endogenous spontaneous mutation, for active in lung carcinogenesis. (Cancer Sci 2008; 99: 287–295)
The p53 tumor suppressor gene plays an important role in prevention of carcinoma development through its apoptotic and cell cycle checkpoint functions.( 1 , 2 ) Mutations of the gene have been widely recognized in many kinds of human tumors, and mutation patterns are considered to offer ‘fingerprints’ for mutagenic substances. The high frequency of G→T transversions in lung cancers has been attributed to the direct mutagenic action of tobacco smoke components, in particular polycyclic aromatic hydrocarbons (PAHs).( 3 , 4 , 5 , 6 , 7 , 8 ) In contrast, most G→A transitions at CpG sites (CpG→CpA) are ascribed to endogenous mechanisms, because they are presumed to arise due to spontaneous hydrolytic deamination of cytosine at methylated CpG sites.( 3 , 9 , 10 , 11 )
However, other notions on the genesis of mutation patterns have recently been presented. Rodin and Rodin have questioned the direct mutagenic action of PAH‐like compounds and have suggested that other factors, such as selection of pre‐existing endogenous mutations by physiological stress aggravated by smoking( 12 ) can better explain the excess of G→T transversions in lung tumors. Two different ideas also exist for the causes of G→T transversions – direct mutagenic action or selection of pre‐existing endogenous mutations – from both in vivo and in vitro studies.( 13 , 14 ) Relationships between p53 mutation patterns and smoking status are critical for judgment of etiological influences.
Many authors have analyzed the International Agency for Research on Cancer (IARC) database for lung cancers to elucidate relationships, but have reached different conclusions. One reason may partly depend on the fact that the database, as described by Hainaut et al. is exclusively a repository for mutations described in peer‐reviewed articles,( 8 ) and some information on smoking status, occupational exposure to known carcinogenic agents, tumor histology, sex, and ethnicity, which confound the relationship between p53 mutation spectra and smoking, is uncertain. As to CpG→CpA transitions, the DNA sequence context may participate in the generation of a mutation pattern, lesions being found to occur predominantly in a 5′‐CGT‐3′ sequence context with activation of benzo[a]pyrene (BaP) in vitro.( 15 ) Adducts such as those formed with alkylating agents are another likely cause of G→A transitions.( 16 , 17 , 18 ) However, to our knowledge there has been no detailed study on the genesis of CpG→CpA transitions using human lung cancers.
It is important to judge whether the p53 mutation patterns in lung cancers (e.g. those involving G→T transversions and CpG→CpA transitions) are induced by direct mutagenic action of inhaled exogenous carcinogens, especially tobacco smoke compounds, or endogenous processes, respectively, for identification of carcinogenic agents and thus, clues to prevention methods.
We have collected a large series of cases of Japanese non‐small cell lung cancers (NSCLCs) with accurate smoking status, undergoing surgery at one hospital that were classified histologically by the same pathologists. The cases were examined for p53 mutation spectra to elucidate relationships, especially for G→T transversions and CpG→CpA transitions, with smoking status to re‐evaluate the validity of fingerprints for mutagenic substances with Japanese NSCLCs. Further, to clarify etiological differences between squamous cell carcinomas (SQCCs) and adenocarcinomas (ADCs) of the lung, a comparison of their mutation spectra was carried out.
Materials and Methods
Tumor samples, clinicopathological data, and smoking history. We examined a large series of 297 Japanese NSCLCs (223 ADCs and 74 SQCCs) that had been consecutively resected from 1989 to 1995 at Cancer Institute Hospital, Tokyo, Japan. All patients analyzed had undergone a potentially curative resection with lobectomy or pneumonectomy, combined with pulmonary hilar and mediastinal systematic lymph node dissection. Out of 223 ADC cases, three received preoperative chemotherapy, and 67 had postoperative therapy (adjuvant chemotherapy for 49, local radiotherapy for 16, and both for two). With the SQCC patients, none had received chemotherapy and radiotherapy before surgery, but 25 underwent postoperative adjuvant chemotherapy. The study population was aged 26–84, with a mean of 62 years, and a mean age of the SQCC cases (66) was higher than that for ADCs (61) (Table 1). The patients were 193 males and 104 females in total, but since the number of females with SQCCs was very small (only five) the figure of 99 ADCs was almost equal to the 124 ADCs in males. Histological diagnosis was carried out on the basis of the 1999 World Health Organization classification of lung tumors by two of the authors (E.T. and Y.I.). The pathological stages (p‐stages) were determined using the Union Internationale Contre le Cancer (UICC) tumor node metastasis staging system, sixth edition. Most cases were in p‐stage I or III, and the same was observed for adenocarcinomas.
Table 1.
p53 mutations and clinicopathological parameters
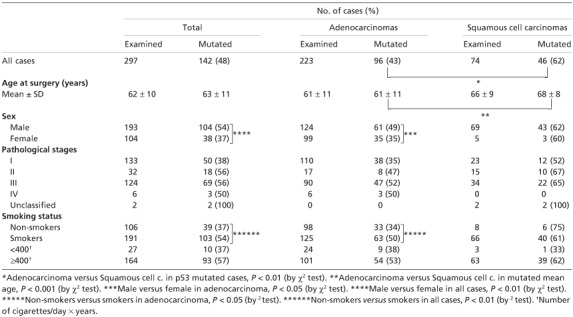
Smoking histories (number of cigarettes per day, starting age, and duration of smoking) were obtained from preoperative personal interviews, with division into never‐smokers and smokers, the latter including both former and current smokers. To evaluate the amount of cigarette consumption, a smoking index (SI) was used: cigarette consumption per day multiplied by smoking years. Referring to the index, smokers were divided into two groups, heavy smokers with indices ≥400, and light ones <400. The percentage of smokers for all cases was 64% (191/297) (87% for males and 22% for females). When looked at by histology, the percentages showed marked differences; 89% (66/74) for SQCCs, compared to 56% (125/223) for ADCs (P < 0.001). Most smokers were heavy smokers, and the rate was higher for SQCCs (95%, 63/66) than for ADCs (81%, 101/125) (P < 0.01).
DNA preparation. Fresh tumor samples were obtained from all patients, quickly frozen in liquid nitrogen, and stored at –80°C until DNA extraction and analysis. Genomic DNAs for SQCC samples were prepared as previously described.( 19 ) For ADCs, the DNAs for part of the tumor samples (the first 124) were prepared as previously described.( 20 ) For the latter part (115) and for 78 samples from the first part that did not show p53 mutations, DNAs were extracted from microdissected tissues. Frozen specimens were cut serially at 25 µm and sections were placed in 99% ethanol. Microdissection was carried out manually under direct observation with a stereoscope for two to four sections stained with Hematoxylin using 18G or 22G needles and the collected tumor cells in 99% ethanol were pelleted by centrifugation at high speed (13 000 g ) for 5 min. DNAs were extracted using a DNA extraction kit (Puregene Kit, Gentra Systems, MN, USA) according to the manufacturer's instructions.
PCR‐SSCP, and Sequencing. Exons 4–8 and 10 of the p53 gene were analyzed in all cases. The polymerase chain reaction (PCR)‐SSCP method and sequencing were carried out for SQCCs and the first half of ADCs with the primers and PCR conditions described previously.( 20 , 21 ) For the second half of the ADCs and those that did not show p53 mutation in the first half, PCR amplification reactions and direct sequencing methods were used.
Sequences of oligonucleotides for PCR were as follows: exon 4 of p53, the sense primer, 5′‐ACC TGG TCC TCT GAC TGC TCT TTT CA and the antisense primer, 5′‐CCA GGC ATT GAA GTC TCA TGG AAG C; exon 5–8, 5′‐CTG TTC ACT TGT GCC CTG ACT TTC AAC and 5′‐TCT GAG GCA TAA CTG CAC CCT TGG TCT; exon 10, 5′‐TAT ACT TAC TTC TCC CCC TCC TCT and 5′‐ATG AGA ATG GAA TCC TAT GGC TTT. PCR amplification of exons 5–8 was carried out together due to their close to proximity. PCR reaction mixtures contained 50 ng genomic DNA, 10 pM of each pair of primers, 10 mM deoxynucleotide triphosphates (dATP, dTTP, dGTP and dCTP), 1.0 U Taq DNA polymerase (Platinum Taq DNA polymerase High Fidelity, Invitrogen), 600 mM Tris‐SO4 (pH 8.9) buffer, 2 mM magnesium sulfate. After initial denaturation (for 2 min at 94°C), 35–40 cycles of PCR were carried out as follows: 94°C for 30 s, adequate annealing temperature 60°C for 30 s and 68°C for 30 s with a T‐personal thermal cycler system (Biometra).
After PCR product purification by centrifugal filtration (Montage MILLIPORE), direct sequencing was carried out for exons 4, 5–8 and 10 of the p53 gene with a CEQ 2000 DNA analyzer (Beckman Coulter Inc.) using CEQ 2000 Dye terminator cycle sequencing and a quick start kit (Beckman Coulter Inc.).
Statistical analysis. To assess any correlations between the p53 mutation status and clinicopathological data, the χ2 test, Fisher's exact probably test, Student's t‐test, and Mann–Whitney's U‐test were used, with significance concluded at P < 0.05.
Results
p53 mutations. Of the 297 NSCLCs, 142 (48%) had p53 mutations; 46 of the SQCCs and 96 of the ADCs (Table 1). Seven cases (DNA nos. 25, 31 and 43 in SQCCs, and DNA nos. 17, 244, 350 and 360 in ADCs) had two mutations each, making 149 (50%) in total (Table 2). Fourteen mutations (9%) were located in exon 4, 36 (24%) in exon 5, 26 (17%) in exon 6, 33 (22%) in exon 7, 28 (19%) in exon 8, five (3%) in exon 10, and seven (5%) in splicing junctions of exons. The majority (134, 90%) was located in the sequence‐specific DNA binding region (codons 100–293). There were 11 codons, 157, 158, 175, 176, 196, 220, 245, 248, 249, 273 and 282, where the number of mutations were more than three (Fig. 1a,b; Table 2). All six known mutation hotspots for lung cancer were included; 11 at codon 273, nine at 245, six at 248, five at 158, and three each at 157 and 249, and this total of 37 accounted for 25% for all mutations.
Table 2.
p53 mutations in lung adenocarcinomas and squamous cell carcinomas
| Sequential no. | DNA no. | Age (years) | Sex | Smoking index † | Tumor | Exon | Mutation | Amino acid | Mutation pattern | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Codon | Base change by strand | Next letter of the mutation to 3′ on the transcribed strand | ||||||||||||
| Histology | pStage | No. | Letters | Non‐transcribed | Transcribed | |||||||||
| 1 | 198 | 55 | M | 700 | Ad | IA | 4 | 46 | Repeat of 16 bp | − | Frameshift | Insertion | ||
| 2 | 393 | 54 | F | 0 | Ad | IIIB | 4 | 98 | Deletion of 1bp | − | Frameshift | Deletion | ||
| 3 | 360 | 64 | M | 1760 | Ad | IIB | 4 | 104 | CAG→CAT | G→T | − | Gln→His | TV | |
| 4 | 360 | 64 | M | 1760 | Ad | IIB | 4 | 105 | GGC→TGC | G→T | − | Gly→Cys | TV | |
| 5 | 269 | 66 | M | 920 | Ad | IIIB | 4 | 110 | Deletion of 1bp | CGT→GT | − | Frameshift | Deletion | |
| 6 | 138 | 44 | M | 0 | Ad | IIIA | 4 | 120 | AAG→AGG | A→G | − | Lys→Arg | TS | |
| 7 | 316 | 60 | M | 0 | Ad | IA | 4 | 124 | Insertion of 1bp | − | Frameshift | Insertion | ||
| 8 | 136 | 50 | M | 1600 | Ad | IB | 4 | 125 | ACG→ACT | G→T | − | Thr→Thr | TV | |
| 9 | 363 | 63 | M | 540 | Ad | IIIB | 4 | 125 | ACG→ACA | G→A (CpG) | − | Thr→Thr | TS | |
| 10 | 111 | 54 | M | 540 | Ad | IB | 4 | 3′ junction | Deletion of 1bp | CGgt→CG_t | − | Splicing | Deletion | |
| 11 | 368 | 73 | F | 0 | Ad | IIIB | 4 | 41–53 | Deletion of 38bp | − | Frameshift | Deletion | ||
| 12 | 17 | 69 | M | 846 | Ad | IA | 4 | 113–119 | Deletion of 19bp | − | Frameshift | Deletion | ||
| 13 | 90 | 73 | M | 1040 | Ad | IB | 4 | 124 | Deletion of 25bp | − | Frameshift | Deletion | ||
| 14 | 203 | 67 | F | 0 | Ad | IIIB | 5 | 132 | AAG→AGG | A→G | − | Lys→Arg | TS | |
| 15 | 307 | 66 | M | 740 | Ad | IIIA | 5 | 132 | AAG→GAG | A→G | − | Lys→Gln | TS | |
| 16 | 105 | 61 | M | 1600 | Ad | IB | 5 | 135 | TGC→TTC | G→T | − | Cys→Phe | TV | |
| 17 | 255 | 66 | F | 0 | Ad | IIIB | 5 | 136 | CAA→TAA | C→T | G→A (non‐CpG) | − | Gln→Stop | TS |
| 18 | 11 | 72 | F | 0 | Ad | IIIB | 5 | 138 | GCC→GTC | C→T | G→A (non‐CpG) | − | Ala→Val | TS |
| 19 | 19 | 57 | F | 0 | Ad | IIIA | 5 | 138 | GCC→CCC | G→C | − | Ala→Pro | TV | |
| 20 | 208 | 71 | M | 990 | Ad | IIIA | 5 | 157 | GTC→TTC | G→T | − | Val→Phe | TV | |
| 21 | 290 | 69 | M | 2820 | Ad | IIIB | 5 | 157 | GTC→GGC | T→G | A→C | − | Val→Gly | TV |
| 22 | 22 | 72 | M | 800 | Ad | IIIA | 5 | 158 | CGC→CAC | G→A (CpG) | − | Arg→His | TS | |
| 23 | 96 | 60 | M | 800 | Ad | IIA | 5 | 158 | CGC→CAC | G→A (CpG) | − | Arg→His | TS | |
| 24 | 173 | 54 | M | 680 | Ad | IA | 5 | 158 | CGC→CTC | G→T | − | Arg→Leu | TV | |
| 25 | 197 | 47 | M | 1620 | Ad | IIIA | 5 | 158 | CGC→CCC | G→C | − | Arg→Pro | TV | |
| 26 | 389 | 57 | F | 0 | Ad | IIA | 5 | 158 | CGC→CAC | G→A (CpG) | − | Arg→His | TS | |
| 27 | 79 | 56 | M | 720 | Ad | IIIB | 5 | 159 | Deletion of 2bp | GCC→C | − | Frameshift | Deletion | |
| 28 | 364 | 70 | M | 800 | Ad | IB | 5 | 159 | GCC→GTC | C→T | G→A (non‐CpG) | − | Ala→Val | TS |
| 29 | 315 | 81 | M | 0 | Ad | IIIB | 5 | 164 | AAG→TAG | A→T | − | Lys→Stop | TV | |
| 30 | 293 | 60 | M | 1200 | Ad | 5 | 167 | Deletion of 2bp | CAG→ G | − | Frameshift | Deletion | ||
| 31 | 238 | 71 | M | 765 | Ad | IIIB | 5 | 171 | GAG→TAG | G→T | − | Glu→Stop | TV | |
| 32 | 278 | 51 | F | 0 | Ad | IV | 5 | 172 | Deletion of 2bp | GTT→T | − | Frameshift | Deletion | |
| 33 | 103 | 74 | M | 860 | Ad | IA | 5 | 175 | CGC→CAC | G→A (CpG) | − | Arg→His | TS | |
| 34 | 134 | 26 | F | 0 | Ad | IIIA | 5 | 176 | TGC→TTC | G→T | − | Cys→Phe | TV | |
| 35 | 399 | 51 | F | 420 | Ad | IB | 5 | 176 | TGC→AGC | T→A | A→T | − | Cys→Ser | TV |
| 36 | 142 | 70 | M | 561 | Ad | IB | 5 | 181 | CGC→CCC | G→C | − | Arg→Pro | TV | |
| 37 | 352 | 64 | F | 0 | Ad | IIIB | 5 | 169–170 | Insertion of 6bp | − | Inframe | Insertion | ||
| 38 | 191 | 50 | M | 1160 | Ad | IA | 5 | 179–185 | Deletion of 18bp | − | Inframe | Deletion | ||
| 39 | 398 | 59 | F | 0 | Ad | IIA | 6 | 188 | Duplication | − | Frameshift | Duplication | ||
| 40 | 302 | 51 | M | 620 | Ad | IB | 6 | 189 | GCC→GTC | C→T | G→A (non‐CpG) | − | Ala→Val | TS |
| 41 | 160 | 50 | M | 2310 | Ad | IIIB | 6 | 190 | Deletion of 1bp | CCT→CT | − | Frameshift | Deletion | |
| 42 | 380 | 38 | F | 0 | Ad | IIA | 6 | 194 | CTT→CCT | T→C | A→G | − | Leu→Pro | TS |
| 43 | 391 | 81 | F | 0 | Ad | IV | 6 | 196 | CGA→TGA | C→T | G→A (CpG) | − | Arg→Stop | TS |
| 44 | 97 | 54 | M | 960 | Ad | IIIA | 6 | 198 | GAA→TAA | G→T | − | Glu→Stop | TV | |
| 45 | 361 | 69 | M | 920 | Ad | IIIA | 6 | 205 | TAT→TGT | A→G | − | Tyr→Cys | TS | |
| 46 | 122 | 74 | F | 240 | Ad | IIIA | 6 | 209 | AGA→TGA | A→T | − | Arg→Stop | TV | |
| 47 | 205 | 49 | F | 0 | Ad | IB | 6 | 213 | CGA→TGA | C→T | G→A (CpG) | A | Arg→Stop | TS |
| 48 | 329 | 53 | M | 100 | Ad | IIA | 6 | 215 | AGT→ATT | G→T | − | Ser→lle | TV | |
| 49 | 357 | 68 | F | 0 | Ad | IIIA | 6 | 218 | Deletion of 1bp | − | Frameshift | Deletion | ||
| 50 | 350 | 63 | F | 0 | Ad | IIIA | 6 | 219 | CCC→TCC | C→T | G→A (non‐CpG) | − | Pro→Ser | TS |
| 51 | 77 | 41 | F | 0 | Ad | IA | 6 | 220 | TAT→TGT | A→G | − | Tyr→Cys | TS | |
| 52 | 186 | 74 | M | 1060 | Ad | IB | 6 | 3′ junction | AGgt→AGat | G→A (non‐CpG) | − | Splicing | TS | |
| 53 | 89 | 41 | M | 750 | Ad | IB | 6 | 5′ junction | agG→atG | G→T | − | Splicing | TV | |
| 54 | 23 | 58 | M | 25 | Ad | IA | 7 | 234 | TAC→TGC | A→G | − | Tyr→Cys | TS | |
| 55 | 38 | 77 | F | 0 | Ad | IIIB | 7 | 237 | ATG→ATT | G→T | − | Met→lle | TV | |
| 56 | 86 | 56 | M | 900 | Ad | IIIA | 7 | 238 | TGT→AGT | T→A | A→T | − | Cys→Ser | TV |
| 57 | 157 | 49 | M | 20 | Ad | IIA | 7 | 238 | TGT→AGT | T→A | A→T | − | Cys→Ser | TV |
| 58 | 344 | 46 | F | 230 | Ad | IIIB | 7 | 239 | AAC→GAC | A→G | − | Asn→Asp | TS | |
| 59 | 80 | 65 | F | 0 | Ad | IA | 7 | 241 | Deletion of 1bp | TCC→TC | − | Frameshift | Deletion | |
| 60 | 101 | 68 | F | 75 | Ad | IIIA | 7 | 242 | TGC→TAC | G→A (non‐CpG) | − | Cys→Tyr | TS | |
| 61 | 294 | 58 | M | 1440 | Ad | IA | 7 | 244 | GGC→TGC | G→T | − | Gly→Cys | TV | |
| 62 | 28 | 47 | M | 650 | Ad | IIIA | 7 | 245 | GGC→TGC | G→T | − | Gly→Cys | TV | |
| 63 | 66 | 51 | F | 0 | Ad | IIIB | 7 | 245 | GGC→AGC | G→A (CpG) | − | Gly→Ser | TS | |
| 64 | 235 | 61 | F | 300 | Ad | IIIB | 7 | 245 | GGC→CGC | G→C | − | Gly→Arg | TV | |
| 65 | 313 | 61 | M | 2420 | Ad | IB | 7 | 245 | GGC→TGC | G→T | − | Gly→Cys | TV | |
| 66 | 381 | 61 | M | 1600 | Ad | IIIB | 7 | 245 | GGC→GTC | G→T | − | Gly→Val | TV | |
| 67 | 33 | 37 | F | 30 | Ad | IA | 7 | 248 | CGG→TGG | C→T | G→A (CpG) | G | Arg→Trp | TS |
| 68 | 139 | 70 | F | 0 | Ad | IA | 7 | 248 | CGG→CAG | G→A (CpG) | − | Arg→Gln | TS | |
| 69 | 297 | 59 | M | 570 | Ad | IA | 7 | 248 | CGG→CAG | G→A (CpG) | − | Arg→Gln | TS | |
| 70 | 327 | 64 | M | 1020 | Ad | IB | 7 | 249 | AGG→ATG | G→T | − | Arg→Met | TV | |
| 71 | 350 | 63 | F | 0 | Ad | IIIA | 7 | 258 | GAA→GAC | A→C | − | Glu→Asp | TV | |
| 72 | 3 | 73 | M | 700 | Ad | IIIB | 7 | 259 | GAC→AAC | G→A (non‐CpG) | − | Asp→Asn | TS | |
| 73 | 244 | 62 | M | 1000 | Ad | IIIA | 7 | 259 | GAC→TAC | G→T | − | Asp→Tyr | TV | |
| 74 | 282 | 66 | M | 1290 | Ad | IV | 7 | 253‐254 | Deletion of 3bp | − | Inframe | Deletion | ||
| 75 | 49 | 49 | F | 0 | Ad | IIIA | 8 | 273 | CGT→CAT | G→A (CpG) | − | Arg→His | TS | |
| 76 | 50 | 70 | M | 540 | Ad | IIIA | 8 | 273 | CGT→CAT | G→A (CpG) | − | Arg→His | TS | |
| 77 | 69 | 58 | M | 1600 | Ad | IIIA | 8 | 273 | CGT→TGT | C→T | G→A (CpG) | C | Arg→Cys | TS |
| 78 | 100 | 48 | M | 900 | Ad | IA | 8 | 273 | CGT→CTT | G→T | − | Arg→Leu | TV | |
| 79 | 152 | 68 | F | 0 | Ad | IA | 8 | 273 | CGT→CTT | G→T | − | Arg→Leu | TV | |
| 80 | 174 | 72 | M | 510 | Ad | IA | 8 | 273 | CGT→CAT | G→A (CpG) | − | Arg→His | TS | |
| 81 | 182 | 56 | M | 750 | Ad | IA | 8 | 273 | CGT→TGT | C→T | G→A (CpG) | C | Arg→Cys | TS |
| 82 | 215 | 63 | M | 0 | Ad | IA | 8 | 273 | CGT→CTT | G→T | − | Arg→Leu | TV | |
| 83 | 259 | 56 | M | 435 | Ad | IIIB | 8 | 273 | CGT→TGT | C→T | G→A (CpG) | C | Arg→Cys | TS |
| 84 | 362 | 53 | M | 660 | Ad | IIIB | 8 | 273 | CGT→TGT | C→T | G→A (CpG) | C | Arg→Cys | TS |
| 85 | 34 | 59 | M | 480 | Ad | IA | 8 | 274 | GTT→TTT | G→T | − | Val→Phe | TV | |
| 86 | 156 | 65 | M | 0 | Ad | IA | 8 | 275 | TGT→TAT | G→A (non‐CpG) | − | Cys→Tyr | TS | |
| 87 | 244 | 62 | M | 1000 | Ad | IIIA | 8 | 281 | GAC→TAC | G→T | − | Asp→Tyr | TV | |
| 88 | 155 | 63 | F | 0 | Ad | IIIA | 8 | 282 | CGG→TGG | C→T | G→A (CpG) | G | Arg→Trp | TS |
| 89 | 382 | 54 | F | 0 | Ad | IB | 8 | 282 | CGG→TGG | C→T | G→A (CpG) | G | Arg→Trp | TS |
| 90 | 400 | 61 | M | 820 | Ad | IB | 8 | 286 | GAA→GTA | A→T | − | Glu→Val | TV | |
| 91 | 347 | 80 | F | 240 | Ad | IIIA | 8 | 287 | GAG→TAG | G→T | − | Glu→Stop | TV | |
| 92 | 331 | 63 | M | 840 | Ad | IA | 8 | 298 | GAG→TAG | G→T | − | Glu→Stop | TV | |
| 93 | 154 | 72 | M | 2520 | Ad | IIIB | 8 | 274 | Deletion of 2bp | GTT→T | − | Frameshift | Deletion | |
| 94 | 17 | 69 | M | 846 | Ad | IA | 8 | 301 | Deletion of 1bp | CCA→C A | − | Frameshift | Deletion | |
| 95 | 284 | 75 | M | 2120 | Ad | IIIA | 8 | –262 | Deletion of 17bp | − | Splicing | Deletion | ||
| 96 | 15 | 64 | M | 660 | Ad | IA | 8 | 3′ junction | AGgt→AGtt | G→T | − | Splicing | TV | |
| 97 | 185 | 67 | M | 612 | Ad | IIB | 8 | 305–306 | Repeat of 23bp | − | Frameshift | Insertion | ||
| 98 | 83 | 65 | F | 0 | Ad | IA | 10 | 335 | CGT→CAT | G→A (CpG) | − | Arg→His | TS | |
| 99 | 388 | 79 | F | 0 | Ad | IIIB | 10 | 342 | CGA→TGA | C→T | G→A (CpG) | G | Arg→Stop | TS |
| 100 | 148 | 51 | F | 0 | Ad | IIIA | 10 | 341 | Deletion of 1bp | TTC→T C | − | Frameshift | Deletion | |
| 101 | 25 | 71 | M | 1020 | Sq | IIIB | 4 | 88 | GCC→ACC | G→A (non CpG) | − | Ala→Thr | TS | |
| 102 | 24 | 71 | M | 1020 | Sq | IIIB | 4 | 103 | TAC→TAG | C→G | G→C | − | Tyr→stop | TV |
| 103 | 36 | 69 | M | 1020 | Sq | IIB | 5 | 130 | CTC→GTC | C→G | G→C | − | Leu→Val | TV |
| 104 | 16 | 76 | M | 1250 | Sq | IIB | 5 | 144 | CAG→CCG | A→C | − | − | Gln→Pro | TV |
| 105 | 43 | 76 | M | 780 | Sq | IIIB | 5 | 152 | Deletion of 2bp | CCG→G | − | − | Frameshift | Deletion |
| 106 | 33 | 75 | M | 1060 | Sq | IB | 5 | 154 | GGC→GTC | G→T | − | − | Gly→Val | TV |
| 107 | 44 | 79 | M | 0 | Sq | 5 | 157 | GTC→TTC | G→T | − | − | Val→Phe | TV | |
| 108 | 30 | 71 | M | 2550 | Sq | IB | 5 | 163 | TAC→TGC | A→G | − | − | Try→Cys | TS |
| 109 | 21 | 69 | M | 2040 | Sq | IA | 5 | 166 | TCA→TAA | C→A | G→T | − | Ser→stop | TV |
| 110 | 9 | 59 | M | 1200 | Sq | IIIA | 5 | 175 | CGC→CAC | G→A (CpG) | − | − | Arg→His | TS |
| 111 | 28 | 60 | M | 1600 | Sq | IIIB | 5 | 175 | CGC→CAC | G→A (CpG) | − | − | Arg→His | TS |
| 112 | 27 | 67 | M | 470 | Sq | IA | 5 | 176 | TGC→TAC | G→A (non CpG) | − | − | Cys→Tyr | TS |
| 113 | 7 | 70 | M | 1250 | Sq | IIIB | 5 | 149‐175 | Deletion of 79bp | − | − | Frameshift | Deletion | |
| 114 | 37 | 71 | M | 820 | Sq | IB | 5 | 5'junction | agTA→tgTA | a→t | − | − | splicing | TV |
| 115 | 14 | 63 | M | 1505 | Sq | IIIA | 6 | 190 | Deletion of 1bp | CCT→CT | − | − | Frameshift | Deletion |
| 116 | 45 | 66 | M | 0 | Sq | 6 | 190 | CCT→CTT | C→T | G→A (nonCpG) | − | Pro→Leu | TS | |
| 117 | 26 | 81 | M | 1100 | Sq | IB | 6 | 193 | CAT→TAT | C→T | G→A (nonCpG) | − | His→Tyr | TS |
| 118 | 19 | 70 | M | 1590 | Sq | IIIA | 6 | 195 | ATC→ACC | T→C | A→G | − | lle→Thr | TS |
| 119 | 42 | 69 | M | 960 | Sq | IIIA | 6 | 195 | ATC→ACC | T→C | A→G | − | Ile→Thr | TS |
| 120 | 15 | 49 | M | 300 | Sq | IIIA | 6 | 196 | CGA→CCA | G→C | − | − | Arg→Pro | TV |
| 121 | 35 | 65 | M | 1260 | Sq | IIB | 6 | 196 | CGA→TGA | C→T | G→A (CpG) | G | Arg→stop | TS |
| 122 | 2 | 71 | M | 0 | Sq | IA | 6 | 220 | TAT→TGT | A→G | − | − | Try→Cys | TS |
| 123 | 18 | 82 | M | 1170 | Sq | IIIB | 6 | 220 | TAT→TGT | A→G | − | − | Try→Cys | TS |
| 124 | 31 | 62 | M | 1320 | Sq | IIB | 6 | 220 | TAT→TTT | A→T | − | − | Try→Cys | TV |
| 125 | 38 | 61 | M | 1200 | Sq | IB | 6 | 220 | TAT→TGT | A→G | − | − | Tyr→Cys | TS |
| 126 | 41 | 66 | M | 966 | Sq | IIIA | 6 | 220 | TAT→TGT | A→G | − | − | Tyr→Cys | TS |
| 127 | 31 | 62 | M | 1320 | Sq | IIB | 6 | 221 | Deletion of 1bp | GAG→AG | − | − | Frameshift | Deletion |
| 128 | 40 | 69 | M | 490 | Sq | IIIB | 7 | 234 | TAC→TGC | A→G | − | − | stop→Tro | TS |
| 129 | 6 | 64 | M | 0 | Sq | IIIA | 7 | 236 | TAC→TGC | A→G | − | − | Try→Cys | TS |
| 130 | 11 | 65 | M | 760 | Sq | IB | 7 | 244 | GGC→TGC | G→T | − | − | Gly→Cys | TV |
| 131 | 3 | 70 | F | 1000 | Sq | IIIB | 7 | 245 | GGC→TGC | G→T | − | − | Gly→Cys | TV |
| 132 | 5 | 79 | M | 2205 | Sq | IB | 7 | 245 | GGC→CGC | G→C | − | − | Gly→Arg | TV |
| 133 | 10 | 66 | M | 709 | Sq | IIIA | 7 | 245 | GGC→CGC | G→C | − | − | Gly→Arg | TV |
| 134 | 23 | 68 | M | 1440 | Sq | IIIA | 7 | 245 | GGC→GTC | G→T | − | − | Gly→Val | TV |
| 135 | 29 | 43 | M | 440 | Sq | IIIB | 7 | 248 | CGG→CTG | G→T | − | − | Arg→Leu | TV |
| 136 | 43 | 76 | M | 780 | Sq | IIIB | 7 | 248 | CGG→TGG | C→T | G→A (CpG) | G | Arg→Trp | TS |
| 137 | 46 | 81 | M | 1200 | Sq | IIB | 7 | 248 | CGG→CTG | G→T | − | − | Arg→Leu | TV |
| 138 | 12 | 72 | M | 705 | Sq | IA | 7 | 249 | AGG→ATG | G→T | − | − | Arg→Met | TV |
| 139 | 25 | 71 | M | 1020 | Sq | IIIB | 7 | 249 | AGG→AGT | G→T | − | Arg→Ser | TV | |
| 140 | 34 | 74 | M | 2750 | Sq | IIA | 8 | 266 | GGA→TGA | G→T | − | − | Gly→stop | TV |
| 141 | 22 | 53 | M | 1320 | Sq | IIB | 8 | 271 | GAG→TAG | G→T | − | − | Glu→stop | TV |
| 142 | 39 | 65 | M | 760 | Sq | IIIB | 8 | 272 | GTG→TTG | G→T | − | − | Val→Leu | TV |
| 143 | 13 | 68 | M | 1950 | Sq | IIIA | 8 | 273 | CGT→TGT | C→T | G→A (CpG) | C | Arg→Cys | TS |
| 144 | 32 | 65 | F | 400 | Sq | IIB | 8 | 280 | AGA→GGA | A→G | − | − | Arg→Gly | TS |
| 145 | 8 | 59 | M | 1260 | Sq | IIIB | 8 | 282 | CGG→TGG | C→T | G→A (CpG) | G | Arg→Trp | TS |
| 146 | 20 | 75 | F | 0 | Sq | IIB | 8 | 282 | CGG→TGG | C→T | G→A (CpG) | G | Arg→Trp | TS |
| 147 | 1 | 61 | M | 0 | Sq | IIIB | 10 | 337 | CGC→CTC | G→T | − | − | Arg→Leu | TV |
| 148 | 4 | 76 | M | 600 | Sq | IB | 10 | 342 | CGA→TGA | C→T | G→A (CpG) | G | Arg→stop | TS |
| 149 | 17 | 51 | M | 1240 | Sq | IIA | 10 | 5′ junction | agAT→tgAT | a→t | − | − | Splicing | TV |
Smoking index is the number of cigarettes/day × years. TS, transition; TV, transversion.
Figure 1.
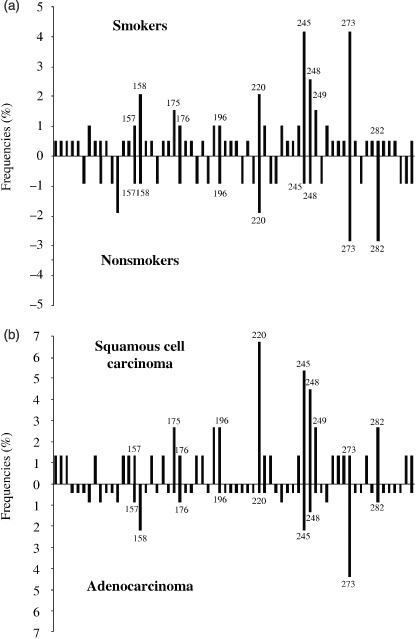
Distribution of all point mutations (except junctional mutations) along the p53 codons in resected Japanese non‐small cell lung cancers (NSCLCs) by smoking status (a), and by histology (b): x axis, codon position; y axis, percentage of p53 mutations among all cases of each group. The labeled peaks indicate codon numbers, in which there were more than three mutations.
Relationships between p53 mutations and clinicopathological parameters. The frequency of p53 mutations for smokers in all cases was 54%, which was higher than those for non‐smokers (37%) with statistical significance (P < 0.01) (Table 1). When compared by an amount of cigarette consumption, the frequency for heavy smokers was higher than that for light smokers with almost borderline significant difference (P = 0.057). Numbers of point mutations along p53 codons except at exon‐intron junctions were 50 for smokers and 25 for non‐smokers, 11 being found in common (Fig. 1a). Comparing the frequencies for the six hot spots, the sum for smokers (35%, 30/86) was higher than that for non‐smokers (23%, 7/31), though no statistical significance was observed (P = 0.21).
When p53 mutation frequencies were compared between histologies, the SQCC value was higher than for the ADCs (P < 0.01) (Table 1). Numbers of mutated codons were 30 for SQCCs and 48 for ADCs with 13 codons involved in common (Fig. 1b; Table 2). Mutations were most frequent in codon 220 for SQCCs, and codons 273 for ADCs, the difference for codon 220 being significant (P = 0.004).
Mean ages of the mutated cases were higher for SQCCs than for ADCs (P < 0.001) (Table 1). By gender, the mutation frequency was higher in males than females overall (P < 0.01). Regarding the pathological stages, the number with p‐stage IV was small, so they were combined with those of p‐stage III (Table 1). For p‐stages III + IV, percentages of mutated cases were 52% for ADCs, 65% for SQCCs, and 55% overall. Frequencies of mutations increased with the advance of p‐stages for all cases and for ADCs (P = 0.015 and 0.017, respectively).
p53 mutational spectra ( Table 3 ). Most p53 mutations were transitions (61/297, 21%) or transversions (61/297, 21%), and deletions/insertions accounted for only 9% (27/297). The frequency of transversions in the smokers was higher than in non‐smokers with statistically significant difference (P < 0.001). By histology, frequencies of the former two were higher in SQCCs than in ADCs with statistical significance (P < 0.05 and < 0.01, respectively), but no difference was evident for deletions/insertions.
Table 3.
p53 mutation spectra by smoking status and by histology, and strand bias
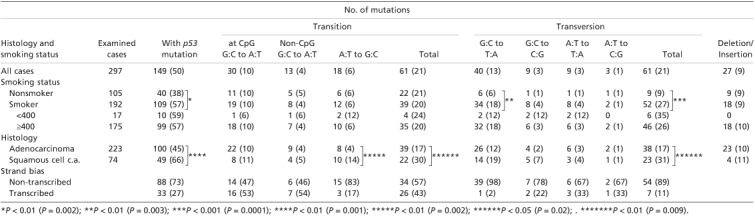
With regard to base substitutions, G→T transversions accounted for 40 out of 297 total cases (13%). By smoking status, the frequency in the smokers was 18%, which was higher than that of non‐smokers (6%) with significance (P = 0.003), although there was no difference between histologies (19% in SQCCs and 12% in ADCs). Distributions of G→T transversions along the p53 codons by smoking status and by histology are shown in Fig. 2(a,b). The total number of mutations was 38, as two mutations observed in intron‐exon junctions were excluded. By smoking status, there were 32 mutations in 24 codons for smokers, and six in five codons for non‐smokers, and only two codons were commonly mutated. For smokers, all five hot spot codons for adduct formation by PAHs (exclusion of codon 249 from the six lung cancer hot spot codons) were included, and the average mutation number in each of the five codons was 2.0 (10/5); two times higher than in the other codons, 1.1 (22/19). In non‐smokers, mutations were observed in only two out of the five hot spot codons, and the average mutation number was 0.6 (3/5), lower than for other codons, 1.0 (3/3) (Fig. 2a; Table 2).
Figure 2.

Distribution of G→T transversions along p53 codons by smoking status (a), and by histology (b): x axis, codon positions; y axis, percentages of G→T mutations among all cases of each group. All hot spots (codons 157, 158, 245, 248 and 273) for adduct formation by polycyclic aromatic hydrocarbons (PAHs) were mutated in smokers (a). The mutations were distributed more widely along p53 codons for adenocarcinomas than for squamous cell carcinomas (b).
The distribution patterns also differed with the histology. Mutations numbered 14 over 11 codons for SQCCs, and 24 over 20 codons for ADCs, with only four commonly mutated codons (Fig. 2b; Table 2). Average mutation numbers for the five hot spot codons was 1.0 (5/5) and almost the same as for other codons, 1.1 (9/8), for SQCCs. For ADCs, the figure for the five hot spots 1.6 (8/5) was higher than for the others, 1.0 (16/16).
CpG→CpA transitions were observed in 10% of all cases, and mutation frequencies were equal across smoking status and between histology types. This base substitution occurred in a 5′‐CGT‐3′ sequence context on a non‐transcribed strand of two smokers (DNA nos. 50 and 174) and two non‐smokers (DNA nos. 49 and 83), all of which had ADCs (Table 2).
A→G transitions were observed in 6% of all cases, and the frequencies were the same for smokers and non‐smokers, but higher in SQCCs than in ADCs (P < 0.01).
Silent mutations of p53 were observed in two ADCs of smokers, case DNA nos. 136 (ACG to ACT; Thr to Thr) and 363 (ACG to ACA; Thr to Thr). Neither of these cases had any other p53 mutations.
Strand bias ( Table 3 ). A marked strand bias was observed for G→T transversions, 98% (39/40) occurring on a non‐transcribed strand. Other transversion mutations, G→C, A→T and A→C, also showed a bias towards the non‐transcribed strand (78%, 67% and 67%, respectively). In addition 83% of A→G transitions were on non‐transcribed strands. In contrast, the CpG→CpA transitions were equally distributed on both strands (14 vs 16).
Discussion
The frequency of p53 mutations for NSCLCs (SQCCs and ADCs) in this study was 48%, approximately the same as in our previous study (45%) in 151 cases and other Japanese studies (48% each).( 20 , 22 , 23 ) Most mutations were found in the DNA binding domain of the gene, as described earlier.( 24 ) The frequencies by histological types and by gender, 62% in SQCCs and 43% in ADCs, and 54% in male and 37% in female, were also similar to the outcomes in past Japanese studies (58–67% vs 35–41%, respectively, for histology, and 50–57% vs 23–36%, respectively, for gender).( 20 , 22 , 23 ) Thus, our examined cases can be considered representative of lung cancer series in Japanese.
The p53 mutation frequencies in smokers were obviously higher than those in non‐smokers in our cases. When compared by tobacco consumption, a trend for more frequent mutations in heavy than in light smokers was also recognized, in line with other studies.( 6 , 22 ) Further, distribution patterns of the mutations along the p53 codons differed with the smoking status. Thus, p53 mutations are strongly related to tobacco smoking as reported.( 25 , 26 )
Metabolites of polycyclic aromatic hydrocarbons (PAHs) and tobacco‐specific N‐nitrosamine (4‐methylnitrosamino‐1‐(3‐pyridyl)‐1‐butanone (NNK) included in tobacco smoke, and several reactive lipid peroxidation products derived from tobacco smoke, lead to the formation of DNA adducts and cause G→T transversions.( 27 , 28 , 29 ) In our study, G→T transversions were observed more frequently in smokers than in non‐smokers with significance (P = 0.003), which was in line with previous reports.( 8 , 30 , 31 ) Although Paschke insisted that he could not find significant differences between smokers and non‐smokers in frequencies of the mutations on analysis of the R3 version of IARC TP53 database, this is an exceptional report.( 32 ) Thus we conclude that the frequency of G→T transversions is higher in smokers than in non‐smokers.
The bulky adducts formation with tobacco carcinogenic chemicals at mutational hotspots of the p53 gene, especially in codons 157, 158, 245, 248, and 273, results predominantly in G→T transversions.( 13 , 26 ) In our study, such mutations were observed in the five codons for smokers, but only two codons for non‐smokers. Furthermore, in smokers, the average incidences at each of the five hot spots was two times higher than in others, and the results were reverse in non‐smokers, although the numbers were small. Similar results have been reported.( 24 , 31 ) In contrast, Rodin claimed that the G→T transversion distribution pattern along the p53 gene was indistinguishable between lung cancers of smokers and non‐lung cancers, and most likely not associated with smoke.( 14 ) However, even in Rodin's analyzed cases, frequencies of the mutations on the five codons were clearly higher in smokers than in non‐smokers. The importance of differences in mutation frequency should be stressed to distinguish lung cancers in smokers from non‐smokers and cancers ‘less accessible to smoke’, because lungs of non‐smokers and tissues of non‐lung cancers may also be either exposed to certain levels of PAHs directly or PAH metabolites indirectly, so the patterns may look somewhat similar to those in lung cancer, if one examines only G→T events.( 8 ) Thus, it is clear that there is a relatively precise correspondence between the mutational hot spots of lung cancer and the hot spots of adduct formation by carcinogens found in tobacco smoke.( 33 , 34 )
In the present study, G→T transversions showed a marked strand bias; 98% were found in a non‐transcribed strand, which is in line with previous reports.( 3 , 5 ) This phenomenon has been considered to be one of the characteristics of mutations caused by DNA adducts derived from exogenous carcinogens, such as smoke mutagens.( 5 , 35 , 36 , 37 , 38 )
Reactive oxygen species (ROS), produced by endogenous mechanisms, such as cell aerobic metabolism and inflammation, or metabolism of PAHs to o‐quinone, are another source of G→T mutations. However, so far, it is uncertain how endogenous mechanisms contribute to the lung cancer specific mutation spectrum, with G→T transversions on non‐transcribed strands.( 29 , 39 , 40 , 41 , 42 , 43 )
Frequencies of the CpG→CpA transition have not been as extensively studied as G→T transversions. Here, they were found in 10% each in smokers and non‐smokers. Such transitions have generally been considered due to elevated susceptibility to spontaneous deamination. However, recently, in vitro evidence was presented indicating that the mutations may originate through adducts formation with metabolites of exogenous agents. Thus the (+)‐anti diol epoxide of BaP gave a preponderance of G→A mutations in a 5′‐CGT‐3′ sequence context,( 15 ) and these mutations are likely to be attributable to the major adduct. We found four human lung cancers with such lesions, all of them involving non‐transcribed strands. However, as a whole, the mutations were observed evenly in both transcribed and non‐transcribed strands, indicating that bulky adduct formation by exogenous carcinogens such as those included in tobacco smoke, do not play an important role in their genesis.
Other mechanisms that cause G→A transitions at CpG sites, other than spontaneous deamination, may be deamination of 5‐methylcytosine by certain chemicals like nitric oxide, oxidative DNA base damage, and enzyme‐catalyzed events.( 44 ) Nitric oxide may be involved in the pathobiology of the airway inflammatory process in chronic obstructive pulmonary disease, but most of our examined cases did not have any history of chronic obstructive disease.( 45 , 46 ) Whether oxidative mechanisms and enzymatic reactions that are endogenous are significant events in vivo for G→A transitions at CpG sites is not clear at present. G→A transitions may also occur at guanine of non‐CpG sites through formation of O6–methylguanine as a consequence of exposure of DNA to tobacco‐specific nitrosamines such as NNK.( 31 , 47 ) However, at CpG sites, most cytosines are methylated, and methylated cytosine in p53 protects neighboring guanine from O6–alkylation by NNK,( 18 ) explaining the preferential occurrence of these adducts at non‐CpG sites. From this discussion, we consider that most GpG→CpA transitions in lung cancers are attributable to endogenous mechanisms.
Regarding A→G transitions, the tobacco‐related differences have been reported only in two papers, so far.( 6 , 31 ) However, in our study, the mutation frequencies did not show any differences between smokers and non‐smokers, although strong strand bias, 83% on non‐transcribed, was found to be described in some papers.( 3 , 6 , 31 ) This observation is an indicator of mutagenesis by exogenous chemical compounds in the environment throughout the formation of bulky adducts. The observed preferential existence of the mutations in squamous cell carcinomas compared to adenocarcinomas has not been documented before. The reasons of why are not clear, but it could be due to differences in carcinogen exposure between distinct mucosal sites, squamous cell carcinomas usually arising at large bronchi or more proximal portions of the bronchioles and adenocarcinomas with more distal bronchiolar portions, or variation in carcinogenic activation or DNA repair by progenitor cells of the histologies.
Ethnic characteristics. There has been a paper reporting frequencies of the CpG→CpA transitions among all p53 mutations of 8% for smokers and 14% for non‐smokers in eastern countries, but 12% and 23%, respectively, in westerners.( 6 ) Our data calculated in the same way were more in line with the latter: 17% (19/109) and 28% (11/40), respectively,. This may also indicate that endogenous mechanisms may be playing a more important role in the genesis of NSCLCs than in other eastern countries.
Histological differences in p53 mutation spectra. The fact that our p53 mutation frequency was higher in SQCCs than in ADCs is in line with other reports,( 3 , 22 ) this presumably being related to the higher smoking rate for the former than the latter.( 6 , 22 ) In addition, compared to ADCs, uniquely high p53 mutations on codon 220, which is not a hot spot for PAHs, and more frequent A→G transitions with strand bias, of which induction were not affected by smoking status, were observed in SQCCs. This indicates that genesis of SQCCs may be more related with both tobacco smoke compounds except PAHs and environmental bulky‐adduct forming carcinogenic agents. Whereas, in ADCs, G→T transversions were more widely dispersed along the p53 gene, which implies roles for other exogenous factors for the genesis of ADCs
We could not find differences between ADCs and SQCCs in frequencies of CpG→CpA transitions and the G→T transversions, which is in agreement with the data of Calvez.( 31 ) However, there was one report of less frequent CpG→CpA transitions for ADCs than SQCCs, and more common G→T transversions in ADCs.( 48 ) This discrepancy may partly be related to differences in frequencies of either histological subtypes of ADCs or locations of SQCCs by ethnicity or geography. In this context it should be stressed that we have reported that the frequencies of these mutations significantly differ among subtypes of ADCs or among locations of SQCCs.( 19 , 20 )
Increase of p53 mutation frequency with progression of carcinoma. Adenocarcinomas at advanced stages showed more frequent p53 mutation than those at early stages (stage I) with significant differences. To our knowledge, this is the first report to note an increase of mutation rates with advance in the stages in ADCs of the lung. This may be for the following reason. Usually patients are continuously exposed to carcinogenic agents that can induce p53 mutation, even after their carcinoma development, so that cancer cells without p53 mutation at an early development stage remain at risk of mutation of the gene. If these mutations occur in the cancer cells, the mutated cells might gain a growth advantage superior to other cancer cells, and this could account for the greatest volume of carcinomas.
Acknowledgments
We thank T. Yanagisawa for technical assistance. This work was supported in part by grants‐in‐aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Kinzler KW, Vogelstein B. Life (and death) in a malignant tumour. Nature 1996; 379: 19–20. [DOI] [PubMed] [Google Scholar]
- 2. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–31. [DOI] [PubMed] [Google Scholar]
- 3. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54: 4855–78. [PubMed] [Google Scholar]
- 4. Kato S, Bowman ED, Harrington AM, Blomeke B, Shields PG. Human lung carcinogen‐DNA adduct levels mediated by genetic polymorphisms in vivo. J Natl Cancer Inst 1995; 87: 902–7. [DOI] [PubMed] [Google Scholar]
- 5. Denissenko MF, Pao A, Pfeifer GP, Tang M. Slow repair of bulky DNA adducts along the nontranscribed strand of the human p53 gene may explain the strand bias of transversion mutations‐in cancers. Oncogene 1998; 16: 1241–7. [DOI] [PubMed] [Google Scholar]
- 6. Bennett WP, Hussain SP, Vahakangas KH, Khan MA, Shields PG, Harris CC. Molecular epidemiology of human cancer risk: gene–environment interactions and mutation spectrum in human lung cancer. J Pathol 1999; 187: 8–18. [DOI] [PubMed] [Google Scholar]
- 7. Besaratinia A, Van Straaten HW, Godschalk RW et al . Immunoperoxidase detection of polycyclic hydrocarbom‐DNA adducts in mouth floor and buccal mucosa cells of smokers and non‐smokers. Environ Mol Mutagen 2000; 36: 127–33. [DOI] [PubMed] [Google Scholar]
- 8. Hainaut P, Pfeifer GP. Patterns of p53 G→T transversions in lung cancers reflect the primary mutagenic signature of DNA‐damage by tobacco smoke. Carcinogenesis 2001; 22: 367–74. [DOI] [PubMed] [Google Scholar]
- 9. Holliday R, Grigg GW. DNA methylation and mutation. Mutant Res 1993; 285: 61–7. [DOI] [PubMed] [Google Scholar]
- 10. Ehrlich M, Zhang XY, Inamdar NM. Spontaneous deamination of cytosine and 5‐methylcytosine residues in DNA and replacement of 5‐methylcytosine residues with cytosine residues. Mutant Res 1990; 238: 277–86. [DOI] [PubMed] [Google Scholar]
- 11. Rideout WM III, Coetzee GA, Olumi AF, Jones PA. 5‐Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990; 249: 1288–90. [DOI] [PubMed] [Google Scholar]
- 12. Rodin SN, Rodin AS. Human lung cancer and p53. The interplay between mutagenesis and selection. Proc Natl Acad Sci USA 2000; 97: 12 244–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pfeifer GP, Hainaut P. On the origin of G→T transversions in lung cancer. Mut Res 2003; 526: 39–43. [DOI] [PubMed] [Google Scholar]
- 14. Rodin SN, Rodin AS. Origins and selection of p53 mutations in lung carcinogenesis. Semin Cancer Oncol 2005; 15: 103–12. [DOI] [PubMed] [Google Scholar]
- 15. Shukla R, Liu T, Geacintov NE, Loechler EL. The major, N2‐dG adduct of (+)‐anti‐B[a]PDE shows a dramatically different mutagenic specificity (predominantly, G→A) in a 5′‐CGT‐3′ sequence context. Biochemistry 1997; 36: 10 256–61. [DOI] [PubMed] [Google Scholar]
- 16. Singer B, Essigmann JM. Site‐specific mutagenesis: retrospective and prospective. Carcinogenesis 1991; 12: 949–55. [DOI] [PubMed] [Google Scholar]
- 17. Tornaletti S, Pfeifer GP. Complete and tissue‐independent methylation of CpG sites in the p53 gene: implications for mutations in human cancers. Oncogene 1995; 10: 1493–9. [PubMed] [Google Scholar]
- 18. Ziegel R, Shallop A, Upadhyaya P, Jones R, Tretyakova N. Endogenous 5‐methylcytosine protect neighboring guanines from N7 and O6‐methylation and O6‐pyridyloxobutylation by the tobacco carcinogen 4‐(methylnitrosamino)‐1‐(3‐pyidyl)‐1‐butanone. Biochemistry 2004; 43: 540–9. [DOI] [PubMed] [Google Scholar]
- 19. Shimmyo T, Hashimoto T, Kobayashi Y et al . p53 mutation spectra for squamous cell carcinomas at different levels of human bronchial branches. Int J Cancer 2006; 119: 501–7. [DOI] [PubMed] [Google Scholar]
- 20. Hashimoto T, Tokuchi Y, Hayashi M et al . Different subtypes of human lung adenocarcinoma caused by different etiological factors. Am J Pathol 2000; 157: 2133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tokuchi Y, Hashimoto T, Kobayashi Y et al . The expression of p73 is increased in lung cancer, independent of p53 gene alteration. Br J Cancer 1999; 80: 1623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki H, Takahashi T, Kuroishi T et al . p53 Mutation in non‐small cell lung cancer in Japan: association between mutation and smoking. Cancer Res 1992; 52: 734–6. [PubMed] [Google Scholar]
- 23. Kishimoto Y, Murakami Y, Shiraishi M, Hayashi K, Sekiya T. Aberrations of the p53 tumor suppressor gene in human non‐small cell carcinomas of the lung. Cancer Res 1992; 52: 4799–804. [PubMed] [Google Scholar]
- 24. Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking‐associated cancers. Oncogene 2002; 21: 7435–51. [DOI] [PubMed] [Google Scholar]
- 25. Shen YM, Troxel AB, Vedantam S, Penning TM, Field J. Comparison of p53 mutations induced by PAH o‐quinones with those caused by anti‐benzo[a]pyrene diol epoxide in vitro: role of reactive oxygen and biological selection. Chem Res Toxicol 2006; 19: 1441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Besaratinia A, Pfeifer GP. Investigating human cancer etiology by DNA lesion footprinting and mutagenicity analysis. Carcinogenesis 2006; 27: 1526–37. [DOI] [PubMed] [Google Scholar]
- 27. Hoffmann D, Hoffmann I, Bayoumy KE. The less harmful cigarette: a controversial issue. A tribute to Ernst L. Wynder. Chem Res Toxicol 2001; 14: 767–90. [DOI] [PubMed] [Google Scholar]
- 28. Hecht SS. Tobacco smoke carcinogenesis and lung cancer. J Natl Cancer Inst 1999; 21: 1194–210. [DOI] [PubMed] [Google Scholar]
- 29. Martinez GR, Loureiro AP, Marques SA et al . Oxidative and alkylating damage in DNA. Mutat Res 2003; 544: 115–27. [DOI] [PubMed] [Google Scholar]
- 30. Robles AI, Linke SP, Harris CC. The p53 network in lung carcinogenesis. Oncogene 2002; 7: 6898–907. [DOI] [PubMed] [Google Scholar]
- 31. Calvez F, Mukeria A, Hunt JD et al . TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res 2005; 15: 5076–83. [DOI] [PubMed] [Google Scholar]
- 32. Paschke T. Analysis of different versions of the IARC p53 database with respect to G→T transversion mutation frequencies and mutation hotspots in lung cancers of smokers and non‐smokers. Mutagenesis 2000; 15: 457–8. [DOI] [PubMed] [Google Scholar]
- 33. Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in. P53. Science 1996; 274: 430–2. [DOI] [PubMed] [Google Scholar]
- 34. Smith LE, Denissenko MF, Bennett WP et al . Targeting of lung cancer mutational hotspots by polycyclic aromatic hydrocarbons. J Natl Cancer Inst 2000; 92: 803–11. [DOI] [PubMed] [Google Scholar]
- 35. Chen RH, Maher VM, Brouwer J, Van De Putte P, McCormick JJ. Preferential repair and strand‐specific repair of benzo[a]pyrene diol epoxide adducts in the HPRT gene of diploid human fibroblasts. Proc Natl Acad Sci 1992; 89: 5413–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rodin SN, Rodin AS. On the origin of p53 G:C→T: a transversions in lung cancer. Mut Res 2002; 508: 1–19. [DOI] [PubMed] [Google Scholar]
- 37. Rodin SN, Rodin AS. On the excess of G→T transversions in the p53 gene in lung cancer cell lines. Mut Res 2004; 545: 141–4. [DOI] [PubMed] [Google Scholar]
- 38. Hanawalt PC. Role of transcription‐coupled DNA repair in susceptibility to environmental carcinogenesis. Environ Health Perspect 1996; 104: 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Penning TM, Ohnishi ST, Ohnishi T, Harvey RG. Generation of reactive oxygen species during the enzymatic oxidation of polycyclic aromatic hydrocarbon dihydrodioles catalyzed by dihydrodiole dehydrogenase. Chem Res Toxicol 1996; 9: 84–92. [DOI] [PubMed] [Google Scholar]
- 40. Penning TM, Burczynski ME, Hung C‐F, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenase and policyclic aromatic hydrocarbon activation. generation of reactive and redox active o‐quinones. Chem Res Toxicol 1999; 12: 1–18. [DOI] [PubMed] [Google Scholar]
- 41. Page FL, Klungland A, Barnes DE, Sarasin A, Boiteux S. Transcription coupled repair of 8‐oxoguanonine in murie cells: the oggl protein is required for repair in non‐transcribed sequences but not in transcribed sequences. Proc Natl Acad Sci USA 2000; 97: 8397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anson RM, Croteau DL, Stierum RH, Filburn C, Parsell R, Bohr VA. Homogeneous repair of singler oxygen‐induced DNA damage in diferentailly transcribed regions and strands of human mitochondrial DNA. Nucl Acids Res 1998; 26: 662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thorslund T, Suneson M, Bohr VA, Stevnsner T. Repair of 8‐oxoG is slower in endogenous nuclear genes than in mitochondrial DNA and is without strand bias. DNA Repair 2002; 1: 261–73. [DOI] [PubMed] [Google Scholar]
- 44. Pfeifer GP. Mutagenesis at methylated CpG sequences. Curr Top Microbiol Immunol 2006; 301: 259–81. [DOI] [PubMed] [Google Scholar]
- 45. Ambs S, Bennett WP, Merriam WG et al . Relationship between p53 mutations and inducible nitric oxide synthase expression in human colorectal cancer. J Natl Cancer Inst 1999; 91: 86–8. [DOI] [PubMed] [Google Scholar]
- 46. Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med 2000; 162: 701–6. [DOI] [PubMed] [Google Scholar]
- 47. Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 2004; 38: 445–76. [DOI] [PubMed] [Google Scholar]
- 48. Gao WM, Mady HM, Yu GY et al . Comparison of p53 mutations between adenocarcinoma and squamous cell carcinoma of the lung: unique spectra involving G to A transitions in both histologic types. Lung Cancer 2003; 40: 141–50. [DOI] [PubMed] [Google Scholar]