

Abstract
Enzastaurin is an oral serine/threonine kinase inhibitor that targets the protein kinase C (PKC) and phosphoinositide 3‐kinase/AKT pathways to induce apoptosis and suppress proliferation of various cancer cell lines. This phase I study evaluated the tolerability and pharmacokinetics of enzastaurin in Japanese patients with advanced solid tumors and determined the recommended dose for phase II. Eligible patients had advanced solid tumors and an Eastern Cooperative Oncology Group performance status of 0–2. Patients received enzastaurin orally once daily until disease progression (PD) or unacceptable toxicity occurred. Enzastaurin was started at 250 mg/day followed by stepwise dose increases based on the incidence of dose‐limiting toxicities (DLT). Twenty‐three patients (seven patients: 250 mg; six patients: 375 mg; six patients: 500 mg; four patients: 750 mg) were enrolled and received enzastaurin. The major tumor types were non‐small‐cell lung cancer (n = 5) and breast cancer (n = 3). No DLT was reported at doses of 500 mg or less. Because two DLT (grade 2 QTc prolongation lasting for a week) were observed at 750 mg enzastaurin, this was determined as the maximum tolerated dose. Multiple daily doses at 500 mg achieved the target plasma concentration to inhibit PKC activity (1400 nmol/L). Enzastaurin was well tolerated up to 500 mg in Japanese patients with advanced solid tumors. The recommended dose for phase II was determined to be 500 mg daily for a 28‐day cycle on the basis of safety and plasma exposures. (Cancer Sci 2010);
Protein kinase C (PKC) is overexpressed and its activity is increased in many cancers including colon cancer,( 1 ) non‐small‐cell lung cancer (NSCLC)( 2 ) and diffuse large B‐cell lymphoma (DLBCL).( 3 ) Protein kinase C is stimulated on activation of receptors on the cell membrane, including vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF).( 4 ) Recent evidence suggests a link between PKC and phosphoinositide 3‐kinase (PI3K)/AKT, the main pathway responsible for cell survival and proliferation.( 5 , 6 )
Enzastaurin is an oral serine/threonine kinase inhibitor that targets the PKC and PI3K/AKT pathways to induce apoptosis and suppress proliferation of various cancer cell lines.( 1 , 7 , 8 ) In animal models, enzastaurin showed antitumor and antiangiogenic activity in various malignancies, including NSCLC, colon cancer, renal cell cancer, hepatocellular cancer and glioblastoma.( 1 , 9 , 10 ) Enzastaurin has been well tolerated in an early clinical study conducted in the USA at doses from 20 to 750 mg/day in patients with advanced solid tumors.( 11 )
Enzastaurin is primarily metabolized by cytochrome P450 3A (CYP3A), leading to the formation of multiple metabolites, including three active metabolites: a desmethylenepyrimidyl metabolite (LY326020); a desmethyl metabolite (LY485912); and a hydroxymethyl intermediate (LSN2406799). These metabolites are pharmacologically active, inhibiting PKC‐β with similar potencies to enzastaurin (IC50 approximately 6 nmol/L) (data on file).( 12 ) Results from in vitro studies have shown that enzastaurin inhibits 90% of PKC‐β enzymatic activity (IC90) at 70 nmol/L (data on file). Since preliminary data suggest that plasma protein binding of enzastaurin is approximately 95%, total plasma concentrations above 1400 nmol/L would be required to achieve 70 nmol/L or higher of the free concentration.
Here we report the results of a phase I study that was conducted to evaluate the tolerability of enzastaurin and its pharmacokinetic profile in Japanese patients with advanced solid tumors. Based on these results we have also determined the recommended dose of enzastaurin for a planned phase II study, and evaluated the antitumor effect of enzastaurin in these patients.
Patients and Methods
Patients. Patients with histologically or cytologically confirmed malignant solid tumor, refractory to conventional chemotherapy or available non‐standard therapy and without any carry‐over influence of previous therapies, were eligible to enter the study. Inclusion criteria included the following: age between 20 and 75 years; Eastern Cooperative Oncology Group (ECOG) performance status (PS) from 0 to 2; predicted life expectancy of at least 3 months; and adequate hematopoietic, hepatic and renal functions (absolute neutrophil counts ≥ 1.5 × 103/mm3, platelets ≥ 100 × 103/mm3, hemoglobin ≥ 9 g/dL, total bilirubin ≤ 1.5 × upper limit of normal [ULN], aspartate amino‐transferase [AST] and alanine amino‐transferase [ALT] ≤ 2.5 × ULN, and serum creatinine ≤ 1.5 × ULN). Exclusion criteria included the following: present cardiac disorders of grade 3 or greater; myocardial infarction within 6 months after onset; pleural, peritoneal or pericardial effusion requiring drainage; QTc ≥ 0.45 s (QTc = QT/√ (R‐R) (Bazette); dissemination to the meninges; or history of stomach resection. Toxicity was graded in accordance with the National Cancer Institute Common Terminology for Adverse Events (CTCAE) ver. 3.0.
Ethical considerations. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and was consistent with Good Clinical Practice and all applicable laws and regulations. Written informed consent was obtained from all patients and the study was approved by the Institutional Review Board of the participating institution.
Study design. This was an open‐label phase I dose‐escalation study in one clinical center, the National Cancer Center Hospital East, Japan.
Five fixed dose levels of enzastaurin, 250, 375, 500, 750 and 1000 mg/day, were to be examined in the study. The starting dose, 250 mg enzastaurin, was selected based on the results from the previous phase I study of enzastaurin conducted in patients with advanced solid tumors.( 11 ) In that study, 500 mg enzastaurin was the lowest daily dose at which dose‐limiting toxicity (DLT) was observed. We therefore selected half that dose as the starting dose in this study. A six patient cohort was used for each dose level being escalated in a stepwise manner based on DLT incidence. The DLT was defined as any of the following occurring in cycle 1: grade 3, or higher non‐hematological toxicity except nausea, vomiting, anorexia, fatigue, constipation, diarrhea and allergic reactions; grade 4 neutropenia or thrombocytopenia; febrile neutropenia; grade 2 QTc prolongation lasting at least 1 week; or dose omission lasting at least 8 days. Because preclinical study showed that enzastaurin could cause QTc prolongation in dogs and it was actually observed in the preceding phase I study in the USA, we defined sustained QTc prolongation as DLT even when it was grade 2, from a safety perspective. Nausea, vomiting, anorexia, fatigue, constipation and diarrhea were considered as DLT only when they persisted in grade 3 or higher despite adequate symptomatic treatment and the investigator judged them as DLT. The maximum tolerated dose (MTD) was defined as the lowest dose level where the incidence of DLT was 33% or higher. If there was a patient whose compliance was <75% and who experienced no DLT in cycle 1 at a certain dose level, an additional patient was enrolled at that dose level. A patient whose dose omission was <25% or who experienced DLT in cycle 1 was evaluable for DLT analysis. The incidence of DLT at each dose level was summarized using the patients evaluable for DLT.
Treatment plan. Enzastaurin was administered orally once daily after breakfast, because exposures of enzastaurin following administration in the fed state were higher than exposures following administration in the fasted state (data on file).
The study treatment consisted of two periods: cycle 0 and all subsequent cycles. In cycle 0, patients received a single dose of enzastaurin to investigate its pharmacokinetic profile. Cycle 1 was initiated during the period from 8 to 15 days following single‐dose administration in cycle 0. During cycle 1 and the subsequent cycles (1 cycle = 28 days), patients received enzastaurin tablets once daily until disease progression (PD) or unacceptable toxicity occurred.
If any adverse event meeting DLT criteria occurred, the patient was required to omit the administration of enzastaurin until the event resolved or improved to grade 1. If the patient restarted the study treatment, the dose of enzastaurin was reduced to the next lowest level. For each patient, a maximum of two dose reductions were allowed.
Prohibited medications during the study period included Vaughan‐Williams Class Ia, Ic and III antiarrhythmic drugs, CYP3A4 inhibitors, CYP3A4 inducers and warfarin.
Toxicity and response monitoring. The following measures were used to assess patients at pre‐study and during treatment: body weight, PS, laboratory tests, vital signs, electrocardiogram (ECG), chest X‐ray and ophthalmological examination results. Post‐study evaluation was conducted 28 days after the last dose.
Adverse events were graded in accordance with CTCAE ver. 3.0. The antitumor effect was assessed using the Response Evaluation Criteria in Solid Tumors (RECIST)( 13 ) for every other cycle, and was confirmed at least 4 weeks after the initial observation of responses.
Pharmacokinetics. During cycle 0, heparinized blood samples (3 mL) were collected 0.5, 1, 1.5, 2, 3, 4, 6, 9, 24, 48, 72, 96 and 168 h after the dose. During cycle 1, blood samples (3 mL) were collected pre‐dose and 6 h post‐dose on day 1; pre‐dose on days 8, 15 and 22; and pre‐dose, 0.5, 1, 1.5, 2, 3, 4, 6, 9 and 24 h post‐dose on day 28.
High‐performance liquid chromatography with tandem mass‐spectrometry (LC/MS/MS) was used to detect enzastaurin and its metabolites, LY326020, LY485912 and LSN2406799, in plasma (Advion BioSciences, Ithaca, NY, USA).
The PK parameters for enzastaurin and its metabolites were analyzed using noncompartmental methods with WinNonlin Version 5.0.1. (Pharsight, Mountain View, CA, USA). The primary parameters, such as maximal concentration (Cmax), time of maximal concentration (tmax), area under the plasma concentration vs time curve from 0 to 24 h (AUC0–24 h) and to infinity (AUC0–∞), apparent clearance (CL/F), apparent volume of distribution (Vz/F) and the apparent terminal half‐life (t1/2) were calculated during cycle 0. Similarly, during cycle 1, the maximum concentration at steady‐state during a dosing interval (Cmax,ss) and the time taken to reach Cmax,ss (tmax,ss) were identified. The area under the plasma concentration vs the time curve over the dosing interval at steady‐state (AUCτ,ss) was determined. The linear/log trapezoidal method was used to compute AUC values. Other non‐compartmental parameters such as predicted average drug concentration under steady‐state conditions during a dosing interval (Cav,ss), apparent clearance (CLss/F) and apparent volume of distribution at steady‐state (Vss/F) were also calculated. The accumulation ratio (RA) was assessed as AUCτ,ss to AUC0–24. The metabolic ratio (MR) was calculated as metabolite AUC to parent AUC.
Cmax and tmax values of total analytes (enzastaurin + LY326020 + LY485912 + LSN2406799) were reported by summing the concentration–time data for enzastaurin together with its metabolites, because these metabolites inhibit PKC‐β with potencies similar to enzastaurin. If two or more of the individual values were not present, the value to be computed was indicated as missing.
The analysis to estimate intra‐subject and inter‐subject variability used data from cycle 0 and cycle 1. AUC0–∞ from single dosing was combined with AUCτ,ss after multiple dosing. This combination strategy was based on linear pharmacokinetic principles that have shown “AUCτ,ss≈ AUC0–∞”.
Results
Patient characteristics. This study was conducted from November 2005 to January 2008 at the National Cancer Center Hospital East in Japan and 23 patients were enrolled (Table 1). The most common types of cancer were NSCLC (n = 5) and breast cancer (n = 3).
Table 1.
Patient characteristics (N = 23)
| Patient characteristics | N = 23 |
|---|---|
| Gender | |
| Male | 15 |
| Female | 8 |
| Age (years) | |
| Median | 55.0 |
| Range | 27–71 |
| ECOG performance status | |
| 0 | 10 |
| 1 | 13 |
| Diagnosis | |
| Non‐small‐cell lung cancer | 5 |
| Breast cancer | 3 |
| GIST | 2 |
| Renal cancer | 2 |
| Brain tumor | 1 |
| Gastric cancer | 1 |
| Intrahepatic bile duct cancer | 1 |
| Liposarcoma | 1 |
| Large intestine carcinoma | 1 |
| Malignant neoplasm of urachus | 1 |
| Malignant neoplasm of retromolar area | 1 |
| Pleural mesothelioma | 1 |
| Rectal cancer | 1 |
| Thyroid tumor and leiomyosarcoma | 1 |
| Carcinoma of unknown primary | 1 |
ECOG, Eastern Cooperative Oncology Group; GIST, gastro‐intestinal stromal tumor.
Maximum tolerated dose and toxicity. All 23 patients received at least one enzastaurin dose. The median number of treatment cycles was two cycles at 250 mg, four at 375 mg, 2.5 at 500 mg and 2.5 at 750 mg. Twenty‐one patients discontinued the study due to progressive disease and two patients discontinued due to adverse events. One patient at 250 mg experienced progressive disease before the end of the first cycle, so that patient was added to the cohort. One patient at 375 mg discontinued the study due to drug‐related QTc prolongation and the other patient at 750 mg discontinued because of anorexia, which was not drug related.
Of 23 treated patients, four patients were excluded from DLT analysis because of early progression of their disease. The incidence of DLT was summarized by the enzastaurin dose in 19 patients (five patients at 250 mg, six patients at 375 mg, five patients at 500 mg and three patients at 750 mg). No DLT was reported at doses of 500 mg or less. Two DLT were reported at 750 mg; both were grade 2 QTc prolongations lasting at least 1 week. Based on the results, 750 mg of enzastaurin was determined as the MTD and 500 mg as the recommended dose for phase II studies.
Table 2 shows toxicities possibly related to enzastaurin that were observed in more than 10% of patients. The most common toxicities in the treated patients were chromaturia (n = 15) and somnolence (n = 9), which were both grade 1. The incidence of somnolence appeared to increase dose‐dependently. The only grade 3 toxicity was lymphocytopenia (n = 1). There were no drug‐related deaths.
Table 2.
Toxicity possibly related to enzastaurin observed in >10% of patients
| Preferred term (MedDRA ver.10.0) | Dose of enzastaurin (mg) | Total (N = 23) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 250 (N = 7) | 375 (N = 6) | 500 (N = 6) | 750 (N = 4) | |||||||||
| G1 | G2 | G1 | G2 | G1 | G2 | G1 | G2 | G3 | G1 | G2 | G3 | |
| Chromaturia | 4 | 0 | 4 | 0 | 4 | 0 | 3 | 0 | 0 | 15 | 0 | 0 |
| Somnolence | 0 | 0 | 2 | 0 | 4 | 0 | 3 | 0 | 0 | 9 | 0 | 0 |
| Faeces discoloured | 2 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 5 | 0 | 0 |
| Fatigue | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 2 | 2 | 0 |
| ECG QTc interval prolonged | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 2 | 0 | 0 | 4 | 0 |
| Blood albumin decreased | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 2 | 1 | 0 |
| Blood urine present | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 2 | 1 | 0 |
| Lymphocyte count decreased | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 1 | 0 | 2 | 1 |
| Rash | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 3 | 0 |
ECG, electrocardiogram; G, grade of CTCAE v3.0; MedDRA, the Medical Dictionary for Regulatory Activities.
Three patients (one at 375 mg and two at 750 mg) required dose omission due to grade 2 QTc prolongation, which was deemed as DLT in two of them. One patient at 375 mg experienced grade 2 QTc prolongation lasting for 8 days in cycle 2. All QTc prolongation events were reversible in these patients and they recovered from these events after dose omission. However, QTc prolongation events continued to occur again until sufficient dose reduction was taken (Fig. 1). The patient at 375 mg discontinued due to drug‐related prolonged QTc interval, which occurred again even after a dose reduction to 250 mg (Fig. 1). The plasma concentration of enzastaurin and its active metabolites at the point of patients’ experiencing QTc prolongation is not available.
Figure 1.
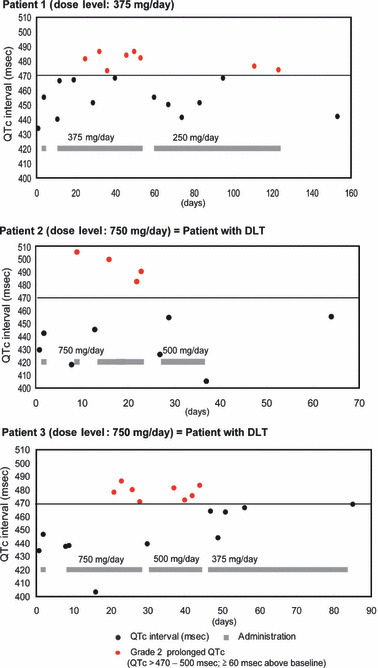
Patients who required dose omission because of QTc prolongation. DLT, dose‐limiting toxicities.
Pharmacokinetics. Plasma concentration–time data and dosing information (dose date and time) for pharmacokinetic evaluation were available from 23 patients. Two patients at 750 mg were excluded from the analysis for steady‐state because their enzastaurin doses were reduced during cycle 1 due to adverse events.
Figure 2 shows the mean plasma concentration–time profile of enzastaurin. The mean pharmacokinetic parameters of enzastaurin and its active metabolites following single and multiple dosing are summarized in 3, 4, respectively. Mean concentration–time profile of enzastaurin after a single dose monophasically declined following Cmax and the mean t1/2 of enzastaurin ranged from 10.1 to 13.3 h independent of the dose administered. Cmax and AUC0–∞ of enzastaurin did not increase dose‐proportionally after a single dose or during multiple dosing. The RA of enzastaurin ranged from 1.19 to 3.56 and did not appear to be dose dependent.
Figure 2.
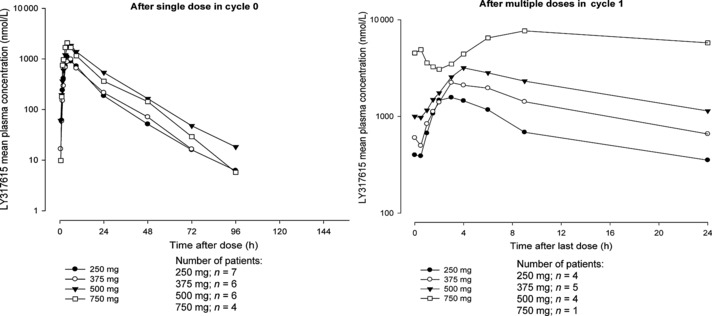
Enzastaurin (LY317615) mean plasma concentration–time profile.
Table 3.
Pharmacokinetic parameters of enzastaurin and its active metabolites in cycle 0
| Analyte | Parameters | Geometric mean (coefficient of variation %) | |||
|---|---|---|---|---|---|
| Dose of enzastaurin (mg) | |||||
| 250 | 375 | 500 | 750 | ||
| Enzastaurin | N | 7 | 6 | 6 | 4 |
| Cmax (nmol/L) | 1090 (57.5) | 968 (61.3) | 2070 (40.2) | 1910 (73.3) | |
| tmax (h)* | 4.07 (2.87–8.92) | 3.96 (3.00–6.10) | 5.96 (3.00–9.03) | 3.92 (3.15–4.00) | |
| AUC0–∞ (h μmol/L) | 12.7 (78.7) | 12.9 (77.6) | 29.5 (70.2) | 23.5 (96.6) | |
| CL/F (L/h) | 38.0 (78.7) | 56.4 (77.6) | 32.9 (70.2) | 61.9 (96.6) | |
| Vz/F (L) | 651 (70.7) | 820 (69.1) | 632 (39.1) | 957 (68.5) | |
| t1/2 (h)† | 11.9 (7.6–17.0) | 10.1 (7.5–16.4) | 13.3 (8.4–20.3) | 10.7 (8.2–14.1) | |
| LY326020 | N | 7 | 6 | 6 | 4 |
| Cmax (nmol/L) | 285 (36.7) | 308 (24.3) | 494 (39.9) | 548 (53.1) | |
| tmax (h)* | 5.77 (2.87–8.97) | 5.01 (4.00–9.02) | 9.02 (4.00–24.02) | 5.00 (3.92–6.00) | |
| AUC0–∞ (h μmol/L) | 12.2 (50.2) | 14.6 (46.8) | 27.8 (61.1) | 26.3 (65.5) | |
| t 1/2 (h)† | 38.2 (27.2–55.6) | 34.8 (28.9–40.5) | 37.8 (26.6–55.6) | 38.4 (33.2–41.1) | |
| MR | 0.961 (60.7) | 1.13 (37.3) | 0.942 (46.9) | 1.12 (23.7) | |
| LY485912 | N | 7 | 6 | 6 | 4 |
| Cmax (nmol/L) | 122 (57.3) | 107 (48.8) | 258 (50.2) | 218 (77.7) | |
| tmax (h)* | 5.77 (3.93–8.97) | 6.10 (4.02–9.02) | 9.02 (6.20–24.02) | 5.00 (3.92–9.07) | |
| AUC0–∞ (h μmol/L) | 2.25 (88.4) | 2.17 (91.3) | 5.88 (103) | 4.54 (132) | |
| t 1/2 (h)† | 12.0 (7.7–19.7) | 10.5 (9.4–14.0) | 12.6 (8.7–19.6) | 11.5 (6.8–17.7) | |
| MR | 0.176 (15.3) | 0.168 (15.8) | 0.199 (33.3) | 0.193 (19.8) | |
| LSN2406799 | N | 7 | 6 | 6 | 4 |
| Cmax (nmol/L) | 145 (34.0) | 143 (41.3) | 276 (31.8) | 242 (92.8) | |
| tmax (h)* | 4.02 (2.87–6.07) | 3.96 (3.00–6.10) | 4.96 (3.00–9.03) | 3.92 (3.15–4.00) | |
| AUC0–∞ (h μmol/L) | 1.40 (68.4) | 1.49 (60.8) | 3.25 (49.6) | 2.71 (92.6) | |
| t 1/2 (h)† | 10.5 (7.1–13.7) | 8.70 (7.1–11.9) | 11.6 (6.1–17.1) | 10.3 (5.7–19.7) | |
| MR | 0.110 (9.2) | 0.115 (15.3) | 0.110 (18.7) | 0.115 (12.5) | |
*Median (range). †Geometric mean (range). AUC0–∞, area under the concentration vs time curve from 0 to infinity; CL/F, apparent total body clearance calculated after oral dosing; Cmax, maximum plasma concentration; MR, metabolic ratio; tmax, median time to reach maximum concentration; t1/2, half‐life associated with the terminal rate constant; Vz/F, apparent volume of distribution after oral administration.
Table 4.
Pharmacokinetic parameters of enzastaurin and its active metabolites in cycle 1
| Analyte | Parameters | Geometric mean (coefficient of variation %) | |||
|---|---|---|---|---|---|
| Dose of enzastaurin (mg) | |||||
| 250 | 375 | 500 | 750 | ||
| Enzastaurin | N | 4 | 5 | 4 | 1 |
| Cmax,ss (nmol/L) | 1460 (62.9) | 1920 (83.1) | 3230 (36.2) | 7680† | |
| tmax (h)* | 2.48 (1.92–3.95) | 3.08 (3.00–6.08) | 4.00 (3.00–6.07) | 9.08† | |
| AUCτ,ss (h μmol/L) | 15.00 (98.1) | 22.20 (122) | 40.90 (61.6) | 147.00† | |
| Cav,ss (nmol/L) | 624 (98.1) | 927 (122) | 1710 (61.6) | 6140† | |
| CL/F (L/h) | 32.4 (98.1) | 32.7 (122) | 23.7 (61.6) | 9.87† | |
| RA | 1.19 (88.4) | 2.24 (45.0) | 1.96 (48.4) | 3.56† | |
| LY326020 | N | 4 | 5 | 4 | 1 |
| Cmax,ss (nmol/L) | 470 (38.3) | 754 (48.0) | 1260 (45.8) | 2640† | |
| tmax (h)* | 3.93 (1.42–4.00) | 6.00 (3.08–9.13) | 5.00 (0.52–24.00) | 0.50† | |
| AUCτ,ss (h μmol/L) | 9.33 (39.4) | 15.1 (55.9) | 27.1 (52.9) | 53.00† | |
| Cav,ss (nmol/L) | 389 (39.4) | 629 (55.9) | 1130 (52.9) | 2210† | |
| MR | 0.623 (60.0) | 0.679 (54.6) | 0.663 (23.4) | 0.359† | |
| RA | 2.18 (52.8) | 2.90 (26.8) | 3.87 (51.6) | 3.78† | |
| LY485912 | N | 4 | 5 | 4 | 1 |
| Cmax,ss (nmol/L) | 188 (90.7) | 253 (108) | 490 (52.5) | 1480† | |
| tmax (h)* | 4.97 (3.92–9.93) | 6.00 (4.03–9.00) | 7.52 (6.00–24.00) | 0.00† | |
| AUCτ,ss (h μmol/L) | 2.92 (125) | 4.29 (148) | 8.99 (67.5) | 29.60† | |
| Cav,ss (nmol/L) | 121 (125) | 179 (148) | 374 (67.5) | 1230† | |
| MR | 0.195 (20.6) | 0.193 (15.8) | 0.220 (9.8) | 0.201† | |
| RA | 1.38 (119) | 2.96 (48.5) | 2.60 (38.5) | 3.60† | |
| LSN2406799 | N | 4 | 5 | 4 | 1 |
| Cmax,ss (nmol/L) | 166 (53.2) | 197 (51.2) | 331 (17.6) | 574† | |
| tmax (h)* | 3.46 (1.92–3.95) | 3.97 (3.00–9.13) | 4.00 (3.00–6.07) | 9.08† | |
| AUCτ,ss (h μmol/L) | 1.60 (75.9) | 2.22 (85.3) | 3.81 (43.1) | 10.50† | |
| Cav,ss (nmol/L) | 66.6 (75.9) | 92.4 (85.3) | 159 (43.1) | 436† | |
| MR | 0.107 (21.3) | 0.0997 (28.8) | 0.0931 (17.2) | 0.0710† | |
| RA | 1.09 (63.1) | 1.78 (40.7) | 1.62 (38.2) | 2.44† | |
*Median (range). †Individual values, mean and CV% were not calculated because N ≤ 2. AUCτ,ss, area under the concentration vs time curve over the dosing interval at steady state; Cav,ss, the predicted average drug concentration under steady‐state conditions during multiple dosing; Cmax,ss, maximum plasma concentration at steady‐state; MR, metabolic ratio; RA, accumulation ratio; tmax, median time to reach maximum concentration.
LY326020 has a longer half‐life than enzastaurin, indicating that this metabolite is elimination‐rate limited. The RA of LY326020 ranged from 2.18 to 3.87, which tended to be higher due to its longer half‐life. The MR of LY326020 ranged from 0.942 to 1.13 after single dosing and from 0.359 to 0.679 after multiple dosing. These results suggest that systemic exposure of LY326020 was similar to or slightly lower than that of enzastaurin. The mean t1/2 of LY485912 and LSN2406799 were similar to that of enzastaurin. This indicates that elimination may be formation‐rate limited. The MR of LY485912 and LSN2406799 ranged from 0.193 to 0.220 and from 0.0710 to 0.107, respectively, after multiple dosing. Systemic exposure of these metabolites was smaller than that of enzastaurin.
The intra‐patient (within‐subject) variability assessed using AUC for enzastaurin and its metabolites ranged from 17% to 40%. The inter‐subject variability was high and ranged from 56% to108% as assessed by AUC. Figure 3 shows the average drug concentration of enzastaurin and its active metabolites in individual patients across all dose levels. Multiple daily doses at 500 mg, the recommended dose for phase II studies, achieved the target plasma concentration, 1400 nmol/L, in all Japanese patients. The predicted average drug concentration of enzastaurin and its active metabolites at steady‐state ranged from 1680 to 5040 nmol/L.
Figure 3.
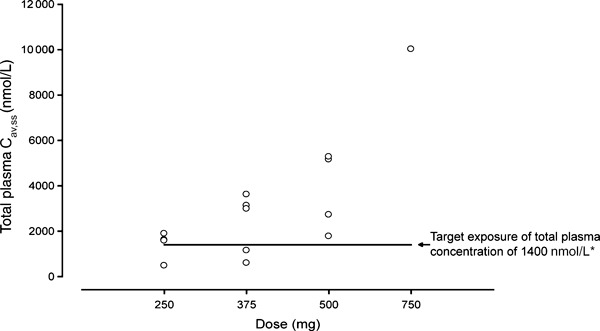
Steady‐state average plasma concentrations of enzastaurin and its active metabolites. *The target exposure of total plasma concentration of 1400 nmol/L was determined based on the IC90 (70 nmol/L) for protein kinase C‐β inhibition by enzastaurin, assuming approximately 95% protein binding.
Antitumor effect. All 23 patients were assessed for tumor response. No formal objective response occurred in accordance with RECIST. Three patients (two at 375 mg and one at 500 mg) had been treated without progressive disease for at least six cycles. Of these, one patient with gastro‐intestinal stromal tumor continued therapy at 375 mg for 21 cycles. Another patient with mesothelioma showed radiographical improvement in positron emission tomography imaging using the glucose analog 2‐deoxy‐2‐[18F]fluoro‐D‐glucose (18FDG) at 7 weeks after baseline (Fig. 4).
Figure 4.
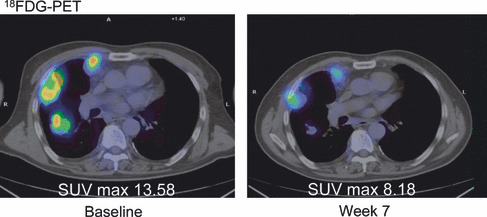
Metabolic response in mesothelioma. PET, positron emission tomography; SUV, standardized uptake value.
Discussion
In this study, we showed that enzastaurin up to 500 mg was well tolerated in Japanese patients with advanced solid tumors. Five hundred mg of enzastaurin achieved the target exposure, 1400 nmol/L, as measured by the total plasma concentration of enzastaurin and its active metabolites. In xenograft models the growth of glioblastoma and colon carcinoma were significantly suppressed by oral dosing with enzastaurin to yield plasma concentrations similar to those achieved in clinical trials.( 1 ) On the basis of safety data and plasma exposures, we recommend a daily dose of 500 mg of enzastaurin for phase II studies in Japan. This result is consistent with results from the phase I study in non‐Japanese patients.( 11 )
In the current study, no clinically significant toxicities other than QTc prolongation were reported. QTc prolongation was the DLT, and drug‐related reversible grade 2 QTc prolongations were observed in four patients (one patient at 375 mg, one patient at 500 mg and two patients at 750 mg). The onset of each event was in cycle 0 or 1, but not on the first day of either cycle. None of these events were exacerbated during the course of treatment. Although the definitive cause of QTc prolongation is undefined, the study results are consistent with the results of a preclinical study in dogs and of the preceded phase I study in the USA. In a preclinical study in which dogs were given high daily doses (exposures higher than those expected to occur in most patients), QT and QTc prolongation was observed after 5 weeks, and cataracts were seen after 13 weeks (data on file). In the phase I study in the USA, QTc prolongation was the DLT. Events were observed at the 700 and 525 mg dose levels although they did not require treatment or discontinuation.( 11 ) Asymptomatic QTc prolongations are reported in several novel molecular targeting oncology agents including multi‐targeted tyrosine kinase inhibitors, histone deacetylase inhibitors and PKC inhibitors, and their mechanisms are being explored.( 14 ) In recent phase II and III studies, an initial high‐loading dose of enzastaurin has been used to attain steady‐state plasma total concentrations of enzastaurin sooner. In such studies, it is necessary to monitor the QTc interval carefully. We are evaluating the safety of the high initial loading dose for Japanese patients in a separate study. In this study, as in another study of enzastaurin,( 11 ) the most common toxicity observed in the treated patients was chromaturia. This is most likely due to the reddish‐orange color of the active ingredient of enzastaurin.
The plasma exposures of enzastaurin in this study as assessed by AUC indicate high inter‐patient variability for enzastaurin and its metabolites (56–108%) compared with intra‐patient variability (17–40%). Since dose escalation was not performed within the same patients and the number of patients in each dose level was small with high variability, dose proportionality for enzastaurin and its metabolites could not be concluded.
Because this was a phase I study, efficacy was not a primary endpoint. Although formal tumor responses were not identified, one mesothelioma patient showed radiographical improvement and three patients were treated for at least six cycles without progressive disease. Phase II studies designed to measure tumor response and/or stable disease over a specified period will provide more information on the expected antitumor activity of enzastaurin.
A recent report showed that treatment with enzastaurin was well tolerated and associated with prolonged freedom from progression in a small subset of patients with relapsed or refractory DLBCL,( 15 ) and a large global phase III trial of standard induction therapy (CHOP/rituximab) with or without enzastaurin consolidation has been initiated in patients with newly diagnosed, high‐intermediate/high‐risk DLBCL. Based on the results of the current study, medical institutes in Japan have joined this phase III study and investigators have been carefully monitoring all clinical aspects, including ECG.
In summary, the current study showed that enzastaurin was well tolerated up to 500 mg in Japanese patients with advanced solid tumors. The only DLT observed was reversible QTc prolongation. The recommended dose for phase II was determined to be 500 mg daily for a 28‐day cycle, the same as that determined in Caucasian patients.
Disclosure Statement
Financial support for this work was provided by Eli Lilly Japan, KK. Minori Koshiji and Kenichi Yoshizawa are employees of Eli Lilly Japan. None of the other authors have a conflict of interest to disclose.
Abbreviations
- AUC0–∞
area under the concentration vs time curve from 0 to infinity
- AUCτ,ss
area under the concentration vs time curve over the dosing interval at steady state
- Cav,ss
the predicted average drug concentration under steady‐state conditions during multiple dosing
- CL/F
apparent total body clearance calculated after oral dosing
- Cmax
maximum plasma concentration
- Cmax,ss
maximum plasma concentration at steady‐state
- CTCAE
common terminology criteria for adverse events
- ECG
electrocardiogram
- ECOG
Eastern Cooperative Oncology Group
- G
grade of CTCAE v3.0
- GIST
gastro‐intestinal stromal tumor
- MedDRA
the Medical Dictionary for Regulatory Activities
- MR
metabolic ratio
- RA
accumulation ratio
- t1/2
half‐life associated with the terminal rate constant
- tmax
median time to reach maximum concentration
- Vz/F
apparent volume of distribution after oral administration
Acknowledgments
The authors acknowledge staff at the investigator site, Luna Musib for pharmacokinetics, and Megumi Sugiura for editorial and technical assistance.
References
- 1. Graff JR, McNulty AM, Hanna KR et al. The protein kinase Cß‐selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res 2005; 65: 7462–9. [DOI] [PubMed] [Google Scholar]
- 2. Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non‐small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistence. Cancer Res 2003; 63: 780–6. [PubMed] [Google Scholar]
- 3. Shipp MA, Ross KN, Tamayo P et al. Diffuse large B‐cell lymphoma outcome prediction by gene‐expression profiling and supervised machine learning. Nat Med 2002; 8: 68–74. [DOI] [PubMed] [Google Scholar]
- 4. McMahon G. VEGF receptor signaling in tumor angiogenesis. Oncologist 2000; 5( Suppl 1): 3–10. [DOI] [PubMed] [Google Scholar]
- 5. Balendran A, Hare GR, Kieloch A, Williams MR, Alessi DR. Further evidence that 3‐phosphoinositide‐dependent protein kinase‐1 (PDK1) is required for the stability and phosphorylation of protein kinase C (PKC) isoforms. FEBS Lett 2000; 484: 217–23. [DOI] [PubMed] [Google Scholar]
- 6. Partovian C, Simons M. Regulation of protein kinase B/Akt activity and Ser473 phosphorylation by protein kinase Calpha in endothelial cells. Cell Signal 2004; 16: 951–7. [DOI] [PubMed] [Google Scholar]
- 7. Wu E, Aguiar R, Savage K et al. PKCbeta: a rational therapeutic target in diffuse large B‐cell lymphoma. Blood 2002; 100: 202a. [Google Scholar]
- 8. Rossi RM, Henn AD, Conkling R et al. The PKCbeta selective inhibitor, enzastaurin (LY317615), inhibits growth of human lymphoma cells. Blood 2005; 106: 427a. [Google Scholar]
- 9. Liu Y, Su W, Thompson EA, Leitges M, Murray NR, Fields AP. Protein kinase CbetaII regulates its own expression in rat intestinal epithelial cells and the colonic epithelium in vivo. J Biol Chem 2004; 279: 45556–63. [DOI] [PubMed] [Google Scholar]
- 10. Keyes K, Cox K, Treadway P et al. An in vitro tumor model: analysis of angiogenic factor expression after chemotherapy. Cancer Res 2002; 62: 5597–602. [PubMed] [Google Scholar]
- 11. Carducci MA, Musib L, Kies MS et al. Phase I dose escalation and pharmacokinetic study of enzastaurin, an oral protein kinase C beta inhibitor, in patients with advanced cancer. J Clin Oncol 2006; 24: 4092–9. [DOI] [PubMed] [Google Scholar]
- 12. Welch PA, Sinha VP, Cleverly AL, Darstein C, Flanagan SD, Musib LC. Safety, tolerability, QTc evaluation, and pharmacokinetics of single and multiple doses of enzastaurin HCl (LY317615), a protein kinase C‐beta inhibitor, in healthy subjects. J Clin Pharmacol 2007; 47: 1138–51. [DOI] [PubMed] [Google Scholar]
- 13. Therasse P, Arbuck SG, Eisenhauer EA et al. New guidelines to evaluate the response to treatment in solid tumors: European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 14. Strevel EL, Ing DJ, Siu LL. Molecularly targeted oncology therapeutics and prolongation of the QT interval. J Clin Oncol 2007; 25: 3362–33671. [DOI] [PubMed] [Google Scholar]
- 15. Robertson MJ, Kahl BS, Vose JM et al. Phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory diffuse large B‐cell lymphoma. J Clin Oncol 2007; 25: 1741–6. [DOI] [PubMed] [Google Scholar]