

Abstract
Photodynamic therapy (PDT) is considered to be effective treatment for many cancers including lung cancer, head and neck cancers, and prostate cancer. It uses the combination of nontoxic photosensitizers and harmless visible light to generate reactive oxygen species and kill cells. However, DNA repair and reactive oxygen species‐induced signaling pathway activation play crucial roles in cellular response to PDT and may also result in therapeutic limitation of PDT. To improve the cancer therapeutic efficacy of PDT, we targeted apurinic/apyrimidinic endonuclease (APE1), which is essential for both DNA repair and redox regulation of gene transcription, as a potential candidate for PDT combined gene therapy. In our study, an adenovirus‐mediated APE1 silencing strategy was introduced to test its therapeutic enhancement for the non‐small cell lung cancer cell line A549 both in vitro and in vivo after hematoporphrphyrin derivative (HpD)‐mediated PDT. The adenovirus vector Ad5/F35‐shAPE1 was validated to significantly suppress the protein expression of APE1 in cultured A549 cell and in its xenograft of nude mice. Ad5/F35‐shAPE1 effectively inhibited APE1 protein upregulation induced by PDT and resulted in an increase in A549 cell killing by photoirradiation compared with the hematoporphrphyrin derivative‐PDT alone group. Ad5/F35‐shAPE1 suppressed the DNA repair capacity for single‐strand breaks and abolished the activation of some stress‐related transcription factors such as hypoxia‐induced factor (HIF)‐1 that consequently lead to increased cell apoptosis after PDT. Additionally, knock down of APE1 enhanced the tumor suppression efficacy of PDT on the A549 xenograft. Our study indicated that APE1‐targeted gene therapy combined with PDT is a promising strategy for enhancement of the efficacy of PDT in treatment of non‐small cell lung cancer. (Cancer Sci 2009)
Photodynamic therapy is a promising antitumor therapeutic regimen developed recently, which requires accumulation of photosensitizers (PS) in tumors that are locally activated by low‐power laser light, which is delivered using optical fibers. PDT is currently being permitted to treat many cancers including lung cancer, head and neck cancers, and prostate cancer.( 1 ) This unique therapy has dual selectivity because PS tend to accumulate in tumors or other tissue lesions( 2 ) and a light beam can be accurately focused on the tumor mass, with less side effects compared with cancer radiotherapy and chemotherapy. The most important biochemical effect of PDT is that PS combined with light irradiation at a specific wavelength generates a cytotoxic oxygen singlet( 3 ) and ROS, which can consequently result in oxidative stress. The end result is efficient induction of cell death, primarily through apoptosis, microvascular damage, and an antitumor immune response.
However, repair of oxidative DNA damage and activation of stress‐induced signaling pathways following PDT‐induced oxidative stress play a vital role in cellular rescue responses, which are considered as the main factors limiting the therapeutic effect of PDT.( 4 , 5 , 6 , 7 ) DNA is one of the most important cellular targets for oxidative damage. Oxidative DNA damages caused by PDT‐induced oxidative stress are often single base lesions, which are mainly repaired by BER.( 8 ) Meanwhile, ROS is now considered as an important intracellular messenger molecule to stimulate signaling pathways involved in response to oxidative stress. ( 9 ) Recent studies have shown that many stress‐induced TF are activated in these rescue responses after PDT.( 6 , 7 ) Therefore, the discovery of molecular targets that interplay between DNA damage response and signaling pathway regulation could shed light on better therapeutic outcomes of PDT.
Apurinic/apyrimidinic endonuclease 1 is a dual‐function protein with both DNA repair and redox regulation activities of TF.( 10 ) As a rate‐limiting enzyme, APE1 plays a central role in BER initialized by various DNA glycosylases. Many studies have indicated that downregulation or site mutation at the repair site of APE1 abrogates the integrity and stability of the genome after oxidative stress.( 11 , 12 , 13 ) Moreover, APE1 possesses unique redox activity to regulate the DNA binding affinity of certain TF by controlling the redox status of their DNA binding domains. TF (regulated by APE1), including HIF‐1, nuclear factor (NF)‐κB, and AP‐1, are crucial to the cellular response to oxidative stress.( 14 ) APE1 exerts its function through two distinct domains that are combined evolutionally.( 15 ) Based on the distinguished biological roles, APE1 is considered to be a gene that plays a critical role in cellular response to oxidative stress.( 16 ) The biological importance of APE1 is underlined by the findings that mice nullizygous for the APE1 gene are embryonic lethal at a very early stage, which makes APE1 an important molecular target for regulation of cell survival.( 17 ) A growing body of evidence shows that activation of signaling pathways and DNA BER after oxidative stress are coupled.( 18 , 19 ) With its unique dual function in DNA repair and redox regulation of TF, APE1 is supposed to be a critical linker between these two cellular processes. Some studies have shown that the APE1 protein level together with its DNA repair and redox activities are enhanced after oxidative stress.( 20 , 21 ) These findings highlight APE1 as a potential therapeutic candidate for oxidative stress‐related disease.
In the present study, we have raised the hypothesis that APE1 may be of importance in the cellular response to oxidative stress induced by PDT and hence inhibition of APE1 can enhance photodynamic tumor therapeutic effectiveness. To prove our hypothesis, the alteration of APE1 was first assayed at the protein level after HpD‐mediated PDT pretreatment. Then, a knock‐down study of APE1 was carried out in vitro in the NSCLC cell line A549 to test the possible therapeutic enhancement of PDT, and the dual functions of APE1 were measured to demonstrate the importance of APE1 in cell killing by PDT. Additionally, we tested the tumor suppression efficacy of combined use of ad5/F35‐shAPE1 and HpD‐PDT on A549 xenograft so as to provide a promising and effective therapy strategy for NSCLC.
Materials and Methods
Materials, cell lines and mice. Adenovirus vector Ad5/F35‐shAPE1 carrying the human APE1 siRNA sequence, which was designed and validated in our previous studies, was constructed and purified as described previously.( 22 ) The control adenovirus, Ad5/F35‐EGFP, was purchased from Vector Gene Technology Company (Beijing, China). The monoclonal antibody against human APE1 was purchased from Novus Biological (Littleton, CO, USA). Anti‐β‐actin was purchased from Sigma (St Louis, MO, USA) and anti‐cytochrome c from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The human lung carcinoma cell line A549 was purchased from the American Type Culture Collection (Manassas, VA, USA). A549 cells were maintained in DMEM supplemented with 10% (v/v) FBS, 50 mg/mL penicillin–streptomycin, and 2 mm l‐glutamine in 5% CO2 at 37°C, and passaged two to three times a week. Specific pathogen‐free female athymic nude mice, ∼4–6 weeks old, were purchased from Shanghai SLAC Laboratory Animal Co. (Shanghai, China). The care and use of mice were in accordance with our university’s guidelines.
Photodynamic therapy. Hematoprophyrin derivative (Huading, Chongqing, China) was added to cells to a final concentration of 10 μg/mL in the medium. The cells were incubated for 24 h in the absence of light. Cells were then rinsed twice in PBS and fresh DMEM was added. The cells were irradiated using Diomed 630 PDT laser (Diomed, Cambridge, UK) with fiber‐optic delivery, filtered to give an output of 630 ± 3 nm. The cells were exposed to doses between 0 and 10 kJ/m2 at a fluence of 20 mW/cm2. Control cells exposed to HpD alone were treated in the same way as experimental cells but without light.
Infection with adenoviruses. A549 cells were infected with Ad5/F35‐EGFP or Ad5/F35‐APE1 siRNA with increasing MOI for 2 h and replaced with fresh medium. A549 cells were then cultured for another 48 h and then analyzed for their EGFP intensity using a FACScan (Becton Dickinson, Mountain View, CA, USA) or directly observed with a fluorescence microscope (1200 ECM; Nikon, Dusseldorf, Germany).
Alkaline comet assay. The Ad5/F35‐shAPE1‐treated or control cells were rinsed twice with ice‐cold PBS and harvested, and the cell suspension was exposed to light as described above. Immediately after treatment, the cell suspension was stored on ice to prevent DNA repair. The alkaline comet assay was carried out using the Comet assay kit (Trevigen, Gaithersburg, MD, USA) according to the manufacturer’s instructions with modifications. Briefly, the cell suspension was mixed with prewarmed LMAgarose then applied to CometSlide. The agarose‐coated slides were then placed onto a cold surface and allowed to set. The slides were submerged in 4°C lysis solution for 1 h in the absence of light. The slides were transferred to an electrophoresis tank containing unwinding buffer and incubated in this buffer for 45 min and then electrophoresed at 21 V for 30 min. After electrophoresis, the slides were rinsed twice with PBS and stained using ethidium bromide staining. Comets were analyzed using Komet software (Andor, Belfast, UK). The tail moment, which combines a measure of the length of the comet tail and the proportion of DNA to migrate into the tail, was used as an index of DNA damage.
Western blot analysis. Western blots were carried out as previously described to assay APE1 protein expression and cytochrome c release.( 23 ) The antibody against β‐actin (Sigma) was used as a whole cellular or cytoplasmic maker.
Detection of cytochrome c release. Mitochondria‐excluded cytoplasmic fractions were isolated by differential centrifugation as described previously.( 24 ) Briefly, 5 × 108 cells were washed and collected. The cells were resuspended in 1 mL of Grinding medium (250 mm sucrose, 2 mm EDTA, 1 mg/mL BSA, pH 7.4) and pulse‐sonicated on ice. The lysate was examined with a light microscope to ensure 50–70% cell lysis. The lysate was centrifuged at 800 g and the supernatant was immediately centrifuged at 8500 g to pellet mitochondria. The obtained supernatant was the crude cytosolic fraction. The cytoplasmic release of cytochrome c was detected by western blotting.
Electrophoretic mobility shift assay. Electrophoretic mobility shift assay was accomplished according to the user’s instructions of the LightShift Chemiluminescent EMSA kit (Pierce, Rockford, IL, USA) with minor modifications. Briefly, 5 μg of nuclear protein was incubated with 3′‐biotin‐labeled and purified HIF‐1 consensus probe.( 25 ) After incubation, samples were separated on a pre‐run 5% polyacrylamide gel at 100 V for 90 min and then transferred to a nylon membrane. The probes were detected by horseradish peroxidase (HRP)‐conjugated streptavidin (1:300) and the bands visualized by ECL Advance (Amersham, Uppsala, Sweden).
Real‐time RT‐PCR. Expression of the VEGF‐165 gene was detected by real‐time RT‐PCR. Total RNA was extracted using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) and then reverse transcribed into single‐stranded DNA using the PrimeScript 1st Strand cDNA Synthesis Kit (Takara, Dalian, China). Real‐time RT‐PCR was carried out with a MyiQ real‐time RT‐PCR system (Bio‐Rad Laboratory, Hercules, CA, USA). Primers for VEGF‐165 and β‐actin were designed as previously described.( 26 ) Gene expression was determined by normalization against β‐actin expression.
MTT assay. Cells (2 × 103) were inoculated into 96‐well plates (200 μL/well) and incubated with MTT for 4 h at 48 h post irradiation. Then, the culture medium was removed, and 150 μL DMSO was added into each well. The plates were shaken on a swing bed for 10 min and the OD value at 492 nm was determined using a microplate reader. Cell viability (%) = OD value of treatment group/OD value of control group × 100%.
Clonogenic survival assay. Following irradiation, 5 × 103 cells of each group were plated into another well at 35 mm in diameter and returned to 37°C incubation for 12 days. Colonies were fixed and stained in 0.1% crystal violet in absolute ethanol for cell counting. Clones of at least 50 cells were counted as one colony.
Apoptosis assay. Cells were treated in different ways as described above and measured by flow cytometry using the Pacific Blue‐conjugated Annexin V/7‐aminoactinomycin D apoptosis kit (Molecular Probes, Invitrogen, Carlsbad, CA, USA).
In vivo experiments. A549 cells (5 × 106) suspended in 100 μL PBS were injected subcutaneously into the right flank of nude mice. When the tumors grew to approximately 100 mm3 on days 13–15 after cell injection, 40 tumor‐bearing mice were randomized into the following four treatment groups (10 animals per group): Ad5/F35‐EGFP group (group A), Ad5/F35‐shAPE1 group (group B), Ad5/F35‐EGFP + PDT group (group C), and Ad5/F35‐shAPE1 + PDT group (group D). Mice in groups A and C were injected directly into the tumors with 50 μL (5 × 108 IU) Ad5/F35‐EGFP, and groups B and D with 50 μL (5 × 108 IU) Ad5/F35‐APE1 siRNA. One day later, tumors in groups C and D were injected with 20 mg/kg HpD then maintained in a dark room for another day. Then, the mice were treated with 12 kJ/m2 of photodynamic irradiation. The bodyweight and volume of xenografts were measured in a blinded fashion using callipers every day, and tumor size was calculated according to the formula AB2/2 (A stands for the longest diameter and B for the shortest diameter of the tumor measured in two different planes). One day before PDT irradiation, tumor volume was measured on graphs. In separate experiments, five mice of each group were killed on day 3 post injection for immunohistochemical analysis of APE1 expression. Tumor tissues were fixed in 10% formalin, embedded in paraffin, and sectioned.
Statistical analysis. All quantitative data were obtained from three independent experiments and expressed as mean ± SD. Statistical differences between two groups were determined by one‐way analysis of the variance (ANOVA) with computer software SPSS 10.0 (SPSS, Chicago, IL, USA). All P‐values were two sided, and P < 0.05 was considered as statistical difference.
Results
APE1 was induced by HpD‐mediated PTD. To investigate the change of APE1 after HpD‐mediated PTD, we examined protein and enzymic activity levels of APE1 in the A549 cell line at different time points after the same photodynamic regime (HpD at 10 μg/mL, light at a fluence of 4.5 kJ/m2), which showed that the APE1 protein level in A549 cells was significantly elevated at 6 h after PDT and remained at a high level at 48 h (Fig. 1). DNA repair and redox activities were both upregulated with higher protein levels of APE1 in a time‐course by HpD‐mediated PDT irradiation (data not shown).
Figure 1.
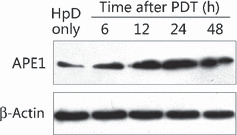
Expression of apurinic/apyrimidinic endonuclease (APE) 1 protein was induced by hematoprophyrin derivative (HpD)‐mediated photodynamic therapy (PDT). The protein level of APE1 in whole A549 cell extracts was analyzed using western blotting, with anti‐β‐actin blots used as a loading control. The APE1 expression level was elevated as early as 6 h after PDT and then gradually increased in a time‐dependent manner. The expression of APE1 reached a peak at 24 h and remained at a high level at 48 h after PDT.
APE1 was critical to the NSCLC cell killing effect in PDT.
APE1 was effectively knocked down by Ad5/F35‐shAPE1 in A549 cells. To investigate the role of APE1 in cell death after PDT, adenovirus carrying an APE1‐targeted siRNA sequence was introduced. Ad5/F35‐shAPE1 was previously constructed and successfully used to knock down APE1 expression in human sporadic colon cancer LOVO cells.( 22 ) To validate the knock‐down effect in human lung carcinoma A549 cells, we examined the transduction efficiency of the adenovirus vector and suppression of APE1 at the protein level after Ad5/F35‐shAPE1 infection. As the EGFP and shAPE1 sequences were expressed in the same vector, we thereby regarded the percentage of positive EGFP cells as putative infectivity of Ad5/F35‐shAPE1 in this study. We observed a dose‐dependent infectivity at 48 h after transduction with different doses of Ad5/F35‐shAPE1 and detected significant infectivity (>90%) with 10 MOI Ad5/F35‐shAPE1 after transduction (Fig. 2). The higher MOI enhanced infectivity, but caused more cell death. Therefore, the dose of 10 MOI was used in suppression of APE1 expression and cellular viability assays. We subsequently investigated the suppression effect of APE1 protein level by Ad5/F35‐shAPE1. APE1 protein level was significantly decreased after 10 MOI of Ad5/F35‐shAPE1 infection in a time course. Figure 3 shows that the suppression rate of APE1 was approximately 85% at 48 h after infection and that the suppression effect was maintained for the next 24 h.
Figure 2.
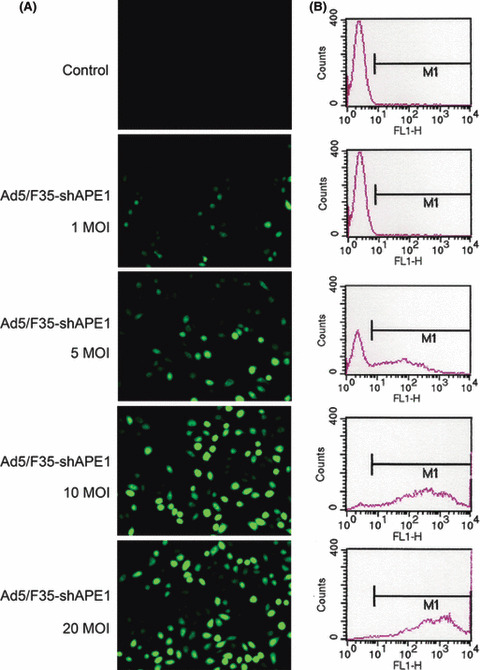
Ad5/F35‐shAPE1 had a high infectivity to A549 cells. Ad5/F35‐shAPE1 carried a CMV promoter‐driven EGFP gene that could be expressed after transduction into eukaryotic cells. Direct detection of EGFP fluorescence by (A) fluorescence microscopy and (B) flow cytometry were used to assay Ad5/F35‐shAPE1 infectivity. It is shown that the adenovirus vector infected A549 cells in a dose‐dependent fashion and 10 MOI rendered >90% infectivity. Higher MOI (such as 20 MOI) enhanced infectivity, but brought more cell death. Therefore, 10 MOI was adopted in all infection experiments of this study.
Figure 3.
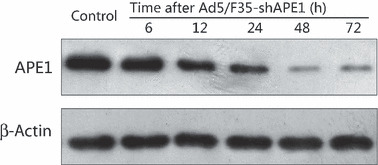
Ad5/F35‐shAPE1 effectively knocked down the apurinic/ apyrimidinic endonuclease (APE) 1 expression in a time course. APE1 protein level was assayed by western blotting at 6, 12, 24, 48 and 72 h after 10 MOI Ad5/F35‐shAPE1 transduced to A549 cells. Ad5/F35‐EGFP carrying no shRNA sequence was also infected at the same dose as a negative control. APE1 expression was inhibited at 12 h and significantly knocked down at 48 h after Ad5/F35‐shAPE1 infection and remained at a low level for another 48 h.
Knock down of APE1 enhanced overall cell killing by PDT. MTT and colony formation assays were used to determine whether the knock down of cellular APE1 level enhanced the cell killing effect of HpD‐mediated PDT. The MTT assay was done on cells treated with various fluences (∼1.5–9 kJ/m2) of PDT, and the percentage of viable cells was determined at 24 h after PDT treatment. A significantly decreased cell survival was observed in Ad5/F35‐shAPE1‐infected cells compared with Ad5/F35‐EGFP‐infected cells, with no statistical differences upon cellular survival between Ad5/F35‐EGFP‐infected and only HpD‐treated cells (Fig. 4A). In addition, there was no statistical difference in aspect of cellular viability under various doses of HpD (data was not shown). Because the MTT assay could provide only overall measurement of cell viability rather than long‐term cell survival and proliferation, we further chose a clonogenic survival assay. Due to sparse plating, these cells were more sensitive to PDT‐induced oxidative stress than confluent cells. Therefore, the clonogenic survival assay required lower fluences of PDT (∼0.5–3 kJ/m2) in comparison with the MTT assay. Figure 4(B) reveals a significant decrease in cell colonies at all tested fluences of PDT in Ad5/F35‐shAPE1‐infected cells compared with Ad5/ F35‐EGFP‐infected cells, but insignificant statistical differences upon cellular survival between Ad5/F35‐EGFP‐infected cells and only HpD‐treated cells.
Figure 4.
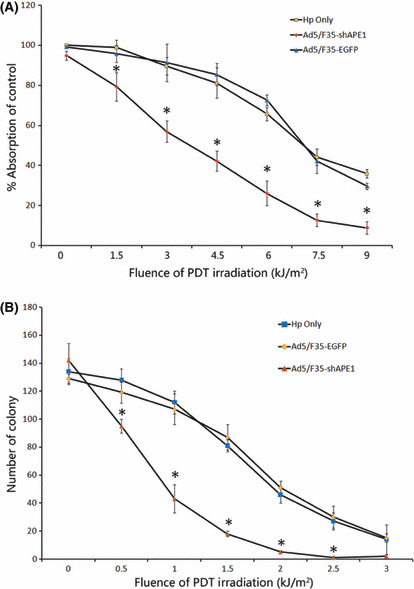
Ad5/F35‐shAPE1 enhanced cell killing after hematoprophyrin derivative (HpD)‐based photodynamic irradiation. Early cell death and proliferative cell death after photodynamic irradiation were detected by (A) MTT and (B) colony formation assay. The HpD‐only and Ad5/F35‐EGFP groups were also tested as controls. MTT assay showed that Ad5/F35‐shAPE1 effectively enhanced the cell killing effect at 24 h after photodynamic therapy (PDT) irradiation under 9 kJ/m2 compared with the HpD‐only and Ad5/F35‐EGFP groups. Colony formation assay showed that Ad5/F35‐shAPE1 decreased by >50% colony formation at 2 week after 1 kJ/m2 PDT compared with the HpD‐only and Ad5/F35‐EGFP groups. In both assays, there was no significant difference between the HpD‐only and Ad5/F35‐EGFP groups. Results were obtained from three independent experiments. *Cell death differed significantly from the HpD‐only and Ad5/F35‐EGFP groups (P < 0.05).
Knock down of APE1 enhanced apoptosis induced by PDT. We investigated the effect of APE1 knock down by Ad5/F35‐shAPE1 on PDT‐induced apoptosis. As shown in Figure 5(A,B), the percentage of apoptotic cells in the Ad5/F35‐shAPE1‐infected group was significantly higher than that in the Ad5/ F35‐EGFP‐infected groups at fluencies of 3 and 9 kJ/m2. Previous studies revealed that the programmed cell death caused by PDT irradiation was mainly via a mitochondria‐mediated pathway,( 27 ) and release of cytochrome c was considered to be a hallmark of mitochondria‐dependent apoptosis. Therefore, we detected the release of cytochrome c by western blotting, which indicated that the Ad5/F35‐shAPE1 infection promoted mitochondrial dysfunction and release of cytochrome c and consequently triggered the mitochondrial pathway of apoptosis, compared with the Ad5/F35‐EGFP infection (Fig. 5C).
Figure 5.
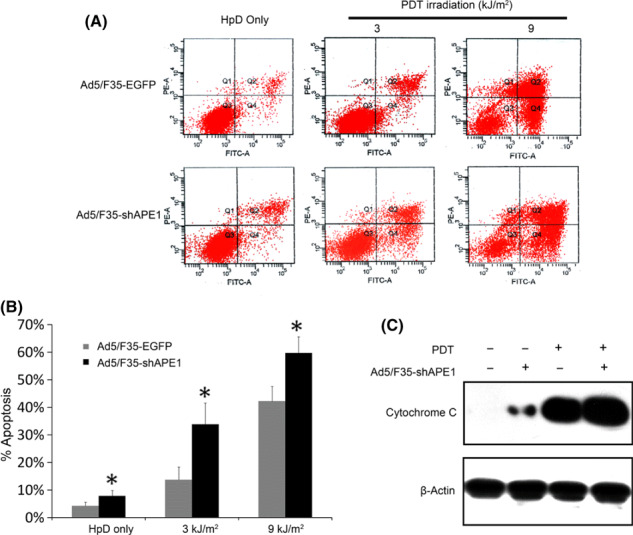
Knock down of apurinic/apyrimidinic endonuclease (APE) 1 by Ad5/F35‐shAPE1 promoted photodynamic therapy (PDT)‐induced apoptosis of A549 cells in a mitochondria‐dependent way. (A) Dot plot of the apoptotic assay of the Ad5/F35‐shAPE1 group at 6 h after PDT irradiation, with the Ad5/F35‐EGFP group as a negative control. The x‐axis is Annexin‐V–Pacific Blue and the y‐axis is 7‐aminoactinomycin D (7‐AAD) for all graphs represented. The cells undergoing early apoptosis are Annexin‐V‐positive and 7‐AAD‐negative in the lower right quadrants. (B) The apoptotic cell percentages of each group are shown in a graphic pattern. After photodynamic irradiation, there were significantly more cells undergoing apoptosis in the Ad5/F35‐shAPE1 group than in the Ad5/F35‐EGFP group. The cytoplasmic fractions were subjected to immunoblotting with anti‐cytochrome c antibody, and the anti‐β‐actin antibody was used as a cytoplasmic fraction loading control. (C) In the Ad5/F35‐shAPE1 group the mitochondria‐excluded cytoplasmic fraction contained more cytochrome c before or 6 h after PDT irradiation compared with the Ad5/F35‐EGFP group. *The apoptotic cell percentage differed significantly from Ad5/F35‐EGFP controls (P <0.05).
Ad5/F35‐shAPE1 inhibited the upregulation of APE1 induced by PDT. To investigate the possible mechanisms leading to increased cellular sensitivity to PDT treatment, we first assayed the APE1 protein level of the Ad5/F35‐shAPE1‐infected group and HpD‐alone group at 12 h after 4.5 kJ/m2 PDT irradiation. As shown in Figure 6(A), upregulation of APE1 expression induced by PDT was significantly inhibited by Ad5/F35‐shAPE1 pretreatment. As a dual‐functional factor, APE1 plays important roles in both DNA repair and redox regulation of TF. We then examined single‐strand DNA damage of the Ad5/F35‐shAPE1‐pretreated and untreated groups after PDT using an alkaline comet assay (Fig. 6B).( 28 ) Initial DNA single base lesions in the Ad5/F35‐shAPE1 group were slightly increased compared with the control group. The HpD‐alone control group was able to gradually repair a significant proportion of the DNA lesions, while the Ad5/F35‐shAPE1‐infected group did not repair an appreciable amount of damage for 24 h after PDT treatment. These data suggested that the repair capacity of APE1 for single base DNA lesions was partly suppressed by Ad5/F35‐shAPE1 infection. HIF‐1 is one of the TF regulated by APE1 through a redox‐dependent mechanism and HIF‐1 activation is considered to be a critical biological issue in response to PDT irradiation.( 29 ) We then measured the HIF‐1 transcriptional activity by quantification of VEGF expression, which is the most important downstream gene of HIF‐1. Quantification of the RT‐PCR results showed that VEGF expression was increased by PDT irradiation and could be effectively blocked by Ad5/F35‐shAPE1 (Fig. 6C,D).
Figure 6.
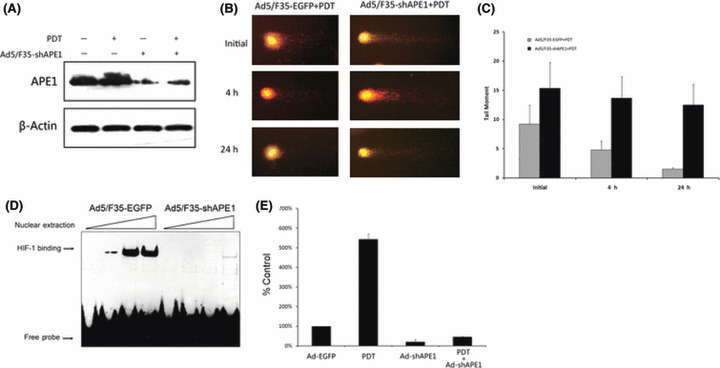
Both DNA repair and redox activities were involved in cellular survival after photodynamic irradiation. The Ad5/F35‐shAPE1‐infected or Ad5/F35‐EGFP‐infected groups received 4.5 kJ/m2 photodynamic irradiation at 48 h after infection. Then, APE1 protein level was assayed by western blotting at 12 h after irradiation. (A) The upregulation of APE1 induced by photodynamic therapy (PDT) was significantly abolished after Ad5/F35‐shAPE1 infection. (B) DNA single‐strand break at initial (immediately after irradiation), 4, and 24 h after PDT in the Ad5/F35‐shAPE1 and control groups using alkaline comet assay. (C) The ‘tail’ of the comet, which was quantified by Komet software, was considered as the impaired DNA fragment. It demonstrated that the repair capacity to this type of DNA lesion was significantly suppressed by Ad5/F35‐shAPE1 infection and the DNA damage caused by PDT was not completely repaired at 24 h post‐irradiation, which was repaired at 4 h in the control group. We then assayed the transcription factor redox regulation activity of APE1 by (D) EMSA and (E) RT‐PCR 12 h after PDT. Oligonucleotide containing HIF‐1 consensus was used to assay the DNA binding activity of HIF‐1, which is regulated by APE1. It was shown that the DNA binding activity was significantly elevated after PDT and inhibited by Ad5/F35‐shAPE1 infection. The expression of VEGF, an important downstream gene of HIF‐1, was then assayed by real‐time RT‐PCR. In accordance with EMSA results, real‐time RT‐PCR indicated that the expression of VEGF was significantly upregulated by PDT and downregulated by Ad5/F35‐shAPE1. Results were obtained from three independent experiments.
Knock down of APE1 potentiated the inhibition of tumor growth by PDT in vivo.
Ad5/F35‐shAPE1 could inhibit PDT‐induced APE1 increase in vivo. We investigated the APE1 protein levels in A549 xenografts on day 3 post injection of Ad5/F35‐shAPE1 by immunohistochemistry. In the adenovirus‐treated group, both nuclear and cytoplasmic immunostaining levels of APE1 were significantly decreased compared with the control group (Fig. 7A,B). To determine whether the adenovirus infection further abolished upregulation of APE1 upon PDT treatment, we also examined the APE1 protein level on day 3 post irradiation by immunohistochemistry. The results showed that APE1 protein immunostaining was increased after irradiation (Fig. 7C), whereas it remained at a lower level in the adenovirus infection group (Fig. 7D).
Figure 7.
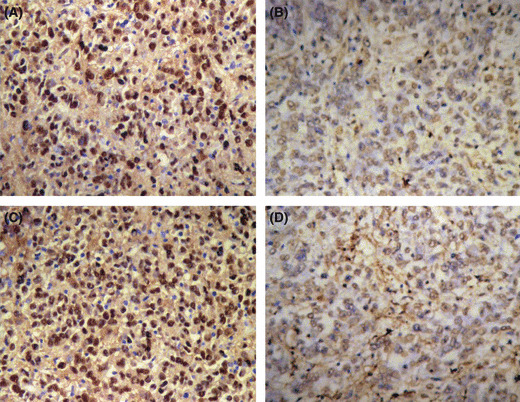
Ad5/F35‐shAPE1 inhibited the upregulation of apurinic/apyrimidinic endonuclease (APE) 1 protein induced by photodynamic therapy (PDT) in vivo. Tumor tissue samples from (A) group A, (B) 3 days after Ad5/F35‐shAPE1 infection, (C) 48 h post‐PDT irradiation of group A, and (D) group B were subjected to APE1 antibody using immunohistochemistry (IHC). (A,C) APE1 had strong positive staining and was mainly localized to the nucleus. (B) Tumor cells had APE1‐negative staining. (D) PDT irradiation failed to induce APE1 expression.
Knock down of APE1 potentiated the inhibition of tumor growth by PDT in vivo. To further verify the hypothesis that suppression of the APE1 expression level in A549 xenografts potentiated enhanced tumor killing effects after PDT irradiation, tumor‐bearing mice were injected intratumorally with Ad5/F35‐shAPE1 or Ad5/F35‐EGFP and treated with the PDT regime 3 d later. On day 21, A549 xenografts from each group were completed isolated and tumor volumes were then examined exactly (Fig. 8A). The results showed that the tumor‐inhibition rates in groups A, C, and D at day 21 were 34.85%, 52.19%, and 93.97%, respectively (Fig. 8B).
Figure 8.
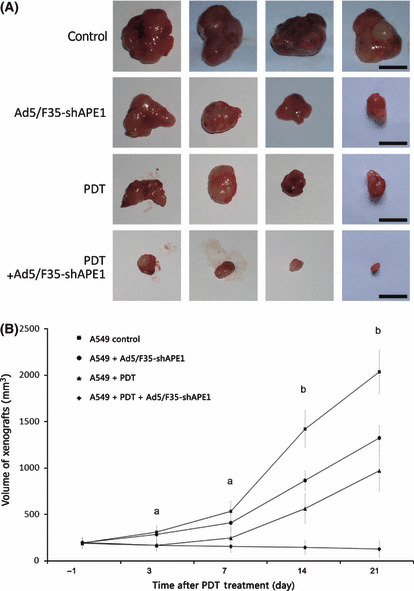
Ad5/F35‐shAPE1 enhanced the suppression effect of photodynamic therapy (PDT) on tumor growth of the A549 xenograft. The tumors of tumor‐bearing mice were injected directly with 5 × 108 IU Ad5/F35‐shAPE1 or Ad5/F35‐EGFP. Three days later, tumors were irradiated at 12 kJ/m2. (A) Isolated xenografts from each group at day 21 post‐irradiation. (B) Tumor growth was measured in two dimensions and tumor volume was recorded. From 1 day before PDT, tumor volume was measured on graphs. Scale bar = 10 mm. aThe tumor volume of the Ad5/F35‐shAPE1 + PDT group differed significantly from Ad5/F35‐EGFP only controls, but there were no statistical differences between the Ad5/F35‐shAPE1 + PDT and PDT‐only groups or between the Ad5/F35‐shAPE1 and Ad5/F35‐EGFP only group. bThe tumor volume differed significantly among all treated groups (P < 0.05).
Discussion
In the present study, we first found that the dual‐functional protein APE1 was induced by HpD‐mediated PDT, which made us presume that APE1 may play a crucial role in cellular response to PDT. To validate this hypothesis, we used an RNAi adenovirus against APE1 to downregulate the expression of APE1 in the cultured human lung carcinoma A549 cell as well as in a nude mice xenograft and found a significant decrease in APE1 expression along with its enzymic level after Ad5/F35‐shAPE1 infection. The suppression of APE1 enhanced the cancer cell killing effect of PDT both in vitro and in vivo, indicating that the dual functional protein APE1 is of biological importance in cellular response to PDT irradiation. In accordance with previous studies by us and other groups, the downregulation of cellular APE1 protein level can enhance cell sensitivity to many DNA damage agents including methyl methanesulfonate (MMS), ionizing radiation, and cisplatin.( 22 , 30 , 31 ) All of these DNA damage agents can cause single base lesions such as alkylation or oxidation, which are mainly repaired by BER.
It is widely accepted that the PDT‐mediated DNA damage is limited, which is because the photogenerated singlet oxygen has a very short life and very limited diffusion in biological systems (half‐life <0.04 μs, radius of action <0.02 μm), indicating that singlet oxygen less likely causes DNA damage unless it either binds or localizes close to the DNA.( 32 ) Previous studies using the alkaline comet assay revealed that HpD‐based photodynamic irradiation could cause obvious initial DNA damage that could be completely repaired by an intact DNA repair system within 4 h.( 28 ) The major DNA damage after PDT is oxidative base lesion induced by photogenerated oxidative stress, which is the typical lesion for BER. APE1 is the rate‐limited enzyme of BER, which makes it a promising target for cancer gene therapy. In the present study, we further explored the possible mechanism of the enhanced cell killing effect of PDT after suppression of APE1 expression and found that APE1 silencing could effectively slow down the repair of initial photogenerated DNA damage, which was thought to be caused by the loss of APE1 DNA repair activity, resulting in a number of impaired AP sites. Accumulation of genetoxic AP sites can further lead to proliferative cellular death, which was detected by colony formation assay in this research.
APE1 is a special dual‐functional protein that possesses two distinct activities important to cell survival after stress. Our previous studies successfully used APE1‐specific RNAi vectors to inhibit APE1 protein levels in various cell lines. These studies had shown that knock down of APE1 significantly enhanced cellular sensitivity to cancer therapeutic agents including ionizing radiation( 22 ) and cisplatin.( 31 ) Recently, our unpublished data showed that suppression of APE1 sensitized the colorectal cancer cell LOVO line to 5‐fluorouracil. In our research, we noticed that APE1 also plays an important role in cellular response to photodynamic irradiation via regulation of TF by redox activity of APE1. ROS generated by oxidative stress have recently been considered an intracellular signal that can activate many TF including HIF‐1. As one of the classic oxidative stress‐related TF, HIF‐1’s activation requires expression of HIF‐1α and its heterodimerization with HIF‐1β,( 33 ) and leads to stimulated expression of its downstream genes such as VEGF, Heme Oxygenase 1 (HO‐1), and platelet‐derived growth factor (PDGF) that enhance the cellular survival rate after oxidative stress. APE1 participates in this procedure by regulating the transcriptional activity of HIF‐1 though its redox domain. In the present study, we observed a significant decrease of the DNA motif binding affinity of HIF‐1 when knocking down the APE1 protein. Consequently, expression of VEGF was dramatically downregulated, which also contributed to the enhanced cell killing effect by PDT. Our results elucidated the importance of both functions of APE1 in cell response to PDT, which makes it a leading target for gene therapy combined with PDT. In comparison with the mechanism of knock down of APE1 enhancing sensitivity to cisplatin‐based cancer chemotherapy, the mechanisms of photodynamic cell killing and ionizing radiation are more similar. Photodynamic irradiation and ionizing radiation are inclined to produce extensive oxidative stress, which further induces DNA oxidative lesions and stress‐related pathway activation. Although ionizing radiation can directly cause DNA double‐strand breaks, APE1 merely has a minor role in the repair activity of this type of damage. Meanwhile, cisplatin mainly causes DNA adducts and interstrand crosslink damage, which is mainly repaired by DNA repair mechanisms. Based on the dual functions of APE1, we considered that both activities participated in the tumor resistance to photodynamic irradiation and ionizing radiation, while its DNA repair may be the major activity involved in cellular resistance to cisplatin.
We also assayed apoptosis after PDT and found that the apoptotic rate in the Ad5/F35‐shAPE1‐infected group was elevated. Further research indicated that the mitochondria‐dependent apoptotic pathway was stimulated by PDT alone, knock down of APE1, or both. Recently, Vascotto et al. also demonstrated that APE1 silencing leads to mitochondria‐associated apoptosis and postulated that the mitochondria‐associated apoptosis was caused by loss of expression of APE1 and directly related to its mtDNA repair.( 34 ) Our previous study showed scarce mitochondrial expression of APE1, which weakened the mtDNA repair capacity.( 23 ) APE1 was mostly localized to the nucleus of A549 cells and knock down of APE1 mainly decreased its nuclear expression. Therefore, we think that APE1 may play an indirect role in the functional regulation of mitochondria. Our unpublished data indicate that APE1 could regulate some important mitochondria‐related TF including nuclear respiratory factor 1 (NRF‐1) and mitochondrial transcription factor A (TFAM) to mainly exert their activity in the nucleus, indicating the correlation between APE1 expression and mitochondrial function.
To the best of our knowledge, this is the first study to explore APE1‐targeted gene therapy combined with PDT both in vitro and in vivo. Our results firmly demonstrate that the dual functions of APE1 play an important role in cellular response to HpD‐mediated PDT and that APE1 silencing through an adenovirus vector may be a promising strategy for PDT combined with gene therapy. However, the extent of DNA damage and oxidative stress varied when different photosensitizers and photodynamic irradiation strengths were used. Future studies should be done to work out an optimal regime for PDT combined with APE1‐targeted gene therapy and to test its effectiveness in other types of cancers.
Abbreviations
- AP
apurinic/apyrimidinic
- APE
apurinic/apyrimidinic endonuclease
- BER
base excision repair
- EGFP
enhanced green fluorescent protein
- EMSA
electrophoretic mobility shift assay
- HIF
hypoxia‐induced factor
- HpD
hematoprophyrin derivative
- MOI
multiplicities of infection
- NSCLC
non‐small cell lung cancer
- PDT
photodynamic therapy
- PS
photosensitizers
- ROS
reactive oxygen species
- TF
transcriptional factors
- VEGF
vascular endothelial growth factor
Acknowledgments
We thank Dr Xiao‐Jing Cao, Mrs Yu‐Xin Yang, and Miss Ling Liao for their kind and excellent technical assistance. This study was supported by Grants from the National Natural Science Foundation of China (No. 30571849) to Professor Zhen‐Zhou Yang.
References
- 1. Harrod‐Kim P. Tumor ablation with photodynamic therapy: introduction to mechanism and clinical applications. J Vasc Interv Radiol 2006; 17: 1441–8. [DOI] [PubMed] [Google Scholar]
- 2. Solban N, Rizvi I, Hasan T. Targeted photodynamic therapy. Lasers Surg Med 2006; 38: 522–31. [DOI] [PubMed] [Google Scholar]
- 3. Plaetzer K, Krammer B, Berlanda J et al. Photophysics and photochemistry of photodynamic therapy: fundamental aspects. Lasers Med Sci 2009; 24: 259–68. [DOI] [PubMed] [Google Scholar]
- 4. Oleinick NL, Evans HH. The photobiology of photodynamic therapy: cellular targets and mechanisms. Radiat Res 1998; 150: S146–56. [PubMed] [Google Scholar]
- 5. Buytaert E, Dewaele M, Agostinis P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta 2007; 1776: 86–107. [DOI] [PubMed] [Google Scholar]
- 6. Mennel S, Meyer CH, Callizo J. Combined intravitreal anti‐vascular endothelial growth factor (Avastin) and photodynamic therapy to treat retinal juxtapapillary capillary haemangioma. Acta Ophthalmol 2009. doi:DOI: 10.1111/j.1755-3768.2008.01449.x [DOI] [PubMed] [Google Scholar]
- 7. Nowis D, Legat M, Grzela T et al. Heme oxygenase‐1 protects tumor cells against photodynamic therapy‐mediated cytotoxicity. Oncogene 2006; 25: 3365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maynard S, Schurman SH, Harboe C et al. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009; 30: 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther 2008; 7: 1875–84. [DOI] [PubMed] [Google Scholar]
- 10. Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref‐1 as a therapeutic and chemopreventive target. Mol Aspects Med 2007; 28: 375–95. [DOI] [PubMed] [Google Scholar]
- 11. McNeill DR, Lam W, Deweese TL et al. Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol Cancer Res 2009; 7: 897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farkasova T, Gurska S, Witkovsky V et al. Significance of amino acid substitution variants of DNA repair genes in radiosusceptibility of cervical cancer patients: a pilot study. Neoplasma 2008; 55 (4): 330–7. [PubMed] [Google Scholar]
- 13. McNeill DR, Wilson DM III. A dominant‐negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA‐damaging agents. Mol Cancer Res 2007; 5: 61–70. [DOI] [PubMed] [Google Scholar]
- 14. Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP‐endonuclease (APE1/Ref‐1), an essential multifunctional protein. Antioxid Redox Signal 2009; 11: 621–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Georgiadis MM, Luo M, Gaur RK et al. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res 2008; 643: 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luo M, Delaplane S, Jiang A et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref‐1 in cancer and endothelial cells: small‐molecule inhibition of the redox function of Ape1. Antioxid Redox Signal 2008; 10: 1853–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xanthoudakis S, Smeyne RJ, Wallace JD et al. The redox/DNA repair protein, Ref‐1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A 1996; 93: 8919–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Breit JF, Ault‐Ziel K, Al‐Mehdi AB et al. Nuclear protein‐induced bending and flexing of the hypoxic response element of the rat vascular endothelial growth factor promoter. FASEB J 2008; 22: 19–29. [DOI] [PubMed] [Google Scholar]
- 19. Ziel KA, Grishko V, Campbell CC et al. Oxidants in signal transduction: impact on DNA integrity and gene expression. FASEB J 2005; 19: 387–94. [DOI] [PubMed] [Google Scholar]
- 20. Jiang Y, Guo C, Vasko MR et al. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res 2008; 68: 6425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paap B, Wilson DM III, Sutherland BM. Human abasic endonuclease action on multilesion abasic clusters: implications for radiation‐induced biological damage. Nucleic Acids Res 2008; 36: 2717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiang DB, Chen ZT, Wang D et al. Chimeric adenoviral vector Ad5/F35‐mediated APE1 siRNA enhances sensitivity of human colorectal cancer cells to radiotherapy in vitro and in vivo . Cancer Gene Ther 2008; 15: 625–35. [DOI] [PubMed] [Google Scholar]
- 23. Li MX, Wang D, Zhong ZY et al. Targeting truncated APE1 in mitochondria enhances cell survival after oxidative stress. Free Radic Biol Med 2008; 45: 592–601. [DOI] [PubMed] [Google Scholar]
- 24. Abou‐Khalil S, Abou‐Khalil WH, Planas L et al. Interaction of rhodamine 123 with mitochondria isolated from drug‐sensitive and ‐resistant Friend leukemia cells. Biochem Biophys Res Commun 1985; 127: 1039–44. [DOI] [PubMed] [Google Scholar]
- 25. Kim KS, Rajagopal V, Gonsalves C et al. A novel role of hypoxia‐inducible factor in cobalt chloride‐ and hypoxia‐mediated expression of IL‐8 chemokine in human endothelial cells. J Immunol 2006; 177: 7211–24. [DOI] [PubMed] [Google Scholar]
- 26. Rashid G, Bernheim J, Green J et al. Parathyroid hormone stimulates the endothelial expression of vascular endothelial growth factor. Eur J Clin Invest 2008; 38: 798–803. [DOI] [PubMed] [Google Scholar]
- 27. Hilf R. Mitochondria are targets of photodynamic therapy. J Bioenerg Biomembr 2007; 39: 85–9. [DOI] [PubMed] [Google Scholar]
- 28. Haylett AK, Ward TH, Moore JV. DNA damage and repair in Gorlin syndrome and normal fibroblasts after aminolevulinic acid photodynamic therapy: a comet assay study. Photochem Photobiol 2003; 78: 337–41. [DOI] [PubMed] [Google Scholar]
- 29. Mitra S, Cassar SE, Niles DJ et al. Photodynamic therapy mediates the oxygen‐independent activation of hypoxia‐inducible factor 1alpha. Mol Cancer Ther 2006; 5: 3268–74. [DOI] [PubMed] [Google Scholar]
- 30. Luo M, Kelley MR. Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res 2004; 24: 2127–34. [PubMed] [Google Scholar]
- 31. Wang D, Xiang DB, Yang XQ et al. APE1 overexpression is associated with cisplatin resistance in non‐small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells, 2009. doi: 10.1016/j.lungcan.2009.02.019. [DOI] [PubMed]
- 32. Jarvi MT, Niedre MJ, Patterson MS et al. Singlet oxygen luminescence dosimetry (SOLD) for photodynamic therapy: current status, challenges and future prospects. Photochem Photobiol 2006; 82: 1198–210. [DOI] [PubMed] [Google Scholar]
- 33. Hofer T, Desbaillets I, Höpfl G et al. Dissecting hypoxia‐dependent and hypoxia‐independent steps in the HIF‐1alpha activation cascade: implications for HIF‐1alpha gene therapy. FASEB J 2001; 15: 2715–7. [DOI] [PubMed] [Google Scholar]
- 34. Vascotto C, Cesaratto L, Zeef LA et al. Genome‐wide analysis and proteomic studies reveal APE1/Ref‐1 multifunctional role in mammalian cells. Proteomics 2009; 9: 1058–74. [DOI] [PMC free article] [PubMed] [Google Scholar]