

Abstract
Cyclooxygenase 2 (COX‐2) and retinoid X receptor α (RXRα) are suggested to have roles in carcinogenesis. COX‐2 inhibitors have been reported to suppress growth of hepatocellular carcinoma (HCC) cell lines in vitro. However, little is known about the preventive effect of these drugs on spontaneous hepatocarcinogenesis in vivo. Etodolac exists in a racemic mixture containing S‐ and R‐etodolac. S‐etodolac is responsible for COX‐2 inhibitory activity and R‐etodolac is related to the downregulation of RXRα. Here, the effect of etodolac on spontaneous development of HCC in fatty liver Shionogi mice is evaluated. Etodolac was administered at a low (2 mg/kg) or high (10 mg/kg) dose three times a week for 16 months starting at the age of 3 months. The development of HCC was suppressed slightly in the high‐dose group, and suppressed markedly in the low‐dose group, although the development of fatty liver was not inhibited in either group. Plasma prostaglandin E2 levels were also decreased significantly in the low‐dose group, consistent with the suppression of HCC. The expression of RXRα and proliferating cell nuclear antigen in non‐tumorous liver tissues was decreased significantly in both the low‐dose and high‐dose groups. These findings show that etodolac treatment at an optimum dose suppresses hepatocarcinogenesis in vivo, and may be useful for preventing the development of HCC in humans. (Cancer Sci 2006; 97: 768–773)
Abbreviations:
- BW
bodyweight
- COX‐2
cyclooxygenase‐2
- E‐HD
high‐dose treatment group
- E‐LD
low‐dose treatment group
- FLS
fatty liver Shionogi
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- PCNA
proliferating cell nuclear antigen
- PGE2
prostaglandin E2
- RT‐PCR
reverse transcription–polymerase chain reaction
- RXRα
retinoid X receptor α.
Hepatocellular carcinoma is a common malignancy worldwide, accounting for approximately 6% of all human cancers and up to 1 million deaths per year.( 1 , 2 ) Epidemiological studies and clinical observations have indicated that some medicines, such as vitamin A, vitamin K2 and interferon‐α, have chemopreventive effects for hepatocarcinogenesis.( 3 , 4 , 5 ) Because these medicines are not enough to prevent hepatocarcinogenesis in humans, other efficient preventive tools are needed urgently.( 6 )
The use of COX‐2 inhibitors is associated with a reduced development of certain types of tumors, such as colorectal cancer and prostate cancer.( 7 , 8 , 9 ) COX‐2 inhibitors suppress the growth of human HCC implants in nude mice and lung metastasis of HCC in F344 rats, and show preventive effects on chemically induced hepatocarcinogenesis in rats.( 6 , 10 , 11 , 12 , 13 ) We reported previously that a specific COX‐2 inhibitor, etodolac ([±]‐1,8‐diethyl‐1,3,4,9,‐tetrahydropyrano‐[3,4‐b] indole‐1‐acetic acid), decreases the levels of PGE2 and inhibits the expression of PCNA in several HCC cell lines in vitro.( 14 ) Etodolac exists in a racemic mixture containing S‐ and R‐etodolac. S‐etodolac has been shown to possess COX‐2 inhibitory activity and R‐etodolac was recently reported to bind RXRα and to inhibit the development of prostate cancer.( 15 , 16 , 17 ) RXRα, which plays an important role in regulating cell proliferation and differentiation, is expressed abundantly in the liver and is involved in hepatic steatosis and hepatocarcinogenesis in HBV and HCV infection in humans.( 18 , 19 , 20 ) However, little is known about the chemopreventive effect of COX‐2 inhibitors on spontaneous hepatocarcinogenesis in vivo.
Fatty liver Shionogi mouse is an inbred strain that shows neither hyperphagia nor obesity but has an abnormal triglyceride accumulation in hepatocytes after birth.( 21 , 22 ) Fifty percent of the mice show fatty liver grade I and II 9 weeks after birth, and all mice develop fatty liver grade III and IV after 15 weeks.( 21 ) FLS mice develop severe fatty liver (hepatic steatosis) and chronic HCC under normal conditions, in which the incidence of HCC is reached to 52% at 16 months of age.( 22 ) To explore the mechanism involved and to find a specific and effective medicine for the prevention of hepatocarcinogenesis, we studied the effect of a COX‐2 inhibitor, etodolac, on spontaneous development of HCC in FLS mice.
Materials and Methods
Animals and experimental design
Thirty male FLS mice aged 2 months were obtained from Aburahi Laboratories, Shionogi Company (Shiga, Japan). They were housed, one per cage, under specific pathogen‐free conditions in a 12 : 12 h L : D cycle at 23 ± 1°C and 50 ± 10% humidity, and fed a standard CE‐2 diet (CLEA Japan, Tokyo, Japan) and tap water ad libitum. The mice were divided randomly into three groups of 10 mice each. All animals received humane care and all experiments followed the Guidelines for Animal Experimentation of the Japanese Association for Laboratory Animal Science.( 23 ) Two doses of etodolac (Nihon Shinyaku Company, Tokyo, Japan) were used: 2 mg/kg BW for the E‐LD group and 10 mg/kg BW for the E‐HD group. Etodolac was dissolved in 100% ethanol and diluted to suitable concentrations with a 5% aqueous solution of arabic gum. The solutions of etodolac were given to mice by oral gavage, three times per week (Monday, Wednesday and Friday) for 16 months from age 3–18 months. The control group was treated with the same amounts of 0.7% ethanol and 5% arabic gum. The mice were observed weekly for BW, skin damage and general condition. The animals that were still alive at 18 months were anesthetized with diethyl ether and blood was collected from the heart. The livers were immediately removed and weighed. Tumor nodules that had developed were measured for diameter and cut for formalin fixation and paraffin embedding or frozen storage.
Measurement of prostaglandin E2
Prostaglandin E2 levels in the plasma were assayed using the PGE2 High Sensitivity Immunoassay Kit (R & D Minneapolis, MN, USA) as described previously.( 14 )
Histological examination
Tumor and non‐tumorous liver tissues were fixed in 10% neutral‐buffered formalin, embedded in paraffin, cut into sections 5 µm thick, and stained with hematoxylin and eosin. The classification of liver histology was based on the criteria described by Frith and Ward.( 24 )
Immunohistochemical analysis
Paraffin sections from HCC and non‐tumorous liver tissues were deparaffinized in xylene, rehydrated with graded concentrations of ethanol, and treated with antibodies against COX‐2, RXRα and PCNA, as described previously.( 25 ) All antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and used at a dilution of 1/100. The RXRα‐positive and PCNA‐positive cells were counted microscopically in five high‐power fields at magnitude × 400. The labeling index of RXRα and PCNA was expressed as the proportion of cells with positive RXRα and PCNA nuclear activity.
RNA extraction and reverse transcription–polymerase chain reaction
Total RNA was extracted from liver tissues using Isogen (Nippon Gene, Toyama, Japan), and mRNA was prepared using an Oligotex‐dT30 mRNA Purification Kit (Takara Bio, Otsu, Japan) according to the manufacturer's instructions. cDNA was synthesized using random 9‐mers and an RNA PCR Kit (version 2.1; Takara). The primers for polymerase chain reaction were as follows: COX‐2 forward, 5′‐GGTCT GGTGC CTGGT CTGAT GATG‐3′; COX‐2 reverse, 5′‐GTCCT TTCAA GGAGA ATGGT GC‐3′;( 9 ) RXRα forward, 5′‐CTTTG ACAGG GTGCT AACAG AGC‐3′; RXR α reverse, 5′‐ACGCT TCTAG TGACG CATAC ACC‐3′;( 26 )β‐actin forward, ATGGT GGGAA TGGGT CAGAA GGAC‐3′; and β‐actin reverse, 5′‐CTCTT TGATG TCACG CACGA TTTC‐3′.( 27 ) cDNA amplification was carried out under the conditions 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min, using β‐actin as an internal control. The products were analyzed on a 3% NuSieve 3 : 1 agarose gel (FMC BioProducts, Rockland, ME, USA), stained with ethidium bromide and photographed under ultraviolet light.
Statistical analysis
Statistical analysis for the development of HCC was carried out using the Student's t‐test or Fisher's exact test. Values are expressed as mean ± SE. P < 0.05 was considered statistically significant.
Results
Effects of etodolac on HCC development
Histological findings of fatty liver and HCC spontaneously developed in the liver of 18‐month‐old FLS mice are shown in Fig. 1A,B. The expression of COX‐2 protein in the non‐tumorous liver tissues was determined by immunohistological staining (Fig. 1C,D). The mRNA expression of COX‐2 was confirmed by RT‐PCR (Fig. 1E).
Figure 1.
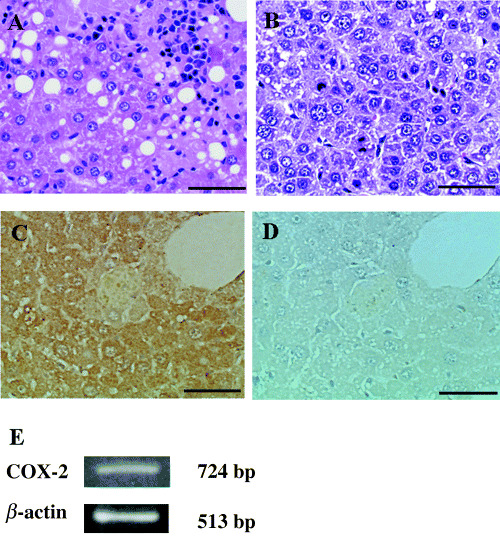
Histopathological features of hepatocellular carcinoma (HCC) and expression of cyclooxygenase‐2 (COX‐2) in non‐tumorous fatty liver of fatty liver Shionogi mice. (A) Non‐tumorous liver tissue, (B) HCC, (C) expression of COX‐2 protein in non‐tumorous liver tissue by immunohistological staining, (D) negative control of (C). Scale bar = 50 µm. (E) Expression of COX‐2 mRNA in a non‐tumorous liver tissue by reverse transcription–polymerase chain reaction.
The incidence of HCC was evaluated after the administration of etodolac. The total numbers of HCC nodules were 11 in the control group (10 mice), 0 in the E‐LD group (eight mice) and three in the E‐HD group (nine mice). The numbers of mice that developed HCC were 5, 0 and 3 in the control, E‐LD and E‐HD groups, respectively. Development of HCC was suppressed completely by the administration of low‐dose etodolac. The administration of high‐dose etodolac also showed a suppressive effect, although it was not statistically significant (Table 1).
Table 1.
Incidence of hepatocellular carcinoma (HCC) in fatty liver Shionogi mice
| Group | No. mice | Etodolac (mg/kg) | Grade of steatosis | No. HCC nodules | No. mice that developed HCC | ||
|---|---|---|---|---|---|---|---|
| II | III | IV | |||||
| Control | 10 | 0 | 0 | 4 | 6 | 11 | 5 (50%) |
| E‐LD | 8 | 2 | 1 | 2 | 5 | 0* | 0* |
| E‐HD | 9 | 10 | 0 | 3 | 6 | 3 | 3 (33%) |
P < 0.05 by Fisher's exact test. Grades of classification are according to the size and distribution pattern of the vesicles in the hematoxylin–eosin‐stained sections.( 24 ) E‐HD, high dose of etodolac; E‐LD, low dose of etodolac.
All of the 27 mice used in the present study developed fatty liver from grades II to IV at the end of experiments with no remarkable difference in the degree of fatty or inflammatory changes. A small number of mice in each group developed yellow nodules of 1–2 mm in diameter and reddish cysts, which were identified microscopically as fatty nodules and peliosis hepatis, respectively (data not shown). Liver cirrhosis was not observed in any of the mice. Liver weights also did not show significant differences among the control, E‐LD and E‐HD groups (data not shown). One mouse in the E‐HD group died at the age of 17 months, and two mice in the E‐LD group died at 13 and 14 months. No HCC was found in these mice and the cause of death was not clear.
Except for the livers, no abnormal findings were observed macroscopically in heart, lung, kidney, intestines and large vessels in any of the FLS mice. Five mice in the E‐HD group had skin damage, including depilation and rash, and two among them had skin ulcers. The mean BW of mice at 18 months of age were 37.9 ± 1.11 g (n = 10), 36.88 ± 1.2 g (n = 8) and 35.56 ± 0.93 g (n = 9) in the control, E‐LD and E‐HD groups, respectively. No significant difference was found in BW among the control, E‐LD and E‐HD groups.
Plasma PGE2 levels after etodolac administration
The activity of etodolac can be estimated by analyzing the concentration of PGE2 in the plasma. The concentrations of PGE2 in the plasma were 1010.15 ± 120.22 pg/mL (n = 10), 443.33 ± 116.99 pg/mL (n = 8) and 773.8 ± 137.67 pg/mL (n = 9) in the control, E‐LD and E‐HD groups, respectively. We found that the plasma levels of PGE2 of the E‐LD group, but not the E‐HD group, were significantly lower than in the control group (P < 0.05) (Fig. 2).
Figure 2.
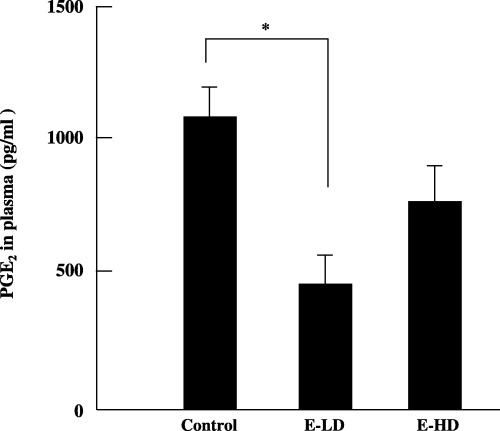
Effect of etodolac on plasma prostaglandin E2 (PGE2) levels in fatty liver Shionogi mice. A marked decrease in plasma PGE2 levels was observed after low‐dose (2 mg/kg bodyweight) administration of etodolac (E‐LD). E‐HD, high‐dose (10 mg/kg bodyweight) administration of etodolac. *P < 0.05. Bars indicate ±SE of mean.
Effects of etodolac on RXRα expression
Hepatocarcinogenesis in FLS mice has been attributed to chronic inflammation in fatty liver. Therefore, we investigated the effects of etodolac administration on hepatocytes in non‐tumurous fatty liver of FLS mice. In prostate cancer, R‐etodolac has been shown to bind to RXRα, inducing its degradation via ubiqutin and the proteasome‐dependent pathway.( 17 ) Analysis by immunohistological staining showed that the expression of RXRα in hepatocytes in non‐tumorous liver tissues was significantly lower in E‐LD (10.98 ± 0.87%n = 6) and E‐HD (11.65 ± 1.72%n = 4) groups than in the control group (27.28 ± 2.91%n = 5) (P < 0.01; Fig. 3A–C). In contrast, semiquantitative RT‐PCR analysis showed identical expression of RXRα mRNA among non‐tumorous liver tissue of the control, E‐LD and E‐HD groups (Fig. 3D). These findings suggest that etodolac binds to RXRα and induces its degradation in the non‐tumorous liver of FLS mice.
Figure 3.
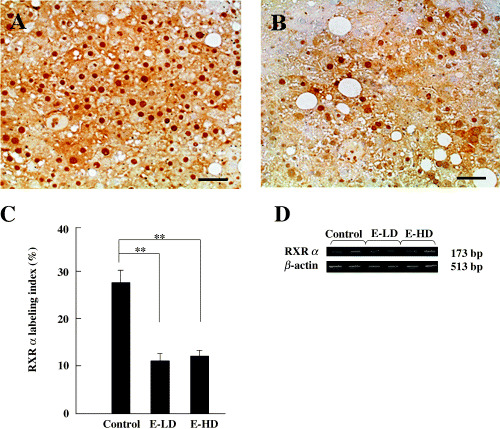
Effect of etodolac on retinoid X receptor α (RXRα) expression in non‐tumorous liver tissues. (A) RXRα in the non‐tumorous liver tissue of a control mouse. (B) RXRα in the non‐tumorous liver tissue of the low‐dose administration group (E‐LD). Scale bars = 50 µm. (C) The RXRα labeling index in the non‐tumorous tissues was significantly lower in the low‐dose and high‐dose (E‐HD) groups than in the control group. **P < 0.01. Bars indicate ±SE of mean. (D) reverse transcription–polymerase chain reaction analysis of RXRα mRNA. RXRα expression from two mice is shown in each group (control, E‐LD and E‐HD). β‐Actin was used as an internal control. Expression of RXRα mRNA was not decreased by the administration of etodolac.
Effects of etodolac on PCNA expression
Proliferating cell nuclear antigen is expressed throughout the cell cycle, except during G0 phase, and plays an important role in cell proliferation. The labeling index of PCNA in the non‐tumorous liver tissues of the E‐LD (2.15 ± 0.11%, n = 8) and E‐HD groups (2.2 ± 0.27%, n = 9) was significantly lower than the control group (3.13 ± 0.26%, n = 10) (P < 0.05, Fig. 4). These findings show that the growth of hepatocytes in non‐tumorous tissue was inhibited by etodolac administration.
Figure 4.
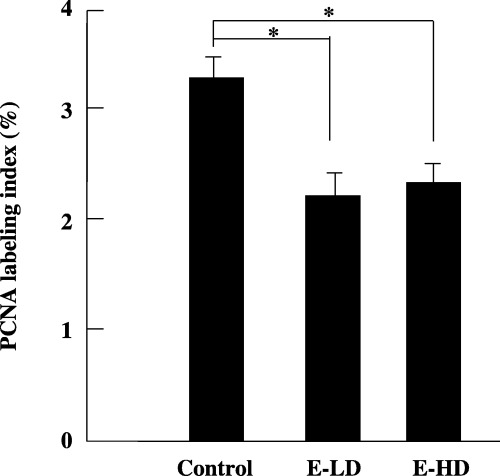
Proliferating cell nuclear antigen (PCNA) labeling index in non‐tumorous liver tissues. The PCNA labeling index was significantly suppressed by low‐dose (E‐LD) and high‐dose (E‐HD) administration of etodolac. *P < 0.05. Bars indicate ±SE of mean.
Discussion
Fatty liver Shionogi mice develop serious fatty liver and HCC with age, providing a good animal model to study hepatocarcinogenesis from fatty liver in vivo.( 21 , 22 ) Using FLS mice, we here examined the in vivo effects of a COX‐2 inhibitor, etodolac, on spontaneous development of HCC.
Etodolac exists in a racemic mixture. S‐etodolac possesses activity to inhibit COX‐2, which catalyzes the conversion of arachidonic acid to PGE2.( 15 , 16 ) COX‐2 and PGE2 have been reported to be involved in carcinogenesis of the colon, prostate and liver.( 7 , 8 , 9 , 28 , 29 , 30 ) Etodolac has been reported to reduce aberrant crypt foci in rat colon, and another selective COX‐2 inhibitor, NS‐398, has been reported to reduce rat colon carcinogenesis.( 31 , 32 , 33 ) In the present study, we found that etodolac was effective in inhibiting PGE2 synthesis and HCC development in FLS mice, particularly at a low concentration. A similar observation has been reported for aspirin, where a low dose has a better preventive effect than a high dose in human colorectal cancer.( 7 ) On the other hand, NS‐398 has been shown to inhibit aberrant crypt foci in F344 rats in a dose‐dependent manner.( 32 )
In the present study, we observed that the plasma concentration of PGE2 was higher in the E‐HD group than in the E‐LD group. The plasma concentration of PGE2 was lower in the E‐HD group than in the control group (not significantly). The plasma levels of etodolac in mice in the E‐HD group were approximately five times higher than those in mice in the E‐LD group (data not shown). Our previous study using HCC cell lines showed that PGE2 generation by etodolac is not inhibited in a dose‐dependent manner. Rather, PGE2 levels in the culture medium were higher with the high‐dose treatment than with the low‐dose treatment.( 14 ) The inhibition of PGE2 generation by NS‐398 was also dose‐independent at doses higher than 100 mM in some HCC cell lines.( 34 ) The dose‐independency of plasma PGE2 suppression by etodolac in vivo is compatible with these findings in the in vitro experiments. However, the precise mechanism of dose‐independency has not yet been clarified. Another suggested explanation is that the higher levels of PGE2 in the E‐HD group may be attributable to adverse effects of a high dose of etodolac. Severe skin damage developed in mice in the E‐HD group. This could be responsible for loss of the preventive effect of COX‐2 inhibitor on HCC in the E‐HD group. Furthermore, in the present study, the plasma PGE2 levels were consistent with the HCC incidences. These data suggest that PGE2 plays an important role in the development of HCC in FLS mice. The 2 mg/kg dose of etodolac three times a week used in this study is less than the usual dose in humans (200 mg orally twice a day), and no side effects were observed in this group. Thus, we consider that administration of a low dose of COX‐2 inhibitor should be sufficient for liver cancer prevention in humans. Furthermore, it is necessary to evaluate the efficacy of etodolace doses lower than 2 mg/kg to elucidate the optimum dose for liver cancer prevention.
Recently, R‐etodolac has been reported to bind specifically to RXRα and prevent prostate cancer.( 17 ) In the present study, we found significant decreases in RXRα protein expression in non‐tumorous liver tissue in both the E‐LD and E‐HD groups, whereas RXRα mRNA expression was almost similar among the control, E‐LD and E‐HD groups. These results suggest that the degradation of RXRα induced by R‐etodolac is also responsible for the preventive effect of hepatocarcinogenesis in FLS mice.
Hepatitis C virus stimulates the expression of COX‐2 via oxidative stress.( 35 ) HCV core protein induces fatty liver by binding to the DNA‐binding domain of RXRα.( 20 ) High levels of COX‐2 and RXRα expression in hepatocytes may be involved in hepatocarcinogenesis following HBV and HCV infection.( 18 , 19 , 20 , 36 , 37 ) Vitamin A has been reported to inhibit hepatocarcinogenesis by dephosphorylating RXRα.( 38 ) Our results showed that both PGE2 and RXRα levels were decreased by etodolac, indicated that etodolac may be useful for the prevention of HCC caused by HBV and HCV.
We have reported that COX‐2 inhibitors (etodolac and NS‐398) suppress PCNA expression and induce cell cycle arrest in HCC cell lines.( 14 , 34 ) In the present study, we found that PCNA expression in non‐tumorous fatty liver was significantly lower in both the E‐LD and E‐HD groups compared with the control group. The PCNA labeling index showed no difference between the E‐LD and E‐HD groups. Similar results have been reported with NS‐398 in F344 rats.( 32 , 33 ) These results suggest that low‐dose administration of etodolac is sufficient to suppress cell cycle progression in FLS mice.
The present results suggest that low‐dose administration of etodolac has a strong chemopreventive effect against hepatocarcinigenesis by inhibiting COX‐2 activity, and RXRα and PCNA expression in mice. The prevention of hepatocarcinogenesis in vivo by COX‐2 inhibitor may be caused by the primary suppression of malignant transformation from hepatocytes or inhibition of the growth of HCC cells in early stages, which have already developed in the liver but can not be detected as tumors. Etodolac may also prove to be of value in the prevention of HCC in humans.
References
- 1. Di Bisceglie AM. Epidemiology and clinical presentation of hepatocellular carcinoma. J Vasc Interv Radiol 2002; 13: S169–71. [DOI] [PubMed] [Google Scholar]
- 2. El‐Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med 1999; 340: 745–50. [DOI] [PubMed] [Google Scholar]
- 3. Takai K, Okuno M, Yasuda I et al. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. Updated analysis of the long‐term follow‐up data. Intervirology 2005; 48: 39–45. [DOI] [PubMed] [Google Scholar]
- 4. Habu D, Shiomi S, Tamori A et al. Role of vitamin K2 in the development of hepatocellular carcinoma in women with viral cirrhosis of the liver. JAMA 2004; 292: 358–61. [DOI] [PubMed] [Google Scholar]
- 5. Nishiguchi S, Kuroki T, Nakatani S et al. Randomised trial of effects of interferon‐alpha on incidence of hepatocellular carcinoma in chronic active hepatitis C with cirrhosis. Lancet 1995; 346: 1051–5. [DOI] [PubMed] [Google Scholar]
- 6. Kern MA, Schoneweiss MM, Sahi D et al. Cyclooxygenase‐2 inhibitors suppress the growth of human hepatocellular carcinoma implants in nude mice. Carcinogenesis 2004; 25: 1193–9. [DOI] [PubMed] [Google Scholar]
- 7. Baron JA, Cole BF, Sandler RS et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med 2003; 348: 891–9. [DOI] [PubMed] [Google Scholar]
- 8. Peek RM Jr. Prevention of colorectal cancer through the use of COX‐2 selective inhibitors. Cancer Chemother Pharmacol 2004; 54 (Suppl. 1): 850–6. [DOI] [PubMed] [Google Scholar]
- 9. Gupta S, Adhami VM, Subbarayan M et al. Suppression of prostate carcinogenesis by dietary supplementation of celecoxib in transgenic adenocarcinoma of the mouse prostate model. Cancer Res 2004; 64: 3334–43. [DOI] [PubMed] [Google Scholar]
- 10. Futakuchi M, Ogawa K, Sano M, Tamano S, Takeshita F, Shirai T. Suppression of lung metastasis by aspirin but not indomethacin in an in vivo model of chemically induced hepatocellular carcinoma. Jpn J Cancer Res 2002; 93: 1175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denda A, Endoh T, Kitayama W et al. Inhibition by piroxicam of oxidative DNA damage, liver cirrhosis and development of enzyme‐altered nodules caused by a choline‐deficient, 1‐amino acid‐defined diet in rats. Carcinogenesis 1997; 18: 1921–30. [DOI] [PubMed] [Google Scholar]
- 12. Denda A, Kitayama W, Murata A et al. Increased expression of cyclooxygenase‐2 protein during rat hepatocarcinogenesis caused by a choline‐deficient, l‐amino acid‐defined diet and chemopreventive efficacy of a specific inhibitor, nimesulide. Carcinogenesis 2002; 23: 245–56. [DOI] [PubMed] [Google Scholar]
- 13. Marquez‐Rosado L, Trejo‐Solis MC, Garcia‐Cuellar CM, Villa‐Trevino S. Celecoxib, a cyclooxygenase‐2 inhibitor, prevents induction of liver preneoplastic lesions in rats. J Hepatol 2005; 43: 653–60. [DOI] [PubMed] [Google Scholar]
- 14. Cheng J, Imanishi H, Liu W et al. Involvement of cell cycle regulatory proteins and MAP kinase signaling pathway in growth inhibition and cell cycle arrest by a selective cyclooxygenase 2 inhibitor, etodolac, in human hepatocellular carcinoma cell lines. Cancer Sci 2004; 95: 666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Demerson CA, Humber LG, Abraham NA, Schilling G, Martel RR, Pace‐Asciak C. Resolution of etodolac and antiinflammatory and prostaglandin synthetase inhibiting properties of the enantiomers. J Med Chem 1983; 26: 1778–80. [DOI] [PubMed] [Google Scholar]
- 16. Becker‐Scharfenkamp U, Blaschke G. Evaluation of the stereoselective metabolism of the chiral analgesic drug etodolac by high‐performance liquid chromatography. J Chromatogr 1993; 621: 199–207. [DOI] [PubMed] [Google Scholar]
- 17. Kolluri SK, Corr M, James SY et al. The R‐enantiomer of the nonsteroidal antiinflammatory drug etodolac binds retinoid X receptor and induces tumor‐selective apoptosis. Proc Natl Acad Sci USA 2005; 102: 2525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huan B, Siddiqui A. Retinoid X receptor RXR alpha binds to and trans‐activates the hepatitis B virus enhancer. Proc Natl Acad Sci USA 1992; 89: 9059–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moriya K, Yotsuyanagi H, Shintani Y et al. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J General Virol 1997; 78: 1527–31. [DOI] [PubMed] [Google Scholar]
- 20. Tsutsumi T, Suzuki T, Shimoike T et al. Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology 2002; 35: 937–46. [DOI] [PubMed] [Google Scholar]
- 21. Soga M, Kishimoto Y, Kawaguchi J et al. The FLS mouse: a new inbred strain with spontaneous fatty liver. Lab Anim Sci 1999; 49: 269–75. [PubMed] [Google Scholar]
- 22. Soga M, Kishimoto Y, Kawamura Y, Inagaki S, Makino S, Saibara T. Spontaneous development of hepatocellular carcinomas in the FLS mice with hereditary fatty liver. Cancer Lett 2003; 196: 43–8. [DOI] [PubMed] [Google Scholar]
- 23. Japanese Association for Laboratory Animal Science. Guidelines for animal experimentation. Exp Anim 1987; 36: 285–8. [Google Scholar]
- 24. Frith CH, Ward JM. A morphologic classification of proliferative and neoplastic hepatic lesions in mice. J Environ Pathol Toxicol 1979; 3: 329–51. [PubMed] [Google Scholar]
- 25. Cheng J, Imanishi H, Ijima H et al. Expression of cyclooxygenase 2 and cytosolic phospholipase A2 in the liver tissue of patients with chronic hepatitis and liver cirrhosis. Hepatol Res 2002; 23: 185–95. [DOI] [PubMed] [Google Scholar]
- 26. Nishizawa H, Morita M, Sugimoto M, Imanishi S, Manabe N. Effects of in utero exposure to bisphenol A on mRNA expression of arylhydrocarbon and retinoid receptors in murine embryos. J Reprod Dev 2005; 51: 315–24. [DOI] [PubMed] [Google Scholar]
- 27. Friedl R, Brunner M, Moeslinger T, Spieckermann PG. Testosterone inhibits expression of inducible nitric oxide synthase in murine macrophages. Life Sci 2000; 68: 417–29. [DOI] [PubMed] [Google Scholar]
- 28. Koga H, Sakisaka S, Ohishi M et al. Expression of cyclooxygenase‐2 in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Hepatology, 1999; 29: 688–96. [DOI] [PubMed] [Google Scholar]
- 29. Sung YK, Hwang SY, Kim JO, Bae HI, Kim JC, Kim MK. The correlation between cyclooxygenase‐2 expression and hepatocellular carcinogenesis. Mol Cells 2004; 17: 35–8. [PubMed] [Google Scholar]
- 30. Mayoral R, Fernandez‐Martinez A, Bosca L, Martin‐Sanz P. Prostaglandin E2 promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis 2005; 26: 753–61. [DOI] [PubMed] [Google Scholar]
- 31. Kishimoto Y, Takata N, Jinnai T et al. Sulindac and a cyclooxygenase‐2 inhibitor, etodolac, increase APC mRNA in the colon of rats treated with azoxymethane. Gut 2000; 47: 812–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yoshimi N, Kawabata K, Hara A, Matsunaga K, Yamada Y, Mori H. Inhibitory effect of NS‐398, a selective cyclooxygenase‐2 inhibitor, on azoxymethane‐induced aberrant crypt foci in colon carcinogenesis of F344 rats. Jpn J Cancer Res 1997; 88: 1044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshimi N, Shimizu M, Matsunaga K et al. Chemopreventive effect of N‐(2‐cyclohexyloxy‐4‐nitrophenyl) methane sulfonamide (NS‐398), a selective cyclooxygenase‐2 inhibitor, in rat colon carcinogenesis induced by azoxymethane. Jpn J Cancer Res 1999; 90: 406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng J, Imanishi H, Amuro Y, Hada T. NS‐398, a selective cyclooxygenase 2 inhibitor, inhibited cell growth and induced cell cycle arrest in human hepatocellular carcinoma cell lines. Int J Cancer 2002; 99: 755–61. [DOI] [PubMed] [Google Scholar]
- 35. Waris G, Siddiqui A. Hepatitis C virus stimulates the expression of cyclooxygenase‐2 via oxidative stress: role of prostaglandin E2 in RNA replication. J Virol 2005; 79: 9725–34. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Lara‐Pezzi E, Gomez‐Gaviro MV, Galvez BG et al. The hepatitis B virus X protein promotes tumor cell invasion by inducing membrane‐type matrix metalloproteinase‐1 and cyclooxygenase‐2 expression. J Clin Invest 2002; 110: 1831–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng AS, Chan HL, To KF et al. Cyclooxygenase‐2 pathway correlates with vascular endothelial growth factor expression and tumor angiogenesis in hepatitis B virus‐associated hepatocellular carcinoma. Int J Oncol 2004; 24: 853–60. [PubMed] [Google Scholar]
- 38. Matsushima‐Nishiwaki R, Okuno M, Takano Y, Kojima S, Friedman SL, Moriwaki H. Molecular mechanism for growth suppression of human hepatocellular carcinoma cells by acyclic retinoid. Carcinogenesis 2003; 24: 1353–9. [DOI] [PubMed] [Google Scholar]