

Abstract
Recent discovery of mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) gene in lung adenocarcinoma greatly stimulated biomarker research on predictive factors for EGFR tyrosine kinase inhibitors (TKI), such as gefitinib and erlotinib. Although patients with activating mutations of the EGFR generally respond to EGFR TKIs very well, it is natural to assume that there is no sole determinant, considering great complexity and redundancy of the EGFR pathway. Subsequently, roles of different types of EGFR mutations or mutations of genes that are members of the EGFR pathway such as KRAS and HER2 have been evaluated. In this review, we summarize the recent findings about how mutations of the EGFR and related genes affect sensitivity to EFGR‐TKIs. We also discuss molecular mechanisms of acquired resistance to EGFR‐TKIs that is almost inevitable in EGFR‐TKI therapy. The door for genotype‐based treatment of lung cancer is beginning to open, and through these efforts, it will be possible to slow the progression of lung cancer and eventually, to decrease mortality from lung cancer. (Cancer Sci 2007; 98: 1817–1824)
Epidermal growth factor receptor signaling pathways
There are four members of the ERBB family of receptor tyrosine kinases, namely epidermal growth factor receptor (EGFR) (also known as ERBB1/HER1): ERBB2/HER2/NEU, ERBB3/HER3 and ERBB4/HER4. However, ERBB2 does not have its ligand and ERBB3 lacks tyrosine kinase activity because of substitutions in crucial residues in the tyrosine kinase domain. The mechanism of activation of the ERBB signaling pathway is depicted in Fig. 1. Ligand binding results in dimerization, autophosphosphorylation, binding of adaptor proteins and subsequent signal transduction, leading to cancer phenotypes. For reviews see Hynes and Lane( 1 ) and Yarden and Slikowski.( 2 )
Figure 1.
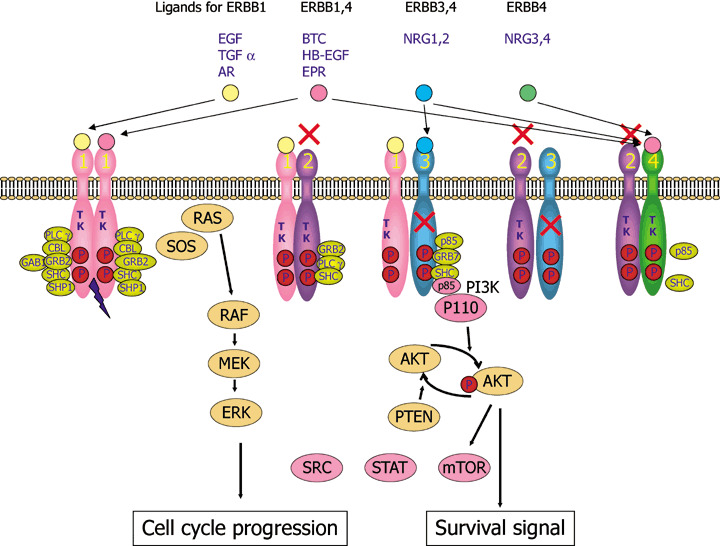
ERBB signaling pathways. Binding of a family of specific ligands to extracellular domain of ERBB leads to formation of homo‐ and heterodimers. In this case, HER2 is a preferred dimerization partner and heterodimers containing HER2 mediate a stronger signal than homodimers. Dimerization consequently stimulates intrinsic tyrosine kinase activity of the receptors and triggers autophosphorylation of specific tyrosine residues within the cytoplasmic domain. These phosphorylated tyrosines serve as specific binding sites for several signal transducers that initiate multiple signaling pathways including mitogen‐activated protein kinase (MAPK), phosphatidyl inositol 3 kinase (PI3K)‐AKT and signal transducer and activator of transcription protein (STAT) 3 and 5 pathways. These eventually result in cell proliferation, migration and metastasis, evasion from apoptosis, or angiogenesis, all of which are associated with cancer. P85 and p110 is a regulatory and catalytic subunit of phosphatidyl inositol 3 kinase (PI3K), respectively. STAT, SRC and mTOR are also activated by ERBB sinaling. AR, amphiregulin; BTC, betacellulin; EPR, epirefulin; ERK, extracellular signal‐regulated kinase; HB‐EGFR, heparin binding EGF; MEK, MAP an ERK kinase; mTOR, mammmalian target of rapamycin; NRG, neuregulin; TGF, transforming growth factor.
Cytotoxic chemotherapy using platinum‐doublet therapy appears to reach a therapeutic plateau.( 3 ) Hence, targeting EGFR is considered to be one of the strategies that improves the outcome of treatment for non‐small cell lung cancer (NSCLC), because EGFR is frequently overexpressed and aberrantly activated in NSCLC. Antibodies against extracellular domain of EGFR (such as cetuximab, matuzumab, and panitumab) and small‐molecule tyrosine kinase inhibitors (TKIs) that target the kinase domain (such as gefitinib and erlotinib) are in clinical use or in late developmental stages.( 4 )
In phase II trials of gefitinib, IDEAL 1 and 2, it was shown that certain patient subgroups appeared to have higher response rates, namely, female, adenocarcinoma and Japanese.( 5 , 6 ) Miller et al. were the first to report that smoking history as well as bronchioloalveolar pathological subtypes predict sensitivity to gefitinib.( 7 ) However, it was not possible to predict gefitinib sensitivity by levels of EGFR overexpression as determined by immunohistochemistry or immunoblotting. The factors that determine gefitinib sensitivity have long been an enigma.
EGFR mutation
In April 2004, two groups of researchers in Boston and subsequently a group in New York reported that activating mutations of the epidermal growth factor receptor gene (EGFR) were present in a subset of NSCLC, and that tumors with EGFR mutations are highly sensitive to EGFR TKI.( 8 , 9 , 10 ) EGFR mutations are predominantly found in female, non‐smoking, adenocarcinoma patients and in patients of East Asian origin. Following this, many groups confirmed and extended these findings. Fig. 2 shows the incidence of EGFR mutations found in 559 mutations in 2880 lung cancer patients in the published reports.( 11 )
Figure 2.
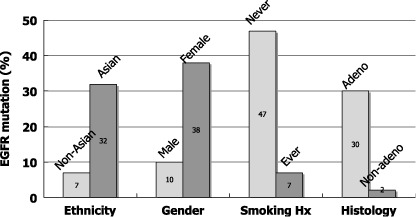
Incidence of epidermal growth factor receptor gene (EGFR) mutations by ethnicity, gender, smoking history and histology. Data were compiled from the published reports (n = 2880).( 11 )
It is of particular interest that EGFR mutations were the first molecular aberrations found in lung cancer that were more frequent among non‐smoking patients than smoking patients. Furthermore, we showed that EGFR mutation frequency is inversely associated with cumulative smoking dosage.( 12 ) When we divided smokers into three categories depending on smoke exposure, there was a trend towards a decrease in the incidence of EGFR mutations as exposure increased.( 12 ) However, it should be noted that EGFR mutations were detected in more than 20% of patients with a heavy smoking history. These findings should not be construed to mean that smoking has a preventive effect on EGFR mutations, rather, they suggest that EGFR mutations are caused by carcinogens other than those contained in tobacco smoke and that the apparent negative correlation with smoking dose is a result of diluting the number of tumors with EGFR mutations with an increased number of tumors with wild‐type EGFR as smoking dose increases. Indeed, this was suggested in our recent case‐control study.( 13 ) In addition, there is a suggestion of association of female gender with EGFR mutations.( 13 )
Type of EGFR mutations. EGFR mutations are mainly present in the first four exons of the gene encoding tyrosine kinase domain (Fig. 3a). About 90% of the EGFR mutations are either small deletions encompassing five amino acids from codons 746 through 750 (ELREA) or missense mutations resulting in leucine to arginine at codon 858 (L858R). There are over 20 variant types of deletion, for example, larger deletion, deletion plus point mutation, deletion plus insertion, etc. Approximately 3% of the mutations occur at codon 719 resulting in the substitution of glycine to cysteine, alanine or serine (G719X). Also, approximately 3% are in‐frame insertion mutations in exon 20.( 11 ) These four types of mutations seldom occur simultaneously. There are many rare point mutations, some of which occur with L858R.
Figure 3.
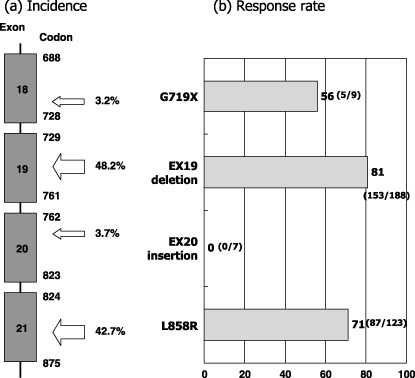
(a) Distribution and frequency of 569 epidermal growth factor receptor gene (EGFR) mutations in the published reports.( 11 ) (b) Response rates to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) according to the type of the EGFR mutations. Data were updated from our review article( 11 ) by adding recent papers.( 24 , 30 , 64 , 65 , 66 )
Biological consequences of the EGFR mutations. Exon 19 deletional mutation and L858R result in increased and sustained phosphorylation of EGFR and other HER family proteins without ligand stimulation. Sordella et al. reported that mutant EGFR selectively activate AKT and signal transducer and activator of transcription protein (STAT) signaling pathways that promote cell survival but no effect on the mitogen‐activated protein kinase (MAPK) pathway that induces proliferation.( 14 )
Two groups of researchers have recently developed transgenic mice that express either exon 19 deletion mutant or the L858R mutant in type II pneumocytes under the control of doxycyclin.( 15 , 16 ) Expression of either EGFR mutant leads to the development of adenocarcinoma similar to human bronchioloalveolar cell carcinoma and withdrawal of doxycyclin to reduce expression of transgene or erlotinib treatment resulting in tumor regression. These experiments showed that persistent EGFR signaling is required for tumor maintenance in human lung adenocarcinoma expressing EGFR mutants.
EGFR mutation and sensitivity to EGFR‐TKIs. When EGFR mutations were first reported, the most exciting finding was that lung cancer harboring this genetic alteration showed a striking response to EGFR‐TKIs.( 8 , 9 , 10 ) According to the data for 1335 patients in the published reports, about 70% of NSCLCs with EGFR mutations respond to EGFR‐TKIs, whereas 10% of tumors without EGFR mutations do so (Fig. 4). High response rates in patients with EGFR mutations to gefitinib were confirmed in the recently published prospective phase II studies.( 17 ) Furthermore, many investigators including us have reported that patients with EGFR mutations have a significantly longer survival than those with wild‐type EGFR when treated with EGFR TKIs.( 18 , 19 , 20 ) However, this point is still controversial, since some investigators clam that EGFR mutations are prognostic rather than predictive because patients with EGFR mutation do better than patients without EGFR treatment even when treated by chemotherapy.( 21 , 22 ) As a result of EGFR mutations being frequent in patients that are female, non‐smokers and good prognostic indicators of lung cancer, survival of patients with EGFR mutations tends to be longer in those who undergo surgery only. To determine whether EGFR mutations indeed predict clinical benefits as assessed by survival, the ongoing randomized controlled trials comparing gefitinib with cytotoxic chemotherapy for patients selected for EGFR mutation in Japan and Spain are of great importance.
Figure 4.
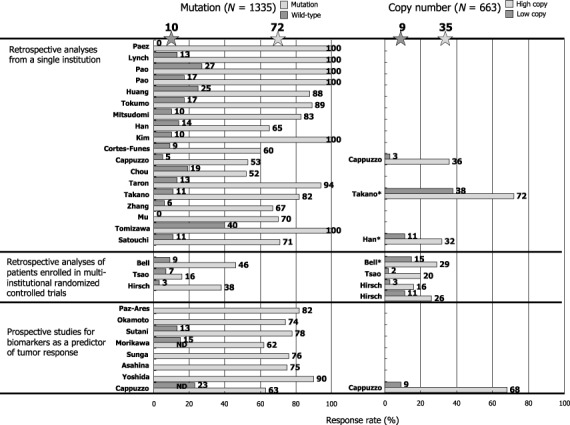
Effect of mutations and copy numbers of the epidermal growth factor receptor gene (EGFR) on response rates in patients treated with gefitinib or erlotinib. Data were updated from our review article( 67 ) by adding recent papers.( 64 , 65 , 68 , 69 )
We were the first to report that the response rate of gefitinib was higher for patients with deletional EGFR mutations than for those with other types of mutations, predominantly L858R( 19 ) and others extended this observation by demonstrating the survival difference between them.( 23 , 24 ) When we compiled data from the published reports, the response rate was highest in exon 19 deletions followed by L858R and G719X (Fig. 3b). In contrast, no reports exist of a single patient with exon 20 insertion mutation who responded to TKIs. In line with these clinical observations, Gleulich et al. showed that one of the insertion mutations (D770insNPG) in exon 20 is associated with in vitro resistance to erlotinib.( 25 ) In this study, G719S of exon 18 showed intermediate sensitivity in vitro.( 25 ) However, they did not observe differences between exon 19 deletion and L858R by their cell‐based assay.( 25 ) On the other hand, biochemical analysis for kinetics of purified wild‐type and mutant kinases revealed that mutant kinases have higher KM for adenosine triphosphate (ATP) (wild‐type 5, L858R 10.9, deletion 129.0 µmol/L) and lower Ki for erlotinib (wild‐type 17.5, L858R 6.25, deletion 3.3).( 26 ) Mulloy et al. showed that Del747–753 kinase had a higher autophosphorylation rate and higher sensitivity to erlotinib than the L858R kinase.( 27 ) These data reflect the differences in clinical response rates between exon 19 deletional mutation and L858R.
EGFR gene copy numbers. Cappuzzo et al. first reported that an increase in the EGFR gene copy number, as determined by fluorescence in situ hybridization (FISH), is more predictive of patient survival after gefitinib treatment than EGFR mutations.( 28 ) However, this report does not necessarily refute the role of EGFR mutations as predictive factors, because EGFR mutations only failed to significantly affect overall survival (P = 0.09), while EGFR mutations were predictive of response rates and time to progression.( 28 ) However, it should be noted that their definition of increased gene copy number included both gene amplification and high polysomy (more than 40% of tumor cells have more than four copies of the EGFR gene). It is biologically unclear whether high polysomy indicates the activation of the EGFR gene, resulting in effects similar to those caused by gene amplification. According to the pooled data from 663 patients, patients with high EGFR copy numbers have response rates of 35% and those with low copy numbers have response rates of 9% (Fig. 4). These magnitudes appear smaller than that of EGFR mutations.
Tsao et al. reported that the increased EGFR gene copy number is most predictive of a longer survival in patients who received erlotinib in a phase III clinical trial (BR.21) that compared erlotinib with best supportive care.( 29 ) They concluded that the detection of EGFR mutations was not necessary in selecting patients who would benefit from erlotinib therapy.( 29 ) However, many investigators refute this point. Han et al. recently reported that EGFR mutation and the high gene copy number were associated with better objective responses in univariate analyses. However, only gefitinib‐sensitive EGFR mutation was independently predictive of both response and survival in multivariate analysis.( 30 ) Furthermore, Tsao et al. report that 53% of the EGFR mutations they found were novel variant mutations, of which 92% were C/G→T/A or A/T→G/C transitions. Marchertti et al. suggested at least some of these mutations could be artifactual due to postmortem deamination that would cause C to T or A to G transitions if a small amount of DNA from paraffin embedded tissues is used.( 31 ) In general, tumors with EGFR mutations tend to have gene amplification. Mutation and amplification are probably both important in determining TKI sensitivity. However, the role of high polysomy is unclear.
KRAS mutation
Activating mutation of the KRAS gene was one of the earliest discoveries of genetic alterations in lung cancer known as a poor prognostic indicator since 1990.( 32 ) We are among the first to report that the occurrence of EGFR and KRAS mutations are strictly mutually exclusive.( 12 , 33 ) This finding can be explained by the fact that that the KRAS–MAPK pathway is one of the downstream signaling pathways of EGFR. Interestingly, KRAS mutations predominantly occur in Caucasian patients with a history of smoking. Pao et al. first reported that lung cancers with KRAS mutations are resistant to EGFR‐TKIs. None of the nine tumors with KRAS mutations responded to EGFR‐TKIs.( 34 ) We( 35 ) and others confirmed that none of the lung cancers with KRAS mutations reported achieved clinical response thus far. However, in their study of adenocarcinoma with bronchioloalveolar cells treated with erlotinib, Miller et al. showed that some of the tumors with KRAS mutations showed minor tumor shrinkage, although the response rate of lung cancer with KRAS mutations was zero by response evaluation criteria in solid tumors (RECIST).( 36 ) Thus, it is not possible to exclude patients with KRAS mutations from the list of patients with potential clinical benefit from EGFR‐TKI therapy.
HER2 mutation
Following the discovery of EGFR mutations, it was reported that mutations of the HER2 are present in a very small fraction of adenocarcinomas and they appeared to target the same population targeted by EGFR mutations, namely, non‐smokers, females, etc.( 37 ) Most of HER2 mutations are insertion mutations in exon 2037. As anticipated, tumors with HER2 mutations are resistant to EGFR‐TKI treatment( 30 ) because constitutively activated HER2 kinase would phosphorylate other HER family proteins resulting in activation of downstream molecules even when EGFR TK is blocked. However, it has been reported that tumors with HER2 mutation are sensitive to HER2 targeted therapy both in vitro and in vivo.( 38 , 39 ) It was also reported that increased HER2 gene copy number is associated with the response to gefitinib therapy in EGFR mutation‐positive NSCLC patients.( 40 ) However the same group reported that the genomic gain for HER3 is not a marker for response or resistance to TKI therapy in advanced NSCLC patients.( 41 )
BRAF and other gene mutations
It has been reported that mutation of the BRAF gene occurs in about 1–3% of lung adenocarcinomas.( 42 , 43 , 44 ) It is also noteworthy that no EGFR, HER2, BRAF, or KRAS mutations have been reported to occur simultaneously in individual patients, suggesting the complementary role of these mutations in lung carcinogenesis. However, because of the low incidence, it is not clear how patients with BRAF mutation respond to EGFR‐TKIs. In mouse lung cancer models driven by BRAF mutation (V600E), tumor regression was induced by a specific MEK inhibitor, CI‐104045. Interestingly, CI‐1040 also led to tumor shrinkage in lung tumors driven by mutant KRAS.( 45 )
Although approximately a third of colon, liver or breast cancers harbor mutations of the catalytic domain of Phosphatidyl inositol 3 kinase (PI3K) catalytic alpha (PIK3CA or p110alpha), it is rare in lung caner (1–4%).( 46 ) We found two PIK3CA E545K mutations in 78 lung cancers. These two female, non‐smoking patients also had EGFR L858R mutation and they showed PR upon gefitinib treatment.( 35 ) Thus, from our limited experience, it appeared that PIK3CA mutation targets patients with similar clinical characteristics as EGFR or HER2 mutation, but there is no mutually exclusionary relationship with other mutations. The role of PIK3CA as a predictive factor for EGFR‐TKI needs further evaluation.
By retrieving transforming genes from mouse 3T3 fibroblasts transfected with cDNA from an expression library constructed from a lung adenocarcinoma arising in a smoking male, Soda et al. very recently identified the transforming echinoderm microtubule‐associated protein‐like 4 (EML4) – anaplastic lymphoma kinase (ALK) fusion gene.( 47 ) This EML4‐ALK fusion gene was a result of a small inversion within chromosome 2p. The EML4‐ALK fusion transcript was detected in five out of 75 NSCLC patients.( 47 ) Interestingly, neither of these five patients harbored EGFR nor KRAS mutations.( 47 ) Whether the activated ALK gene has a role in determining sensitivity to EGFR‐TKI is yet to be examined.
Marks et al. recently carried out mutational profiling of 261 lung adenocarcinomas to uncover other potential somatic mutations in the genes of the EGFR signaling pathway that could contribute to lung tumorigenesis.( 48 ) The coding sequence of 39 genes (including ERBB1‐4, FGFR1‐4, AKT1‐3, FRAP1, RPS6KB1‐2, K‐, H‐, N‐RAS, A‐, B‐, C‐RAF, MAP2K1,‐2,M 4–6, MAPK1,3, 4,6–15) were sequenced. They found13 EGFR, 20 KRAS, two PIK3CA, one BRAF, one FGFR (fibroblast growth factor receptor) and four (Pro712Thr) mutations.( 48 ) One of tumors with PIK3CA mutation also contained KRAS mutation. They concluded that the majority of gain‐of‐function mutations within kinase genes in the EGFR signaling pathway have already been identified.
Acquired resistance
Even in patients whose tumor harbors activating EGFR mutations with an initial dramatic response, acquired resistance to the EGFR‐TKI develops almost without exception after varying periods of time, usually 6–12 months. In 2005, it was reported that the secondary mutation of the EGFR gene resulting in threonine to methionine at codon 790 in exon 20 (T790M) is responsible for this acquired resistance.( 49 , 50 ) Since codon 790 is involved in the binding site of EGFR TKI, substitution of threonine to bulkier methionine is anticipated to cause steric hindrance of TKI binding (Fig. 5a–c). We and others confirmed that T790M occur in about half of patients with acquired resistance to gefitinib or erlotinib.( 51 , 52 ) Interestingly, T790M corresponds to threonine to isoleucine mutation of the ABL gene (T315I) based on amino acid homology that was reported as the mechanism of acquired resistance of chronic myeloid leukemia to imatinib in 2001. Indeed, in 2003 before the discovery of activating mutations of the EGFR gene, it had already been reported that artificially created T790M of the EGFR gene results in gefitinib resistance.( 53 )
Figure 5.
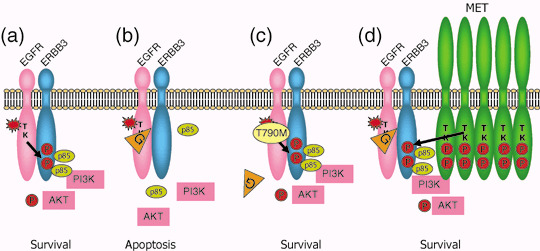
Mechanisms of acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). (a) Activating mutations of the EGFR gene (star) result in constitutive activation of tyrosine kinase without ligand binding. This then turns on survival signal through pathways including the PI3K/AKT pathway. Phosphorylated tyrosine residues of ERBB3 are the main binding sites for p85, a regulatory subunit of PI3K. (b) When gefitinib (G) is administered, EGFR tyrosine kinase is specifically inhibited and survival signal is shut‐won leading to apoptosis of cancer cells. (c) When secondary threonine‐to‐methionine mutation at codon 790 of the EGFR gene (T790M) is acquired, bulkier methionine residue prevents gefitinib from binding EGFR‐TK. (d) Alternatively, when MET is activated by amplification, ERBB3 is phosphorylated by MET. Even when EGFR‐TKI is inhibited by gefitinib, activation of the PI3K/AKT pathway is maintained through ERBB3 phosphorylation. In this case, co‐administration of EGFR‐TKI and MET inhibitor can block survival signal. Modified from Arteaga et al.( 70 )
We reported that T790M is present in a very small fraction (0.5%) of patients without history of gefitinib treatment.( 54 ) One of the patients subsequently received gefitinib treatment, but she did not respond to this treatment and even her tumor harbored L858R activating mutation.( 54 ) Bell et al. reported a family with multiple cases of lung cancer associated with germline transmission of T790M.( 55 ) Four of six tumors analyzed showed a secondary somatic activating EGFR mutation.( 55 ) These lines of evidence suggest that T790M confers some growth advantage and does not only prevent TKI from binding to the EGFR protein. Although initial reports concluded that TK activity of the EGFR with T790M do not change, Mulloy et al. reported that TK activity of L858R plus T790M is higher than that of only L858R.( 27 ) Furthermore, Vikis showed that TK or transforming activity of T790M is higher than those of wild‐type EGFR, but lower than those of exon 19 deletion.( 56 ) However, they failed to find germline T790M mutations in 237 pedigrees associated with multiple lung cancers.( 56 )
Inukai et al. developed an assay for the detection of T790M with a high sensitivity using mutant enriched polymerase chain reaction (PCR) that can detect a single mutant allele in the background of 1000 wild‐type alleles. Using this method, they found that T790M is present in 3.6% of patients before TKI treatment.( 57 ) In seven patients with activating EGFR mutation yet refractory to EGFR TKI, three patients harbored T790M by this assay.( 57 ) In contrast, T790M was not detected in 19 patients who responded to EGFR TKI.( 57 ) In a lung cancer cell line that was made resistant to EGFR TKI by being exposed to increasing concentrations of gefitinib, T790M could be detected.( 58 ) However, the amount of mutant allele was small.( 58 ) These lines of evidence suggest that a very minor population of clones with T790M exist before TKI treatment and that this clone is selected and expanded during TKI treatment, resulting in a resistant phenotype. However, it is not fully understood why T790M allele is present usually as a small fraction compared with the wild‐type allele. In the cases of acquired resistance to imatinib in chronic myeloid leukemia, over 30 secondary mutations of the ABL gene other than T315I are reported.( 59 ) Although we failed to find a novel mutation other than T790M in our 14 patients, Balak et al. found a novel D761Y mutation in addition to 7 T790M in their 16 patients with acquired resistance.( 51 ) Tumors with T790M are highly resistant to reversible TKI such as gefitinib or erlotinib, they are shown to be sensitive to irreversible TKIs that covalently bind to the EGFR kinase domain such as EKB‐569, HKI‐272, CI‐1033, BIBW2992, etc., some of which are in clinical development.( 49 , 50 , 60 )
MET amplification
Recently Engelman et al. reported that amplification of the MET gene is another mechanism of acquired resistance to EGFR TKIs.( 61 ) MET is a receptor for hepatocyte growth factor (HGF)/scatter factor, and overexpression, amplification and mutation of the MET gene have been reported in various human cancers including lung cancer. Engelman et al. established 1000 times‐resistant cell line, HCC827GR by exposinsg it to increasing concentrations of gefitinib.( 61 ) After secondary T790M was ruled out, they found that phosphorylation of MET, ERBB3, and EGFR out of 42 receptor tyrosine kinases remain after gefitinib treatment using receptor tyrosine kinase array.( 61 ) single nucleotide polymorphisms (SNP) array revealed that amplification of 7q31.1‐33.3 where the MET gene is located.( 61 ) In HCC827GR, ERBB3 is phosphorylated through MET and the ERBB3/PI3K/AKT anti‐apoptotic pathway remains activated (Fig. 5d).( 61 ) Inhibition of MET signaling restored the sensitivity to gefitinib. MET amplification has also been detected in four of 18 (22%) clinical lung cancer specimens that developed resistance to gefitinib or erlotinib. Interestingly, MET amplification can occur concurrently with T790M in some specimens.( 61 )
Amplification of a loci containing the MET gene (7q31) is present in a subset of lung adenocarcinoma predominantly without EGFR activating mutation.( 62 ) Overexpression of MET protein was immunohistochemically detected in 24% (13 of 55) of cases including all cases with amplification of the MET gene.( 62 ) This suggest that amplification of MET present before TKI treatment may be associated with inherent resistance to TKI, suggesting the possible strategy of MET inhibitor plus EGFR‐TKI therapy for these type of tumors.
Use of biomarkers and clinical practice
In Japan, a great hindrance to gefitinib treatment in lung cancer is a life‐threatening interstitial lung disease (ILD) or acute lung injury as adverse reaction to gefitinib. A recently published retrospective survey showed prevalence and mortality of 3.5 and 1.6%, respectively.( 63 ) The incidence of ILD was higher in patients with male gender (5.8%), in those with smoking history (6.2%) and pre‐existing interstitial pneumonitis (13.9%). These patient populations are those who are not likely to have activating mutations of the EGFR gene. In contrast, development of ILD is rare in females (1.0%) and non‐smokers (0.8%). However, as described earlier it is also a reality that EGFR mutation is present in over 20% of patients with pack‐year of more than 50 in our country. Therefore, in clinical response, the risk of ILD and the potential benefit of EGFR‐TKI therapy should be well balanced as depicted in Fig. 6.
Figure 6.

Our current view for indication of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in Japanese patients with lung cancer. Numbers in each patient subset indicate response rate or incidence of interstitial lung disease (ILD). For example, when a patient is female with activating EGFR mutation, TKI can be used from the first line therapy (a). If the risk of ILD is estimated to be low, even when the patient does not have EGFR mutation, TKI can be used somewhere in the clinical course, considering the imperfect ability of predictive power of EGFR mutation (b). In contrast, when a male patient with a heavy smoking history and with pre‐existing ILD does not harbor EGFR mutation, there is no indication for EGFR‐TKI (c). It is most difficult when patients with high risk harbor the EGFR mutation. In this case, indication should be considered on individual basis (d).
Conclusion
The development of drug therapies that inhibit receptor tyrosine kinases, especially EGFR‐TK, and the understanding of molecular backgrounds that determine sensitivity and resistance will make genotype‐based medicine (GBM) for treatment of lung cancer possible in the near future. Fig. 7 illustrates a possible GBM approach for Japanese adenocarcinoma of the lung by the currently available knowledge. Through this GBM approach, it will be possible to slow the progression of lung cancer and eventually, to decrease mortality from lung cancer.
Figure 7.
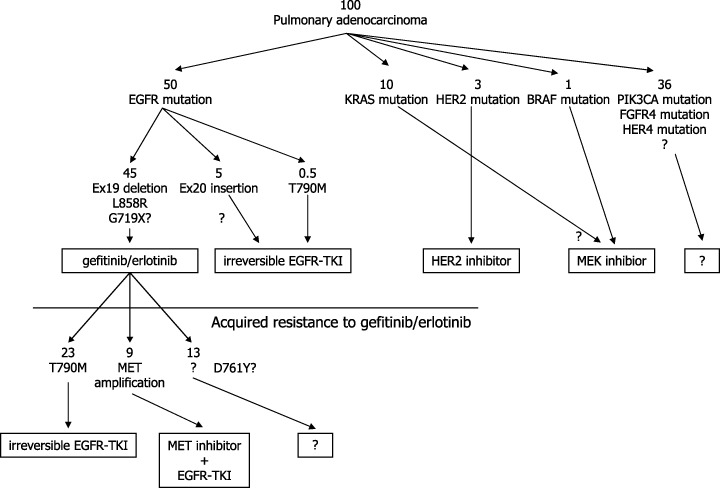
Possible genotype‐based therapeutic approach for lung adenocarcinomas in Japan. Numbers indicate approximate number of patients per 100 patients in Japan.
Acknowledgment
This work was supported in part by a Grant‐in‐Aid (18390386) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5: 341–54. [DOI] [PubMed] [Google Scholar]
- 2. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2: 127–37. [DOI] [PubMed] [Google Scholar]
- 3. Schiller JH, Harrington D, Belani CP et al . Comparison of four chemotherapy regimens for advanced non‐small‐cell lung cancer. N Engl J Med 2002; 346: 92–8. [DOI] [PubMed] [Google Scholar]
- 4. Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res 2001; 7: 2958–70. [PubMed] [Google Scholar]
- 5. Fukuoka M, Yano S, Giaccone G et al . Multi‐institutional randomized phase II trial of gefitinib for previously treated patients with advanced non‐small‐cell lung cancer. J Clin Oncol 2003; 21: 2237–46. [DOI] [PubMed] [Google Scholar]
- 6. Kris MG, Natale RB, Herbst RS et al . Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non‐small cell lung cancer: a randomized trial. JAMA 2003; 290: 2149–58. [DOI] [PubMed] [Google Scholar]
- 7. Miller VA, Kris MG, Shah N et al . Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non‐small‐cell lung cancer. J Clin Oncol 2004; 22: 1103–9. [DOI] [PubMed] [Google Scholar]
- 8. Lynch TJ, Bell DW, Sordella R et al . Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 9. Paez JG, Janne PA, Lee JC et al . EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 10. Pao W, Miller V, Zakowski M et al . EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13 306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitsudomi T, Kosaka T, Yatabe Y. Biological and clinical implications of EGFR mutations in lung cancer. Int J Clin Oncol 2006; 11: 190–8. [DOI] [PubMed] [Google Scholar]
- 12. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004; 64: 8919–23. [DOI] [PubMed] [Google Scholar]
- 13. Matsuo K, Ito H, Yatabe Y et al . Risk factors differ for non‐small‐cell lung cancers with and without EGFR mutation: assessment of smoking and sex by a case‐control study in Japanese. Cancer Sci 2006; 98: 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science 2004; 305: 1163–7. [DOI] [PubMed] [Google Scholar]
- 15. Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down‐regulation of the receptors. Genes Dev 2006; 20: 1496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ji H, Li D, Chen L et al . The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR‐targeted therapies. Cancer Cell 2006; 9: 485–95. [DOI] [PubMed] [Google Scholar]
- 17. Inoue A, Suzuki T, Fukuhara T et al . Prospective phase II study of gefitinib for chemotherapy‐naive patients with advanced non‐small‐cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol 2006; 24: 3340–6. [DOI] [PubMed] [Google Scholar]
- 18. Han SW, Kim TY, Hwang PG et al . Predictive and prognostic impact of epidermal growth factor receptor mutation in non‐small‐cell lung cancer patients treated with gefitinib. J Clin Oncol 2005; 23: 2493–501. [DOI] [PubMed] [Google Scholar]
- 19. Mitsudomi T, Kosaka T, Endoh H et al . Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non‐small‐cell lung cancer with postoperative recurrence. J Clin Oncol 2005; 23: 2513–20. [DOI] [PubMed] [Google Scholar]
- 20. Takano T, Ohe Y, Sakamoto H et al . Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non‐small‐cell lung cancer. J Clin Oncol 2005; 23: 6829–37. [DOI] [PubMed] [Google Scholar]
- 21. Bell DW, Lynch TJ, Haserlat SM et al . Epidermal growth factor receptor mutations and gene amplification in non‐small‐cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol 2005; 23: 8081–92. [DOI] [PubMed] [Google Scholar]
- 22. Eberhard DA, Johnson BE, Amler LC et al . Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non‐small‐cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005; 23: 5900–9. [DOI] [PubMed] [Google Scholar]
- 23. Riely GJ, Pao W, Pham D et al . Clinical course of patients with non‐small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res 2006; 12: 839–44. [DOI] [PubMed] [Google Scholar]
- 24. Jackman DM, Yeap BY, Sequist LV et al . Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non‐small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res 2006; 12: 3908–14. [DOI] [PubMed] [Google Scholar]
- 25. Greulich H, Chen T‐H, Feng W et al . Oncogenic transformation by inhibitor‐sensitive and resistant EGFR mutations. PLoS Med 2005; 2: e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carey KD, Garton AJ, Romero MS et al . Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res 2006; 66: 8163–71. [DOI] [PubMed] [Google Scholar]
- 27. Mulloy R, Ferrand A, Kim Y et al . Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res 2007; 67: 2325–30. [DOI] [PubMed] [Google Scholar]
- 28. Cappuzzo F, Hirsch FR, Rossi E et al . Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non‐small cell lung cancer. J Natl Cancer Inst 2005; 97: 643–55. [DOI] [PubMed] [Google Scholar]
- 29. Tsao MS, Sakurada A, Cutz JC et al . Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med 2005; 353: 133–44. [DOI] [PubMed] [Google Scholar]
- 30. Han SW, Kim TY, Jeon YK et al . Optimization of patient selection for gefitinib in non‐small cell lung cancer by combined analysis of epidermal growth factor receptor mutation, K‐ras mutation, and Akt phosphorylation. Clin Cancer Res 2006; 12: 2538–44. [DOI] [PubMed] [Google Scholar]
- 31. Marchetti A, Felicioni L, Buttitta F. Assessing EGFR mutations. N Engl J Med 2006; 354: 526–8. [DOI] [PubMed] [Google Scholar]
- 32. Slebos RJ, Kibbelaar RE, Dalesio O et al . K‐ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med 1990; 323: 561–5. [DOI] [PubMed] [Google Scholar]
- 33. Shigematsu H, Lin L, Takahashi T et al . Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97: 339–46. [DOI] [PubMed] [Google Scholar]
- 34. Pao W, Wang TY, Riely GJ et al . KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Endoh H, Yatabe Y, Kosaka T, Kuwano H, Mitsudomi T. PTEN and PIK3A expression is associated with prolonged survival after gefitinib treatment in EGFR‐mutated lung cancer patients. J Thorac Oncol 2006; 1: 629–34. [PubMed] [Google Scholar]
- 36. Miller VA, Zakowski M, Riely GJ et al . EGFR mutation and copy number, EGFR protein expression and KRAS mutation as predictors of outcome with erlotinib in bronchioloalveolar cell carcinoma (BAC): Results of a prospective phase II trial. J Clin Oncol (Meeting Abstracts) 2006; 24: 7003. [Google Scholar]
- 37. Shigematsu H, Takahashi T, Nomura M et al . Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 2005; 65: 1642–6. [DOI] [PubMed] [Google Scholar]
- 38. Wang SE, Narasanna A, Perez‐Torres M et al . HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006; 10: 25–38. [DOI] [PubMed] [Google Scholar]
- 39. Cappuzzo F, Bemis L, Varella‐Garcia M. HER2 mutation and response to trastuzumab therapy in non‐small‐cell lung cancer. N Engl J Med 2006; 354: 2619–21. [DOI] [PubMed] [Google Scholar]
- 40. Cappuzzo F, Varella‐Garcia M, Shigematsu H et al . Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor‐positive non‐small‐cell lung cancer patients. J Clin Oncol 2005; 23: 5007–18. [DOI] [PubMed] [Google Scholar]
- 41. Cappuzzo F, Toschi L, Domenichini I et al . HER3 genomic gain and sensitivity to gefitinib in advanced non‐small‐cell lung cancer patients. Br J Cancer 2005; 93: 1334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brose MS, Volpe P, Feldman M et al . BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 2002; 62: 6997–7000. [PubMed] [Google Scholar]
- 43. Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res 2002; 62: 7001–3. [PubMed] [Google Scholar]
- 44. Sasaki H, Kawano O, Endo K et al . Uncommon V599E BRAF mutations in Japanese patients with lung cancer. J Surg Res 2006; 133: 203–6. [DOI] [PubMed] [Google Scholar]
- 45. Ji H, Wang Z, Perera SA et al . Mutations in BRAF and KRAS converge on activation of the mitogen‐activated protein kinase pathway in lung cancer mouse models. Cancer Res 2007; 67: 4933–9. [DOI] [PubMed] [Google Scholar]
- 46. Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 2005; 5: 921–9. [DOI] [PubMed] [Google Scholar]
- 47. Soda M, Choi YL, Enomoto M et al . Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature 2007; 448: 561–6. [DOI] [PubMed] [Google Scholar]
- 48. Marks JL, McLellan MD, Zakowski MF et al . Mutational analysis of EGFR and related signaling pathway genes in lung adenocarcinomas identifies a novel somatic kinase domain mutation in FGFR4. PLoS ONE 2007; 2: e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kobayashi S, Boggon TJ, Dayaram T et al . EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 50. Pao W, Miller VA, Politi KA et al . Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib Is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Balak MN, Gong Y, Riely GJ et al . Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor‐mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006; 12: 6494–501. [DOI] [PubMed] [Google Scholar]
- 52. Kosaka T, Yatabe Y, Endoh H et al . Analysis of epidermal growth factor receptor gene mutation in patients with non‐small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res 2006; 12: 5764–9. [DOI] [PubMed] [Google Scholar]
- 53. Blencke S, Ullrich A, Daub H. Mutation of threonine 766 in the epidermal growth factor receptor reveals a hotspot for resistance formation against selective tyrosine kinase inhibitors. J Biol Chem 2003; 278: 15 435–40. [DOI] [PubMed] [Google Scholar]
- 54. Toyooka S, Kiura K, Mitsudomi T. EGFR mutation and response of lung cancer to gefitinib. N Engl J Med 2005; 352: 2136. [DOI] [PubMed] [Google Scholar]
- 55. Bell DW, Gore I, Okimoto RA et al . Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 2005; 37: 1315–16. [DOI] [PubMed] [Google Scholar]
- 56. Vikis H, Sato M, James M et al . EGFR‐T790M is a rare lung cancer susceptibility allele with enhanced kinase activity. Cancer Res 2007; 67: 4665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Inukai M, Toyooka S, Ito S et al . Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non‐small cell lung cancer. Cancer Res 2006; 66: 7854–8. [DOI] [PubMed] [Google Scholar]
- 58. Engelman JA, Mukohara T, Zejnullahu K et al . Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR‐amplified lung cancer. J Clin Invest 2006; 116: 2695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shah NP, Nicoll JM, Nagar B et al . Multiple BCR‐ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2: 117–25. [DOI] [PubMed] [Google Scholar]
- 60. Carter TA, Wodicka LM, Shah NP et al . Inhibition of drug‐resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA 2005; 102: 11 011–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Engelman JA, Zejnullahu K, Mitsudomi T et al . MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 62. Shibata T, Uryu S, Kokubu A et al . Genetic classification of lung adenocarcinoma based on array‐based comparative genomic hybridization analysis: its association with clinicopathologic features. Clin Cancer Res 2005; 11: 6177–85. [DOI] [PubMed] [Google Scholar]
- 63. Ando M, Okamoto I, Yamamoto N et al . Predicitive factors for interstitial lung disease, anti‐tumor response and survival in non‐smal cell lung cancer patients treated with gefiitinib. J Clin Oncol 2006; 24: 2549–56. [DOI] [PubMed] [Google Scholar]
- 64. Cappuzzo F, Ligorio C, Janne PA et al . Prospective study of gefitinib in epidermal growth factor receptor fluorescence in situ hybridization‐positive/phospho‐Akt‐positive or never smoker patients with advanced non‐small‐cell lung cancer: the ONCOBELL trial. J Clin Oncol 2007; 25: 2248–55. [DOI] [PubMed] [Google Scholar]
- 65. Satouchi M, Negoro S, Funada Y et al . Predictive factors associated with prolonged survival in patients with advanced non‐small‐cell lung cancer (NSCLC) treated with gefitinib. Br J Cancer 2007; 96: 1191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yoshida K, Yatabe Y, Park JY et al . Prospective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non‐small cell lung cancer. J Thorac Oncol 2007; 2: 22–8. [PubMed] [Google Scholar]
- 67. Uramoto H, Mitsudomi T. Which biomarker predicts benefit from EGFR‐TKI treatment for patients with lung cancer? Br J Cancer 2007; 96: 857–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Asahina H, Yamazaki K, Kinoshita I et al . A phase II trial of gefitinib as first‐line therapy for advanced non‐small cell lung cancer with epidermal growth factor receptor mutations. Br J Cancer 2006; 95: 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sunaga N, Tomizawa Y, Yanagitani N et al . Phase II prospective study of the efficacy of gefitinib for the treatment of stage III/IV non‐small cell lung cancer with EGFR mutations, irrespective of previous chemotherapy. Lung Cancer 2007; 56: 383–9. [DOI] [PubMed] [Google Scholar]
- 70. Arteaga CL. HER3 and mutant EGFR meet MET. Nat Med 2007; 13: 675–7. [DOI] [PubMed] [Google Scholar]