

Abstract
c‐Met is the cellular receptor for hepatocyte growth factor (HGF) and is known to be dysregulated in various types of human cancers. Activation of the HGF/c‐Met pathway causes tumor progression, invasion, and metastasis. Vascular endothelial growth factor (VEGF) is also known as a key molecule in tumor progression through the induction of tumor angiogenesis. Because of their key roles in tumor progression, these pathways provide attractive targets for therapeutic intervention. We have generated a novel, orally active, small molecule compound, E7050, which inhibits both c‐Met and vascular endothelial growth factor receptor (VEGFR)‐2. In vitro studies indicate that E7050 potently inhibits phosphorylation of both c‐Met and VEGFR‐2. E7050 also potently represses the growth of both c‐met amplified tumor cells and endothelial cells stimulated with either HGF or VEGF. In vivo studies using E7050 showed inhibition of the phosphorylation of c‐Met and VEGFR‐2 in tumors, and strong inhibition of tumor growth and tumor angiogenesis in xenograft models. Treatment of some tumor lines containing c‐met amplifications with high doses of E7050 (50–200 mg/kg) induced tumor regression and disappearance. In a peritoneal dissemination model, E7050 showed an antitumor effect against peritoneal tumors as well as a significant prolongation of lifespan in treated mice. Our results indicate that E7050 is a potent inhibitor of c‐Met and VEGFR‐2 and has therapeutic potential for the treatment of cancer. (Cancer Sci 2009)
Receptor tyrosine kinases (RTKs) are often dysregulated in human cancers in association with genetic alternations including mutations, translocations, and amplifications. A number of chemical agents that target RTKs have shown promising clinical activity against subsets of cancer patients.( 1 )
c‐Met, the receptor for hepatocyte growth factor (HGF), has become one of the leading molecular targets in cancer therapeutics.( 2 , 3 , 4 ) HGF binds to c‐Met, inducing phosphorylation of its intracellular domain, thereby leading to c‐Met activation.( 5 ) The HGF/c‐Met signal is part of an important pathway in various types of cancer and promotes tumor proliferation, survival, migration, and infiltration.( 6 , 7 , 8 , 9 , 10 ) Dysregulation of c‐Met by its overexpression, mutation, and amplification has been observed in many types of cancers.( 4 , 11 ) In particular, the amplification of the c‐met is seen in gastric, esophageal, and lung cancers as well as in liver metastases of colon cancer.( 12 , 13 , 14 , 15 ) Since c‐met amplification correlates with tumor invasiveness, lymph node metastasis, and peritoneal dissemination in patients with gastric cancer, it is recognized as a marker of poor prognosis in gastric cancer.( 16 , 17 ) Recently, c‐met amplification was observed in acquired resistant tumors of patients with lung cancer who had been treated with epidermal growth factor receptor (EGFR) inhibitors.( 18 , 19 )
Angiogenesis is an essential requirement for tumor growth and metastasis.( 20 , 21 , 22 , 23 ) Among the numerous angiogenic factors that have been identified, vascular endothelial growth factor (VEGF)‐A has been identified as a crucial regulator of both physiologic and pathologic angiogenesis.( 20 , 21 , 22 ) VEGF induces proliferation and migration in endothelial cells through both of its cognate receptors, VEGFR‐1 and VEGFR‐2, although VEGF is thought to act preferentially via VEGFR‐2.( 24 ) Several inhibitors of the VEGF/VEGFR‐2 signaling pathway show clear antitumor activity against many types of tumors, and as a result a number of these inhibitors are now in clinical use.( 25 , 26 , 27 , 28 , 29 ) HGF also known to induce angiogenesis through c‐Met expressed in endothelial cells and acts synergistically with VEGF.( 30 , 31 , 32 )
We propose that if both the c‐Met and VEGFR‐2 pathways were inhibited together by one compound, then this compound would be expected to have more effective antitumor activity than those that inhibit only one pathway. We have generated a novel orally active small molecule compound, E7050, which is a dual inhibitor of both c‐Met and VEGFR‐2. E7050 inhibits the proliferation of tumor and endothelial cells and therefore may exhibit more potent antitumor activity than compounds used in current therapeutic approaches. Here, we describe the remarkably potent antitumor activity of E7050, which induces pronounced tumor regression and prolongation of the lifespan of mice bearing human tumors.
Materials and Methods
Compound. E7050: N‐[2‐Fluoro‐4‐({2‐[4‐(4‐methylpiperazin‐1‐yl)piperidin‐1‐yl] carbonylaminopyridin‐4‐yl} oxy) phenyl]‐N′‐(4‐fluorophenyl) cyclopropane‐1,1‐dicarboxamide (2R,3R)‐tartrate was synthesized at Eisai Co., Ltd, Ibaraki, Japan.
Cell lines and cell cultures. The human gastric cancer cell lines MKN45 and MKN74 were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan). The human lung cancer cell line EBC‐1 was obtained from the Health Science Research Resources Bank (Osaka, Japan). The human gastric cancer cell lines Hs746T, SNU‐5, and SNU‐1; the human lung cancer cell line A549; and the normal human lung fibroblast cell line MRC‐5, were obtained from the ATCC (Manassas, VA, USA). The human pancreatic cancer cell line KP‐1 was a gift from Dr Akihiro Funakoshi at the National Kyushu Cancer Center. KP‐1/VEGF cells were prepared by stable transfection of VEGF expressing plasmids driven by CMV promoters into parental KP‐1 cells. Cells were cultured in RPMI‐1640 with 10% FBS with the exception of SNU‐5 cells, which were maintained in Iscove’s Modified Dulbecco’s medium with 10% FBS. The preparation and maintenance of HUVEC have been described previously.( 32 ) Cells were cultured at 37°C under a humidified atmosphere containing 5% CO2.
Western blot analysis. The phosphorylation status of c‐Met and VEGFR‐2 was detected by Western blot analysis. For c‐Met, MKN45 cells were incubated with a serial dilution of E7050 in complete medium at 37°C for 2 h. For VEGFR‐2, HUVEC were starved with human endothelial serum free medium (Invitrogen, Carlsbad, CA, USA) containing 0.5% FBS for 24 h. Subsequently HUVEC were incubated with a serial dilution of E7050 for 1 h and then incubated with 20 ng/mL of human VEGF (R&D Systems, Minneapolis, MN, USA) for 5 min. Cells were lysed by lysis buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 10% glycerol, 1% Triton X‐100, 1.5 mM MgCl2, 1 mM EDTA [pH 8.0], 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride 1 mM sodium orthovanadate, 10 μg/mL aprotinin, 50 μg/mL leupeptin, and 1 μg/mL pepstatin A). The resected tumor samples were homogenized with lysis buffer containing 25 mM β‐glycerophosphate and 0.5% (v/v) phosphatase inhibitor cocktail 2 (Sigma‐Aldrich, St. Louis, MO, USA) at 4°C. Cellular debris was removed by centrifugation at 17 860 g for 20 min at 4°C. Aliquots of the supernatants containing 5–20 μg of protein were subjected to SDS‐PAGE under reducing conditions. The proteins were then transferred onto PVDF membranes (Millipore, Bedford, MA, USA), blocked with TBS containing 0.05% Tween‐20 and either 5% skim milk or 5% BSA. The membranes were probed with the following antibodies : anti‐c‐Met polyclonal antibody (C‐28) and anti‐VEGFR‐2 polyclonal antibody (C‐20) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); mouse anti‐phosphotyrosine clone 4G10 (Upstate, Charlottesville, VA, USA); and anti‐VEGFR‐2 polyclonal antibody, anti‐phospho‐VEGFR‐2 (Tyr996) polyclonal antibody, and anti‐phospho‐c‐Met (Tyr1234/1235) polyclonal antibody. Detection was performed using a Super Signal enhanced chemiluminescence kit (Pierce, Rockford, IL, USA). Immunoreactive bands were visualized by chemiluminescence with an Image Master‐VDS‐CL detection system (Amersham, Uppsala, Sweden). The intensity of each band was measured by using an image analyzer (1D Image Analysis Software; Eastman Kodak, Rochester, NY, USA).
Cell proliferation assay. Cells (1–3 × 103 cells/100 μL/well) were seeded on 96‐well culture plates with various concentrations of E7050 and cultured for 3 days. Then, 10 μL of WST‐8 reagent (Dojindo, Kumamoto, Japan) was added to each well, and absorbance was measured at 450 nm compared with a reference measurement at 660 nm using a MTP‐500 microplate reader (Corona Electric, Ibaraki, Japan). Proliferation assays using HUVEC were performed as described previously.( 33 ) Briefly, HUVEC (2 × 103 cells/well) were cultured for 3 days in medium containing HGF (30 ng/mL), VEGF (20 ng/mL) (R&D Systems), or basic fibroblast growth factor (bFGF) (20 ng/mL) (Wako Pure Chemicals, Osaka, Japan) together with serially diluted E7050.
Quantitative genomic PCR. Genomic DNA was extracted from tumor cells using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). For the PCR reaction, 5 ng of genomic DNA was mixed with the primer pair and the Power Syber Green PCR Master Mix, and then reaction was performed using an ABI7900 thermal cycler (Applied Biosystems, Foster City, CA, USA). Data were normalized to a LINE‐1 repetitive element internal DNA control and then the relative copy number of c‐met was calculated by calculating the ratio against the MRC‐5 cells. Primer sequences were as follows: c‐met: forward primer: 5′‐TGGCTCATTCACAAGTCTCTCTACC‐3′, reverse primer: 5′‐TTTTGGTGACAGCTTCAGCACT‐3′; LINE‐1: forward primer: 5′‐AAAGCCGCTCAACTACATGG‐3′, reverse primer: 5′‐TGCTTTGAATGCGTCCCAGAG‐3′.
Subcutaneous xenograft models. Nude mice (CAnN.Cg‐Foxn1nu/CrlCrlj, female, 5–6 weeks old) were obtained from Charles River Laboratories Japan (Kanagawa, Japan). Mice were maintained under super pathogen‐free conditions and housed in barrier facilities on a 12‐h light/dark cycle, with food and water ad libitum. Cultured tumor cells were implanted subcutaneously (s.c.) into the flanks of mice. When tumor volume reached 200–300 mm3, mice were randomized into groups. E7050 was dissolved in sterile distilled water and administered orally once a day. The tumor was measured in two dimensions, and the volume was calculated using the formula: tumor volume (mm3) = 1/2 × length (mm) × width (mm)2.
Nude mice bearing MKN45 tumors were used for the pharmacodynamic study of c‐Met phosphorylation. At the indicated time after administration of a single oral dose of E7050, mice were sacrificed and tumors were resected. The tumor was treated with the lysis buffer described above and homogenized at 4°C and analyzed by Western blotting. For the detection of VEGFR‐2 phosphorylation, nude mice bearing the KP‐1/VEGF tumor were used. Four hours after a single administration of E7050, the tumor was resected and the tissue was immediately lysed and homogenized. A 10‐mg aliquot of the lysate was immunoprecipitated using the rabbit anti‐VEGFR‐2 antibody. Western blot analysis was performed and the levels of phosphorylated VEGFR‐2 and total VEGFR‐2 protein were detected using the anti‐phosphotyrosine 4G10 antibody and anti‐VEGFR‐2 antibody, respectively.
Immunohistochemistry. Nude mice bearing the KP‐1/VEGF tumor were treated with vehicle or E7050 for 12 days. On day 13, mice were sacrificed and weights of the tumors were measured. Tumors were embedded in Optimal Cutting Temperature (O.C.T) compound, frozen in dry ice‐acetone, and sectioned (8 μm). Sections were fixed with cold acetone for 10 min and immunostained with rat antimouse CD31 antibody (BD Biosciences, Franklin Lakes, NJ, USA) and visualized using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). Adjacent sections were routinely stained with H&E. All histological specimens were viewed under CCD Hyper Scope (Keyence, Osaka, Japan).
MKN45 peritoneal dissemination model. Cultured MKN45 cells (1 × 107 cells) were inoculated intraperitoneally into nude mice. Six days after inoculation, the treatment was started (day 0). In order to assess the antitumor activity of E7050, the mice were sacrificed at day 7 and an autopsy was performed. Tumors were harvested from the peritoneal cavity and weighed. In the survival study, the mice were euthanized by CO2 asphyxiation when they became moribund. The statistical comparison of survival times between the E7050‐treated group and the vehicle‐treated group was performed by a generalized Wilcoxon test with Bonferroni adjustment. Values of P < 0.01 were considered statistically significant. All of the animal experiments were conducted in accordance with the guideline for animal experiments of Eisai Co., Ltd.
Results
E7050 inhibits tumor cell growth in vitro. We have developed a novel RTK inhibitor, E7050 (Fig. 1) that potently inhibits the autophosphorylation of c‐Met in MKN45 cells, which have constitutively phosphorylated c‐Met.( 34 ) E7050 also inhibits VEGF‐induced phosphorylation of VEGFR‐2 in HUVEC. The IC50 values (14 and 16 nM for c‐Met and VEGFR‐2 respectively, see Table 1) indicate that E7050 is an efficient dual inhibitor of both c‐Met and VEGFR‐2 kinases. E7050 also strongly inhibits the growth of MKN45, EBC‐1, Hs746T, and SNU‐5 tumor cells with IC50 values of 37, 6.2, 23, and 24 nM, respectively (Table 1). The growth of A549, SNU‐1 and MKN74 tumor cells was inhibited by E7050 with much higher IC50 values. Using quantitative genomic PCR, amplification of the c‐met gene was detected in the former four cell lines, but not the latter three cell lines. Similar results were obtained by FISH analysis of the cellular karyotype. Cell lines with c‐met amplification showed a tight gene cluster of c‐met gene copies (data not shown). These data indicate that E7050 selectively inhibits the growth of the c‐met amplified tumor cell lines in vitro, although there is no direct correlation between the amplification levels of the c‐met and the sensitivity of the cells to E7050.
Figure 1.
Structure of E7050.
Table 1.
E7050 inhibits phosphorylation of c‐Met and VEGFR‐2, and cell growth in vitro
| E7050 IC50 (nM) | Relative c‐met copy number | |
|---|---|---|
| In vitro inhibition of kinase phosphorylation | ||
| Receptor tyrosine kinase | ||
| c‐Met (MKN45) | 14 | – |
| VEGFR‐2 (VEGF‐stimulated HUVEC) | 16 | – |
| Inhibition of tumor cell line proliferation | ||
| Cell line (Tumor type) | ||
| MKN45 (gastric) | 37 | 12.7 |
| SNU‐5 (gastric) | 6.2 | 8.7 |
| Hs746T (gastric) | 23 | 5.2 |
| EBC‐1 (lung) | 24 | 11.1 |
| MKN74 (gastric) | 4300 | 1.1 |
| SNU‐1 (gastric) | 4200 | 1 |
| A549 (lung) | 2600 | 1.2 |
| Inhibition of growth factor induced HUVEC proliferation | ||
| Growth factor stimulation | ||
| HGF‐stimulated HUVEC | 17 | – |
| VEGF‐stimulated HUVEC | 84 | – |
| bFGF‐stimulated HUVEC | >1000 | – |
Calculation of the IC50 values for kinase phosphorylation was calculated by chemiluminescence intensity of Western blotting analysis. Relative c‐met copy number was calculated by quantitative genomic PCR method, and was expressed as a ratio to the number of MRC‐5 cells. bFGF, basic fibroblast growth factor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
The growth of endothelial cells is stimulated by many different angiogenic growth factors.( 23 ) We examined the effect of three angiogenic growth factors, HGF, VEGF, and bFGF, on the growth of HUVEC in the presence of E7050. The growth stimulated by HGF or VEGF was inhibited by E7050 with IC50 values of 17 nM and 84 nM respectively, but it did not inhibit bFGF‐stimulated HUVEC growth up to 1000 nM (Table 1). These data indicate that E7050 selectively inhibits the growth of HUVEC using both the HGF/c‐Met and the VEGF/VEGFR‐2 pathways.
Antitumor activities of E7050 in vivo. In order to examine the antitumor efficacy of E7050 in an in vivo model we used a nude mice s.c. xenograft model with four tumor cell lines that were highly sensitive to growth inhibition by E7050 in vitro. Daily oral administration of E7050 inhibited the growth of all tumors in a dose‐dependent manner (Fig. 2). High doses of E7050 caused drastic tumor regression, with 2/5 Hs746T tumors failing to re‐grow after E7050 treatment (50 mg/kg) was terminated for 20 days and 5/5 failing to re‐grow after 100 mg/kg E7050 treatment (data not shown). As a result, we judged that tumor‐bearing mice were cured by treatment with E7050. During the treatment with E7050, no other macroscopic changes or loss of body weight were observed (data not shown).
Figure 2.
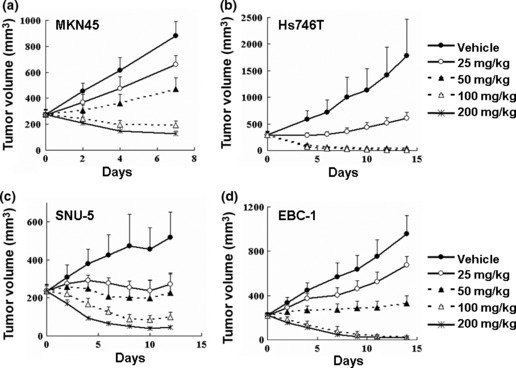
Antitumor activity of E7050 in human tumors in a mouse xenograft model. Nude mice bearing MKN45 (a), Hs746T (b), SNU‐5 (c), or EBC‐1 (d) tumors were administered E7050, at the indicated dose, or vehicle only as a control, once a day. Tumor volume was measured using calipers on the indicated days with the mean tumor volume ± SD indicated for groups of five to eight mice.
E7050 inhibits c‐Met phosphorylation in mouse tumors. To confirm that the antitumor activity of E7050 is caused by the inhibition of c‐Met, the phosphorylation status of c‐Met was assessed in E7050 treated tumors. The phosphorylation of c‐Met in the MKN45 tumor is inhibited by a single oral administration of E7050 (Fig. 3). Duration of the inhibition was dose‐dependent and it was almost completely inhibited for 24 h at the dose of 100 mg/kg. These data suggest that c‐Met inhibition in the tumor accounts for the antitumor activity of E7050.
Figure 3.
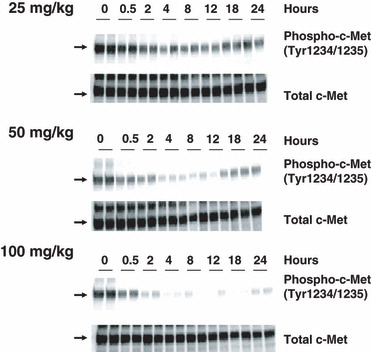
Effect of E7050 on tumor cell c‐Met phosphorylation in vivo. Nude mice bearing MKN45 tumors were given a single administration of E7050 at the indicated dose. At the indicated time, tumors were resected from the mice and the relative levels of phosphorylated c‐Met (Tyr1234/1235) and total c‐Met protein in the tumor lysates were determined by Western blotting.
E7050 inhibits VEGFR‐2 phosphorylation and tumor angiogenesis. In order to investigate the antiangiogenic activity of E7050, we performed xenograft studies using a VEGF‐overexpressing human pancreatic cancer cell line, KP‐1/VEGF. E7050 doses up to 10 μM did not inhibit the growth of KP‐1/VEGF cells in vitro (data not shown), which is consistent with the observation that this cell line does not express VEGFR‐2. Detection of VEGFR‐2 phosphorylation in the vasculature of the KP‐1/VEGF tumor was carried out by immunoprecipitation followed by Western blot analysis. VEGFR‐2 was phosphorylated in the tumor and a single administration of E7050 diminished VEGFR‐2 phosphorylation (Fig. 4a). Immunohistochemical analysis of tumor sections using the endothelial cell marker CD31 revealed that E7050 also decreased the blood vessel density of the tumor (Fig. 4b) and consequently inhibited the growth of the tumor (Fig. 4c). These data indicate that E7050 is able to inhibit tumor growth by inhibiting VEGF/VEGFR‐2 pathway‐mediated tumor angiogenesis.
Figure 4.
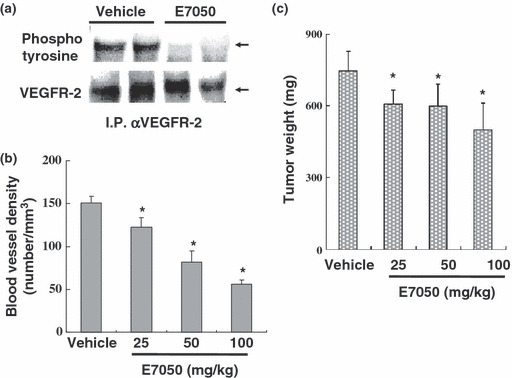
Inhibitory effect of E7050 on vascular endothelial growth factor receptor (VEGFR)‐2 phosphorylation and tumor progression. (a) Nude mice bearing KP‐1/VEGF (vascular endothelial growth factor) tumors were given a single administration of E7050 (100 mg/kg) or vehicle. At 4 h after treatment, the mice were sacrificed and the tumors were resected. VEGFR‐2 was immunoprecipitated with anti‐VEGFR‐2 antibody. The amount of phosphorylated VEGFR‐2 (4G10) and total VEGFR‐2 levels were determined by Western blot analysis. (b) Quantitative analysis of blood vessel density by immunohistochemical staining with anti‐CD31 antibody in KP‐1/VEGF s.c. tumor sections. (mean ± SD *P < 0.05, the significance of the difference from the vehicle‐treated group was determined using the Dunnett‐type multiple comparison test). (c) Tumor weight on the 12th day after treatment with vehicle or different doses of E7050, shows a dose‐dependent decrease with E7050 treatment (mean ± SD *P < 0.05, the significance of the difference from the vehicle‐treated group was determined using the Dunnett‐type multiple comparison test).
E7050 treatment prolongs the lifespan of tumor‐bearing mice. In a clinical setting, the appearance of peritoneal dissemination of tumors is indicative of a poor outcome for patients ( 35 , 36 ) and c‐met amplification significantly correlates with peritoneal dissemination and poor prognosis in gastric cancer patients.( 16 , 17 ) We established a mouse peritoneal tumor dissemination model and evaluated the effect of E7050 on these tumors. Disseminated tumors were observed 6 days after intraperitoneal inoculation of MKN45 cells into nude mice. On that day E7050 treatment was started (day 0). In this model, most of the disseminated tumors were present in the mesenterium (Fig. 5a). At day 0, the mean tumor weight was 186 ± 37 mg, which increased to 586 ± 88 mg at day 7. E7050 inhibited the growth of these tumors in a dose‐dependent manner (Fig. 5b), with an E7050 dose of more than 50 mg/kg completely inhibiting their growth. In this model, mice became moribund within 40 days, suffering from cancer‐induced cachexia, accumulation of bloody ascites, and tumor burden. Daily administration of E7050 significantly prolonged the lifespan of mice at all of the dose levels we tested (Fig. 6). It was particularly apparent that no mice became moribund during E7050 treatment at doses above 100 mg/kg. Although the tumors in these mice were detected in the peritoneal cavity, there were no other macroscopic changes, including accumulation of ascites. These data indicate that E7050 shows antitumor activity against disseminated tumors and also has the potency to prolong the lifespan of the mice without any adverse effects.
Figure 5.
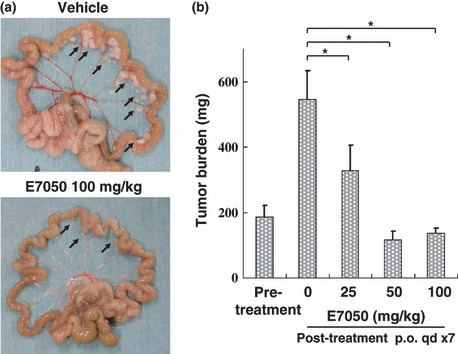
Effect of E7050 on MKN45 tumors in a peritoneal dissemination model. Cultured MKN45 cells were inoculated intraperitoneally into nude mice on day 0 and E7050 was administered orally daily from day 7 to day 13. The mice were sacrificed and autopsies were carried out on day 14. (a) Macroscopic identification of disseminated tumors in the mesenterium (tumors are marked with arrows). (b) Absence of gross peritoneal disseminated tumors in E7050‐treated mice. Data on the total weight of disseminated tumors per mouse are expressed as the mean ± SD for six mice. E7050‐treated groups had significantly fewer tumors than the vehicle‐treated control mice (*P < 0.01 calculated using the Dunnett‐type multiple comparison test).
Figure 6.
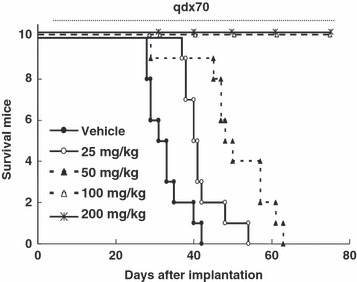
Survival curves of E7050‐treated MKN45 tumor‐bearing mice. Cultured MKN45 cells were inoculated intraperitoneally into nude mice on day 0. Daily administration with either vehicle or E7050 was started on day 7. Each group comprised 10 mice. The lifespans of E7050 treated mice were statistically significantly greater than the vehicle‐treated control mice (P < 0.01, calculated using the generalized Wilcoxon test with Bonferroni adjustment).
Discussion
In this report we have discovered a novel small compound, E7050, which can inhibit both the c‐Met and VEGFR‐2 signaling pathways which contribute to the tumor malignancies. E7050 potently inhibits the growth of tumor and endothelial cells in vitro (Table 1). The growth inhibitory activity of E7050 towards tumor cells was restricted by their c‐Met status, because E7050 potently inhibits the growth of tumor cell lines that have amplified c‐met (Table 1). Amplification of c‐met has been detected and reported in several types of cancer.( 12 , 13 , 15 , 18 , 19 , 37 ) Recently, it has been reported that the growth and survival of gastric and lung cancer cells harboring c‐met amplifications depends on an activated c‐Met signal.( 34 , 38 ) The activity of E7050 against tumor cells is dependent upon c‐met status but does not directly correlate with the copy number of the c‐met gene. E7050 also inhibits the growth of endothelial cells stimulated by HGF or by VEGF but not by bFGF (Table 1). HGF and VEGF independently stimulate angiogenesis and synergistically enhance angiogenesis.( 30 , 31 , 32 ) E7050 potently inhibits these two key growth factor pathways in both tumor and endothelial cells.
E7050 showed antitumor activity with tumor regression against a number of tumors in vivo and was able to completely cure mice bearing a Hs746T cell xenograft (Fig. 2). There are no previous reports of c‐Met inhibitors with antitumor activity that result in tumor disappearance.( 39 , 40 , 41 ) Also VEGF/VEGFR‐2 inhibitors and anti‐VEGF antibody fail to show tumor regression when tested against a number of different of tumor lines.( 42 ) Since E7050 demonstrated inhibitory activity against both c‐Met and VEGFR‐2 in vivo (3, 4), we propose that tumor regression and disappearance depends on its dual inhibitory effects against the HGF/c‐Met and VEGF/VEGFR‐2 signaling pathways.
Dose‐dependent sustained inhibition of c‐Met phosphorylation in tumors was observed after a single administration of E7050 (Fig. 3). Therefore, the level of phosphorylated c‐Met would be the appropriate pharmacodynamic marker for E7050 efficacy, whereas c‐met amplification status would be a suitable predictive marker to identify patients whose tumors are sensitive to E7050.
In the setting of a clinical study, the survival benefit to patients is one of the most important endpoints for a new cancer therapeutic agent. In this report, we have established the peritoneal dissemination model for evaluation of E7050 efficacy. E7050 showed clear antitumor activity against peritoneal tumors, and prolonged the lifespan of tumor‐bearing mice (5, 6). At a dose above 100 mg/kg, all mice survived after 70 days treatment without any macroscopic changes, which suggests that E7050 is well tolerated. We hypothesize that E7050 has dual activity against c‐met amplified tumors, such that the initial direct action on cancer cells is to inhibit c‐Met and thereby inhibit their growth. Secondly, the antiangiogenic activity of E7050 on tumor endothelial cells inhibits c‐Met and VEGFR‐2 stimulation by HGF and VEGF, respectively.
In conclusion, we have developed and described E7050, which is the first kinase inhibitor with dual action against both c‐Met and VEGFR‐2. The dual inhibitory activity of E7050 against tumor growth and angiogenesis results in drastic tumor regression and disappearance and also prolongation of lifespan without adverse effects. We propose that E7050 will be a novel therapeutic agent against cancer patients with aberrant c‐Met signaling. Based on our preclinical rationale, E7050 is currently under evaluation in a phase I clinical trial.
Acknowledgments
We would like to thank Dr Akihiro Funakoshi (National Kyusyu Cancer Center, Fukuoka, Japan) for providing the KP‐1 human pancreatic cancer cell line. We also thank Dr Takashi Owa and Dr Toshimitsu Uenaka for their critical reading of our manuscript.
References
- 1. Laird AD, Cherrington JM. Small molecule tyrosine kinase inhibitors: clinical development of anticancer agents. Expert Opin Investig Drugs 2003; 12 (1): 51–64. [DOI] [PubMed] [Google Scholar]
- 2. Knudsen BS, Vande Woude G. Showering c‐MET‐dependent cancers with drugs. Curr Opin Genet Dev 2008; 18 (1): 87–96. [DOI] [PubMed] [Google Scholar]
- 3. Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov 2008; 7 (6): 504–16. [DOI] [PubMed] [Google Scholar]
- 4. Christensen JG, Burrows J, Salgia R. c‐Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett 2005; 225 (1): 1–26. [DOI] [PubMed] [Google Scholar]
- 5. Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c‐Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev 2002; 13 (1): 41–59. [DOI] [PubMed] [Google Scholar]
- 6. Stabile LP, Rothstein ME, Keohavong P et al. Therapeutic targeting of human hepatocyte growth factor with a single neutralizing monoclonal antibody reduces lung tumorigenesis. Mol Cancer Ther 2008; 7 (7): 1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shinomiya N, Gao CF, Xie Q et al. RNA interference reveals that ligand‐independent met activity is required for tumor cell signaling and survival. Cancer Res 2004; 64 (21): 7962–70. [DOI] [PubMed] [Google Scholar]
- 8. Herynk MH, Stoeltzing O, Reinmuth N et al. Down‐regulation of c‐Met inhibits growth in the liver of human colorectal carcinoma cells. Cancer Res 2003; 63 (11): 2990–6. [PubMed] [Google Scholar]
- 9. Anderson MR, Harrison R, Atherfold PA et al. Met receptor signaling: a key effector in esophageal adenocarcinoma. Clin Cancer Res 2006; 12 (20 Pt 1): 5936–43. [DOI] [PubMed] [Google Scholar]
- 10. Maulik G, Kijima T, Ma PC et al. Modulation of the c‐Met/hepatocyte growth factor pathway in small cell lung cancer. Clin Cancer Res 2002; 8 (2): 620–7. [PubMed] [Google Scholar]
- 11. Trusolino L, Comoglio PM. Scatter‐factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer 2002; 2 (4): 289–300. [DOI] [PubMed] [Google Scholar]
- 12. Kuniyasu H, Yasui W, Kitadai Y, Yokozaki H, Ito H, Tahara E. Frequent amplification of the c‐met gene in scirrhous type stomach cancer. Biochem Biophys Res Commun 1992; 189 (1): 227–32. [DOI] [PubMed] [Google Scholar]
- 13. Miller CT, Lin L, Casper AM et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006; 25 (3): 409–18. [DOI] [PubMed] [Google Scholar]
- 14. Zhao X, Weir BA, LaFramboise T et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res 2005; 65 (13): 5561–70. [DOI] [PubMed] [Google Scholar]
- 15. Di Renzo MF, Olivero M, Giacomini A et al. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res 1995; 1 (2): 147–54. [PubMed] [Google Scholar]
- 16. Tsugawa K, Yonemura Y, Hirono Y et al. Amplification of the c‐met, c‐erbB‐2 and epidermal growth factor receptor gene in human gastric cancers: correlation to clinical features. Oncology 1998; 55 (5): 475–81. [DOI] [PubMed] [Google Scholar]
- 17. Nakajima M, Sawada H, Yamada Y et al. The prognostic significance of amplification and overexpression of c‐met and c‐erb B‐2 in human gastric carcinomas. Cancer 1999; 85 (9): 1894–902. [DOI] [PubMed] [Google Scholar]
- 18. Engelman JA, Zejnullahu K, Mitsudomi T et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316 (5827): 1039–43. [DOI] [PubMed] [Google Scholar]
- 19. Bean J, Brennan C, Shih JY et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007; 104 (52): 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989; 339 (6219): 58–61. [DOI] [PubMed] [Google Scholar]
- 21. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86 (3): 353–64. [DOI] [PubMed] [Google Scholar]
- 22. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000; 407 (6801): 249–57. [DOI] [PubMed] [Google Scholar]
- 23. Folkman J, Klagsbrun M. Angiogenic factors. Science 1987; 235 (4787): 442–7. [DOI] [PubMed] [Google Scholar]
- 24. Kanno S, Oda N, Abe M et al. Roles of two VEGF receptors, Flt‐1 and KDR, in the signal transduction of VEGF effects in human vascular endothelial cells. Oncogene 2000; 19 (17): 2138–46. [DOI] [PubMed] [Google Scholar]
- 25. Kim KJ, Li B, Winer J et al. Inhibition of vascular endothelial growth factor‐induced angiogenesis suppresses tumour growth in vivo. Nature 1993; 362 (6423): 841–4. [DOI] [PubMed] [Google Scholar]
- 26. Wilhelm SM, Carter C, Tang L et al. BAY 43‐9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64 (19): 7099–109. [DOI] [PubMed] [Google Scholar]
- 27. Mendel DB, Laird AD, Xin X et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003; 9 (1): 327–37. [PubMed] [Google Scholar]
- 28. Asano M, Yukita A, Matsumoto T, Kondo S, Suzuki H. Inhibition of tumor growth and metastasis by an immunoneutralizing monoclonal antibody to human vascular endothelial growth factor/vascular permeability factor121. Cancer Res 1995; 55 (22): 5296–301. [PubMed] [Google Scholar]
- 29. Nakamura K, Taguchi E, Miura T et al. KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res 2006; 66 (18): 9134–42. [DOI] [PubMed] [Google Scholar]
- 30. Van Belle E, Witzenbichler B, Chen D et al. Potentiated angiogenic effect of scatter factor/hepatocyte growth factor via induction of vascular endothelial growth factor: the case for paracrine amplification of angiogenesis. Circulation 1998; 97 (4): 381–90. [DOI] [PubMed] [Google Scholar]
- 31. Gerritsen ME, Tomlinson JE, Zlot C, Ziman M, Hwang S. Using gene expression profiling to identify the molecular basis of the synergistic actions of hepatocyte growth factor and vascular endothelial growth factor in human endothelial cells. Br J Pharmacol 2003; 140 (4): 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang YW, Su Y, Volpert OV, Vande Woude GF. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci U S A 2003; 100 (22): 12718–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsui J, Yamamoto Y, Funahashi Y et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer 2008; 122 (3): 664–71. [DOI] [PubMed] [Google Scholar]
- 34. Smolen GA, Sordella R, Muir B et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA‐665752. Proc Natl Acad Sci U S A 2006; 103 (7): 2316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boku T, Nakane Y, Minoura T et al. Prognostic significance of serosal invasion and free intraperitoneal cancer cells in gastric cancer. Br J Surg 1990; 77 (4): 436–9. [DOI] [PubMed] [Google Scholar]
- 36. Kodera Y, Nakanishi H, Yamamura Y et al. Prognostic value and clinical implications of disseminated cancer cells in the peritoneal cavity detected by reverse transcriptase‐polymerase chain reaction and cytology. Int J Cancer 1998; 79 (4): 429–33. [DOI] [PubMed] [Google Scholar]
- 37. Zeng ZS, Weiser MR, Kuntz E et al. c‐Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett 2008; 265 (2): 258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lutterbach B, Zeng Q, Davis LJ et al. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res 2007; 67 (5): 2081–8. [DOI] [PubMed] [Google Scholar]
- 39. Zou HY, Li Q, Lee JH et al. An orally available small‐molecule inhibitor of c‐Met, PF‐2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res 2007; 67 (9): 4408–17. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y, Kaplan‐Lefko PJ, Rex K et al. Identification of a novel recepteur d’origine nantais/c‐met small‐molecule kinase inhibitor with antitumor activity in vivo. Cancer Res 2008; 68 (16): 6680–7. [DOI] [PubMed] [Google Scholar]
- 41. Martens T, Schmidt NO, Eckerich C et al. A novel one‐armed anti‐c‐Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res 2006; 12 (20 Pt 1): 6144–52. [DOI] [PubMed] [Google Scholar]
- 42. Asano M, Yukita A, Suzuki H. Wide spectrum of antitumor activity of a neutralizing monoclonal antibody to human vascular endothelial growth factor. Jpn J Cancer Res 1999; 90 (1): 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]