

Abstract
TrkB is a neurotrophic tyrosine kinase receptor (Trk). To investigate its role in anoikis suppression in human ovarian cancer, we used reverse transcription–polymerase chain reaction and real‐time polymerase chain reaction, immunohistochemistry, and western blotting to compare the expression levels of TrkB and its ligand brain‐derived neurotrophic factor between (i) 20 epithelial ovarian cancers, their multicellular spheroids in ascites or great omentum metastatic lesions, and eight borderline or benign ovarian tumors, as well as four normal ovarian tissues; and (ii) three ovarian cancer cell lines cultured under different conditions: monolayer adhesive culture (adhesive cells), anchorage‐independent culture (cell spheroids), and trypsinized cell spheroids placed in monolayer adhesive dishes (cell spheroids replaced). TrkB and brain‐derived neurotrophic factor were overexpressed in epithelial ovarian cancers, and full‐length TrkB was more often overexpressed in high‐grade carcinomas and multicellular spheroids in ascites. Expression of TrkB mRNA was higher in OVCAR‐3 cell spheroids than in adhesive cells. The expression of full‐length TrkB protein was highest in OVCAR‐3 cell spheroids, but its precursor was expressed highly in OVCAR‐3 cells under all three culture conditions. The relationship between TrkB overexpression and phosphatidylinositol 3′‐kinase (PI3K)–AKT pathway activation in OVCAR‐3 cells was studied by western blotting and RNA interference. The PI3K–AKT pathway was highly activated in anoikis‐survived cells and was inhibited when TrkB was silenced by small interfering RNA. Finally, the chemosensitivity and invasiveness of OVCAR‐3 cells were examined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium, fluorescence‐activated cell sorting, Matrigel invasion assay, and in vivo studies. Adhesive cells showed higher chemosensitivity and lower invasion ability than anoikis‐survived cells. Our study suggests that TrkB might mediate anoikis suppression by activating the PI3K–AKT pathway in ovarian cancer cells. (Cancer Sci 2008; 99: 543–552)
Abbreviations:
- adh‐cells
adhesive cells
- BDNF
brain‐derived neurotrophic factor
- BW
body weight
- CDDP
cis‐diamminedichloride platinum (cisplatin)
- cell‐sph
cell spheroids
- cell‐sph‐adh
cell spheroids replaced
- EDTA
ethylene diamine tetra‐acetic acid
- FACS
fluorescence‐activated cell sorting
- FBS
fetal bovine serum
- FKHR
Forkhead (Drosophila) homolog 1 (rhabdomyosarcoma)
- GAPDH
glyceraldehyde‐3‐phosphate
- HNTG
50 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 1% Triton X‐100, 10% glycerol
- IHC
immunohistochemistry
- MAPK
mitogen‐activated protein kinase
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PI3K
phosphatidylinositol 3′‐kinase
- PKB/AKT
protein kinase B
- PPI
proton pump inhibitor
- RNAi
RNA interference
- RT
reverse transcriptase
- siRNA
small interfering RNA
- Trk
tyrosine kinase receptor
The mortality rate of ovarian carcinoma is the highest among all of the gynecological malignancies. Epithelial ovarian cancer, accounting for nearly 90% of all adult ovarian cancers,( 1 ) presents a complex clinical, diagnostic, and therapeutic challenge because of the difficulty of early detection and the lack of efficient therapeutic measures.
Chemotherapy resistance and metastasis are the major factors responsible for the failure of treatment. Recent studies indicate that chemotherapy resistance is partly mediated by the ability of cancer cells to circumvent apoptosis.( 2 , 3 , 4 , 5 ) Anoikis (apoptosis due to the loss of cell–matrix interactions) has been suggested to act as a physiological barrier to metastasis, and anoikis suppression is the first step taken by metastatic cells.( 3 ) Unlike normal epithelial cells, carcinomas grow, invade, and metastasize as disorganized three‐dimensional cellular spheroids in which cells are deprived of adhesion to the basement membrane but maintain anoikis resistance. Resistance to anoikis may allow the survival of cancer cells during systemic circulation, thereby facilitating secondary formation in distant organs, and anoikis resistance is now thought to be critical for the progression of disease and could therefore serve as a carcinoma‐specific novel therapeutic target. Full‐length TrkB( 6 ) has been investigated as a potent and specific suppressor of anoikis by activating the PI3K–AKT pathway.
The Trk family contains TrkA (NTRK1), TrkB (NTRK2), and TrkC (NTRK3). As single‐pass transmembrane molecules, Trk family proteins possess extracellular glycosylated polypeptides as well as transmembrane and cytoplasmic tyrosine kinase domains. TrkB has two selectively spliced variants: full‐length TrkB and truncated TrkB. The truncated receptor, including T1, T2, and T‐Shc, lacks the cytoplasmic kinase domain, whose functions are not very clear.( 7 ) Full‐length TrkB is a 145‐kDa transmembrane protein that is activated preferentially by BDNF. Following ligand binding, TrkB forms homodimers resulting in autophosphorylation of tyrosine residues, which is required for its catalytic and signaling activities. The first indication that BDNF–TrkB might play a role in human tumors came from studies on the expression patterns of TrkA and TrkB in neuroblastoma, which demonstrated that TrkA is associated with a favorable biology and outcome of neuroblastoma, whereas TrkB is expressed in aggressive neuroblastomas with unfavorable outcomes.( 8 ) Moreover, overexpression of TrkB has been found in several other types of human cancers, such as prostate adenocarcinoma,( 9 ) pancreatic cancer,( 10 ) hepatocellular carcinoma,( 11 ) and pulmonary carcinoid tumor,( 12 ) and higher levels of TrkB generally correlate with more aggressive tumor behavior. In addition, mutations in TrkB were found in colorectal cancer,( 13 ) and the primary mechanism leading to the increase in TrkB activity in cancers seems to occur through overexpression of the full‐length receptor.( 14 )
In neuroblastoma, the BDNF–TrkB rescue of neuroblastoma cells from chemotherapy‐induced cell death comes from the PI3K–AKT pathway.( 8 , 15 ) PI3K acts as an integrator of multiple inputs during tumorigenesis,( 16 ) and the function of the PI3K–AKT signaling pathway( 17 , 18 , 19 ) has been associated with antianoikis and apoptosis in cancers, where the increase in invasiveness and chemoresistance of cancer cells was achieved by regulating the expression of Bcl‐2 family proteins. It has also been reported that the PI3K–AKT pathway is highly activated in ovarian cancer and in the OVCAR‐3 cell line.( 20 , 21 , 22 , 23 )
Anoikis suppression was first reported in ovarian cancer by Frankel et al.( 24 ) It was indicated that the ovarian cancer cells that could survive in vivo as single cells or multicellular spheroids in ascites fluid might escape from anoikis, which was induced by activating the PI3K–AKT pathway through Bcl‐XI or RAB25.( 4 , 24 , 25 ) However, the relationship between the expression of TrkB or BDNF and anoikis suppression in ovarian cancer has not been reported. The present study was designed to detect whether TrkB is overexpressed in ovarian cancer, whether it is associated with the malignant behavior of ovarian cancer, and whether TrkB can mediate anoikis suppression by activating the PI3K–AKT pathway in ovarian cancer cells.
Materials and Methods
Cell culture and anchorage‐independent culture. Three human epithelial ovarian cancer cell lines, OVCAR‐3, SKOV3 (two kinds of serous adenocarcinoma cell lines), and ES2 (a kind of clear‐cell carcinoma cell line), were obtained from the Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences. The human neuroblastoma cell line SK‐N‐DZ was kindly provided by Professor Jinhua Zhang (Department of Hematology, the Second Affiliated Hospital of China Medical University, Shenyang, China). The cells were cultured in RPMI‐1640 medium supplemented with 100 units/mL penicillin, 100 µg/mL streptomycin, and 15% FBS for OVCAR‐3 or 10% calf serum for SKOV3, ES2, and SK‐N‐DZ, and maintained in a 37°C incubator with a humidified atmosphere of 95% O2 and 5% CO2.
For anchorage‐independent culture, 1 × 106 cells were seeded in flat‐bottomed, six‐well plates coated with hydrogel and cultured for 4–5 days in RPMI‐1640 supplemented with 15% FBS. We hypothesized that cell‐sph‐adh (cell spheroids with anchorage‐independent culture that had been trypsinized and reseeded in a monolayer culture dish) could maintain partial antianoikis capacity and imitate the ovarian cancer cells in metastatic lesions.
Tissue samples. Ovarian cancer tissue samples were obtained from patients with ovarian cancer undergoing surgery. Normal ovarian tissue samples (four cases) were obtained from patients with leiomyoma and ovarian cysts undergoing uterus and ovarian resection. Epithelial ovarian cancer samples included eight serous papillary cystadenocarcinoma, six mucinous cystadenocarcinoma, four endometrioid carcinoma, two clear‐cell carcinoma, and six multicellular spheroids in ascites and greater omentum metastatic lesions (corresponding to six serous papillary cystadenocarcinoma). Four borderline ovarian epithelial cancers and four ovarian serous or mucinous cystadenoma were also obtained. Tissue samples were frozen in liquid nitrogen immediately after removal and stored until use. The study protocol followed the ethical guidelines of Shanghai Jiao Tong University Affiliated First People's Hospital. Informed consent was obtained from all subjects.
RNA isolation, RT‐PCR, and real‐time PCR. Total RNA was prepared using TRIzol (Life Technologies). Aliquots of 1–15 µg of total RNA (1–3 µg for BDNF detection, and 7–15 µg for TrkB detection) was used for amplification with the One Step RNA PCR Kit (Takara). Primers for cDNA amplification were as follows: TrkB, F‐AGCATGAGCACATCGTCAAG and R‐ATATGCAGCATCTGCGACTG; BDNF, F‐CTGCAAACATGTCCATGAGG and R‐CCTGCAGCCTTCTTTTGTGT; and β‐actin, F‐CCTTCAACACCCCAGCCAT and R‐TCTTCATTGTGC TGGGTGCCA. The expected fragment lengths of TrkB, BDNF, and β‐actin were 180, 236, and 603 b.p., respectively. The PCR products were detected on 1.5% agarose gels and visualized under ultraviolet transillumination. The mRNA expression of TrkB and BDNF was analyzed using TotalLab software as the grayscale ratio to β‐actin. Real‐time PCR was also used to evaluate TrkB and BDNF expression using SYBR Premix Ex Taq (Takara). All reactions were carried out according to the manufacturer's protocols. The primer sequences for TrkB and BDNF were the same as above, and GAPDH (F‐AAGAAGGTGGTGAAGCAGGC and R‐TCCACCACCCTGTTGCTGTA, 203 b.p.) was used as an internal control for expression level. The annealing temperature for these primer sets was 60°C. The specificity of each PCR reaction was confirmed by melting curve analysis. The level of target gene expression in each sample was normalized to the respective GAPDH expression level.( 26 )
Immunohistochemistry. Tissue samples were fixed, cut, mounted, deparaffinized, and rehydrated. For endogenous peroxidase quenching, the slides were incubated in 0.3% H2O2 for 10 min. After blocking with goat serum, the sections were incubated with the primary antibody solution at 4°C overnight (1:70 dilution for TrkB and 1:100 dilution for BDNF; Santa Cruz Biotechnology). The slides were rinsed twice with PBS and incubated with peroxidase‐conjugated secondary antibodies for 1 h at room temperature. After rinsing, the color was developed by incubating slides with 3,3‐diaminobenzidine for 5 min followed by counterstaining with hematoxylin for 10 s. The slides were dehydrated with a successive washes of dH2O, 95 and 100% ethanol, and xylene before mounting with coverslips. The neuroblastoma specimen was used as a positive control, and the primary antibodies were replaced with PBS as negative control. Immunohistochemical staining results were evaluated using a blind method. Cytoplasmic and cytomembrane immunoreactivity with TrkB and cytoplasmic immunoreactivity with BDNF were considered to be positive. The expression of BDNF and TrkB was evaluated according to the percentage of positive cells and the intensity of staining.
Western blot analysis. After two rinses with ice‐cold PBS, the cells were scraped and lysed in ice‐cold HNTG buffer (50 mmol/L HEPES [pH 7.5], 150 mmol/L NaCl, 10% glycerol, 1% Triton X‐100, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 10 mmol/L sodium PPI, 100 µmol/L sodium orthovanadate, 100 mmol/L NaF, 10 µg/mL aprotinin, 10 µg/mL leupeptin, and 1 mmol/L phenylmethylsulfonyl fluoride) on ice for 30 min. Total protein was measured using Bio‐Rad protein assay reagent according to the manufacturer's protocol. Twenty micrograms of protein was separated on 12% sodium dodecylsulfate–polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes. After blocking with 10% bovine serum albumin in 1× Tris‐buffered saline, the membrane was incubated with various primary antibodies against PI3K, P‐p85PI3K, Bcl‐Xl, and Bad (Cell Signaling Technology) and AKT, P‐AKT, and β‐actin (Kang Chen Biology) at 4°C overnight. The membranes were washed three times with PBS and then incubated with peroxidase‐linked secondary antibody (1:10 000) for 1 h at room temperature. The signals were developed using an ECL kit (Pierce), scanned, and analyzed with TotalLab software. The relative expression of target proteins was presented as the ratio to β‐actin.
RNA interference. Construction of the TrkB–NTRK2 siRNA plasmid (pRNATU6‐NTRK2) was as described by others previously.( 27 ) Three target sequences designed for siRNA of NTRK2 were taken from GenBank (NM_006180): (1) GTAACCTGGTTTCCAAACA 698; (2) GCGCTTCAGTGGTTCTATA 940; and (3) CCACAGACGTCACTGATAA 1256. Three different nucleotide sequences against different target sequence were connected to the plasmid. Before transfection, cells were seeded in six‐well plates at a density of 1.5 × 106 cells/well and cultured for 24 h to 80% confluence. Then, 500 µL transfection medium (Opti‐MEM; Gibco) + 10 µL Lipofectamine 2000 (Invitrogen Life Technologies) mixed with plasmids (4 µg) was used for transfection. After 48 h of culture, the knockdown efficiency was determined by absolute quantification real‐time PCR and western blotting. We selected pRNATU6‐NTRK2 with the second nucleotide sequences for RNAi because of its highest inhibition efficiency. Scrambled siRNA was used as a negative control. The relative expression of TrkB mRNA was determined using the formula:
| (Copy number of TrkB cDNA of TrkB siRNA group1–3/copy numbers of GAPDH cDNA of TrkB siRNA group1–3)/(copy number of TrkB cDNA of cells without any treatment/copy number of GAPDH cDNA of cells without any treatment). |
The relative expression of TrkB protein was presented by the grayscale ratio of TrkB to β‐actin.
Cell viability assay. The MTT tetrazolium assay was used to measure cell viability. Briefly, 1 × 104 cells/well were seeded in 96‐well plates with complete medium. After incubation for 48 h, the cells were rinsed twice and incubated with serum‐free medium supplemented with the drugs. After incubation for the times indicated, 20 µL of MTT solution at 5 mg/mL was added and continually incubated for a further 4 h, and then 150 µL dimethylsulfoxide was added. The optical density was measured at 490 nm on a microplate reader.
Apoptosis assays. For the apoptosis assays, 1.5 × 106 cells/well were seeded in six‐well plates with complete medium. After incubation for 48 h, the cells were rinsed twice and incubated with serum‐free medium supplemented with the drugs. After incubation for the times indicated, the cells were trypsinized and rinsed, fixed in 70% ethanol, and stained with 50 mg/mL propidium iodide (Sigma). The fluorescence intensity was detected using FACScan. The percentage of apoptotic cells was calculated by determining the fraction of cells with sub‐G1 content.
Matrigel invasion assay. Boyden chambers were used to evaluate the invasive ability of the cancer cells. Briefly, Transwell chambers with 8 µm‐pored polycarbonate filters coated with Matrigel were used (BD Bioscience). Five groups of cells with equal quantity (adh‐cells, cell‐sph‐adh, cell‐sph, adh‐cells transfected with scrambled siRNA, and adh‐cells with TrkB silenced by siRNA) were trypsinized and seeded in serum‐free medium in the upper compartment of each chamber. Medium with 10% FBS was added to the lower compartment. After 22 h of culture, the penetrating cells were fixed with ice‐cold methanol, stained with hematoxylin, and visualized microscopically at 200× magnification. The cells were counted at 400× magnification in 10 representative areas per filter. Penetrating cells were represented as the average number of cells of 10 representative areas.
In vivo studies. Thirty female athymic nude mice (5–7 weeks of age) were divided randomly into five groups and inoculated intraperitoneally with 2 × 106 OVCAR‐3 cells. The groups were: (a) adh‐cells; (b) cell‐sph‐adh; (c) cell‐sph; (d) adh‐cells transfected with scrambled siRNA; and (e) adh‐cells with TrkB silenced by siRNA. Abdominal circumference and BW were measured twice weekly. After 8 weeks, the mice were killed. The tumors were excised, weighed, measured, fixed, and embedded.
Statistics. The results are presented as mean ± SE. Data were analyzed with one‐way ANOVA and unpaired Student's t‐test for comparison between groups. P < 0.05 was considered statistically significant. All in vitro experiments were repeated independently at least three times, whereas the in vivo experiments were repeated twice.
Results
Overexpression of TrkB was detected in ovarian cancer tissues, especially in greater omentum metastatic lesions and multicellular spheroids in ascites. TrkB and its high‐affinity ligand BDNF were overexpressed in epithelial ovarian cancers, including greater omentum metastatic lesions, multicellular spheroids in ascites, papillary cystadenocarcinomas, clear‐cell carcinomas, endometrioid carcinomas, and mucinous cystadenocarcinomas. The results showed that the overexpression was significantly higher in greater omentum metastatic lesions and multicellular spheroids in ascites than in the corresponding primary lesions (P < 0.001 and P < 0.01), and the expression levels were also higher in high‐grade serous papillary cystadenocarcinoma than in the other histological types (P < 0.001). Compared with borderline serous cystadenoma, TrkB and BDNF were overexpressed in carcinoma specimens (P < 0.01 and P < 0.001). TrkB and BDNF were also detected in normal ovarian tissue (including cortex and medulla), and were not found in benign epithelial ovarian tumors by RT‐PCR and real‐time PCR (Fig. 1a).
Figure 1.
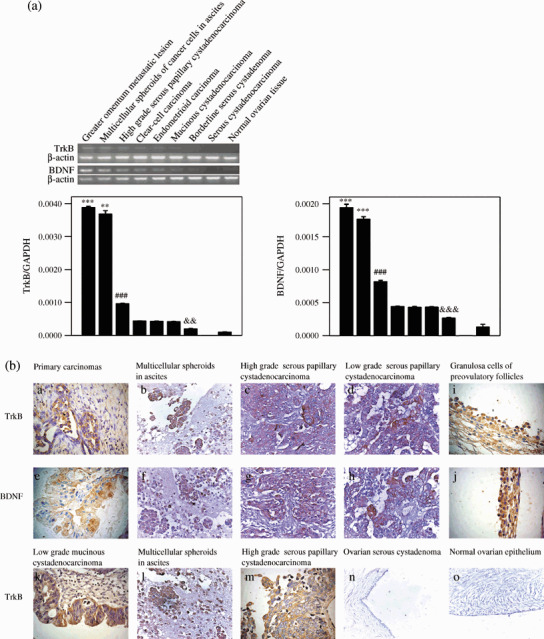
Overexpression of tyrosine kinase receptor (Trk) B and brain‐derived neurotrophic factor (BDNF) in ovarian tumor specimens. (a) TrkB and BDNF mRNA were analyzed by semiquantitative reverse transcription (RT)–polymerase chain reaction (PCR) (upper bands) and real‐time PCR (lower columns). β‐Actin and glyceraldehyde‐3‐phosphate (GAPDH) were used as the internal control for RT‐PCR and real‐time PCR, respectively. Data are expressed as the mean ± SE. ***P < 0.001; **P < 0.01 versus high‐grade serous papillary cystadenocarcinoma; ###P < 0.001 versus ovarian clear‐cell carcinoma, endometrioid, and mucinous cystadenocarcinoma; &&&P < 0.001; &&, P < 0.01 versus ovarian carcinomas and normal ovarian tissue. (b) Expression of the TrkB and BDNF proteins was determined by immunohistochemistry. Expression of (a–d) TrkB in ovarian carcinomas, (e–h) BDNF in ovarian carcinomas, and (i,j) TrkB and BDNF in granulosa cells of adult human preovulatory follicles. (k) Mainly cytoplasmic expression of TrkB in low‐grade mucinous cystadenocarcinoma. (l,m) High‐grade serous papillary cystadenocarcinoma and multicellular spheroids in ascites demonstrating cytoplasmic and intensive membranous expression of TrkB. (n,o) Expression of TrkB in ovarian serous cystadenoma and normal ovarian epithelium (a–n, ×400; m,n, ×200).
Immunohistochemical staining showed cytoplasmic and cytomembrane expression of TrkB,( 28 , 29 ) as well as cytoplasmic expression of BDNF in all borderline and malignant epithelial ovarian tumors. For TrkB, the cytoplasmic staining was seen extensively in all epithelial ovarian carcinomas, whereas cytomembrane staining was more often presented in multicellular spheroids in ascites and high‐grade carcinoma than in corresponding primary carcinomas and low‐grade carcinoma (Fig. 1b). However, none of the benign epithelial ovarian tumor and normal ovarian epithelial tissues displayed any TrkB immunostaining (Fig. 1b,n,o).
Although TrkB and BDNF expression was detected in ovarian tissues by RT‐PCR and real‐time PCR, it was not detected in normal ovarian epithelium by IHC. Consistent with a previous report that TrkB and BDNF were found in oocytes and granulosa cells,( 30 , 31 ) in our study, TrkB and BDNF expression was also detected in granulosa cells of adult human preovulatory follicles by IHC (Fig. 1b,i,j).
Overexpression of TrkB was detected in OVCAR‐3 ovarian cancer cells, especially in anoikis‐survived OVCAR‐3 ovarian cancer cells. The expression of TrkB and BDNF in three epithelial ovarian cancer cell lines (OVCAR‐3, SKOV3, and ES2) and the neuroblastoma cell line (SK‐N‐DZ) was analyzed by RT‐PCR and real‐time PCR. The mRNA expression of TrkB was highest in OVCAR‐3 cells (P < 0.01). We then selected OVCAR‐3 cells as a model for further investigation. Because anoikis suppression is the first step in metastasis, we established different culture conditions to mimic the metastatic status of cancer cells in vitro. The adh‐cells were used to imitate the status of cancer cells in primary lesions; cell‐sph were used to imitate the status of cancer cells deprived of extracellular matrix and surviving in ascites; and cell‐sph‐adh were used to imitate the status of cancer cells in metastatic lesions. Cell‐sph and cell‐sph‐adh were used to represent cells that survived anoikis. Cultured in different conditions, different forms of OVCAR‐3 were developed and detected by RT‐PCR, real‐time PCR, and western blotting. TrkB mRNA was overexpressed more highly in OVCAR‐3 cell‐sph than in adh‐cells (P < 0.001), but BDNF mRNA was the opposite (P < 0.001) (Fig. 2).
Figure 2.
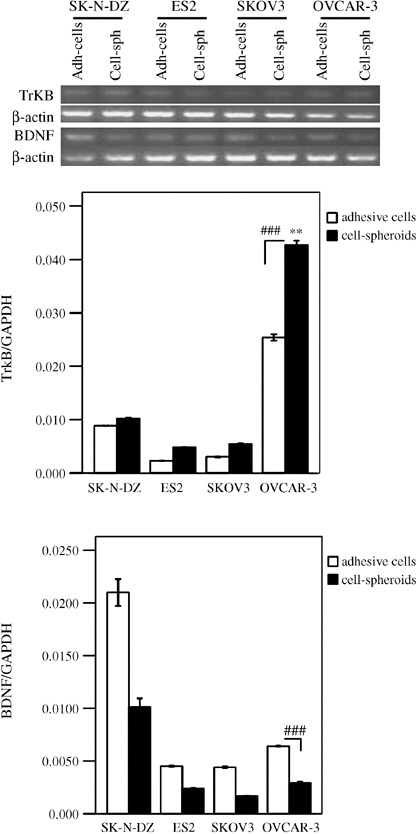
Expression of tyrosine kinase receptor (Trk) B and brain‐derived neurotrophic factor (BDNF) in ovarian cancer cell lines. Expression levels of TrkB and BDNF mRNA in SK‐N‐DZ, ES2, SKOV3, and OVCAR‐3 cells was determined by semiquantitative reverse transcription (RT)–polymerase chain reaction (PCR) (upper bands) and real‐time PCR (lower columns). β‐Actin and glyceraldehyde‐3‐phosphate dehydrogenase were used as the internal control for RT‐PCR and real‐time PCR, respectively. Data are expressed as the mean ± SE. **P < 0.01 versus SK‐N‐DZ, ES2 and SKOV3 cells; ###P < 0.001 cell spheroids versus adhesive cells in the OVCAR‐3 group.
By western blotting with anti‐TrkB antibody two bands were seen in OVCAR‐3 cells. The lower band (approximately 115 kDa) was visible in the three groups with similar density, whereas the upper band (145 kDa) was seen in cell‐sph and cell‐sph‐adh, but hardly seen in the adh‐cells (P < 0.001) (Fig. 3). In addition, TrkB was only shown in cytoplasmic staining in the OVCAR‐3 adh‐cells by IHC (data not shown).
Figure 3.

Expression of tyrosine kinase receptor (Trk) B in OVCAR‐3 cells under different culture conditions. Western blot analysis showing the upper band as the full‐length TrkB (145 kDa, glycosylated receptor form), and the lower band as the TrkB precursor (approximately 115 kDa, non‐glycosylated receptor form). Data are expressed as the mean; bars, ±SE. Mean ± SE densitometric values (normalized to β‐actin) from three independent experiments are shown. ***P < 0.001 versus cell spheroids (cell‐sph) and cell spheroids replaced (cell‐sph‐adh).
Phosphatidylinositol 3′‐kinase pathway was activated in anoikis‐survived OVCAR‐3 cells, but was inhibited and anoikis was increased when TrkB was silenced by siRNA. To determine whether the PI3K pathway was responsible for anoikis suppression in OVCAR‐3 cells, PI3K, AKT, phospho‐PI3K, and phospho‐AKT were detected in the five groups (adh‐cells, cell‐sph‐adh, cell‐sph, adh‐cells transfected with scrambled siRNA, and adh‐cells with TrkB silenced by siRNA), as confirmed by western blotting, and the RNAi technique was used to silence the expression of TrkB (Fig. 4a). The highest level of phosphorylated PI3K was found in cell‐sph and the lowest level in adh‐cells (P < 0.001). Phosphorylated AKT was present at a similar high level in three of the groups (adh‐cells, cell‐sph‐adh, and cell‐sph). However, when TrkB was silenced by siRNA, the expression of phosphorylated PI3K and phosphorylated AKT was inhibited significantly (P < 0.001) (Fig. 4b). These findings clearly point to the possibility that TrkB might contribute to anoikis resistance. Therefore, we evaluated whether downregulation of TrkB results in the increase of ovarian cancer cells to anoikis by apoptosis assay. OVCAR3 adh‐cells transfected with TrkB or scrambled siRNA were plated on hydrogel‐coated plates, which prevented cell attachment. After 48 h of culture, cell‐sph were trypsinized as single cells, stained with propidium iodide and measured by FACS. In the anchorage‐independent culture conditions, the cells transfected with TrkB siRNA showed a significant increase in apoptosis compared to cells transfected with scrambled control siRNA (P < 0.001) (Fig. 4c). These results demonstrate that the downregulation of TrkB sensitizes OVCAR3 ovarian cancer cells to anoikis by inhibiting the PI3K pathway.
Figure 4.
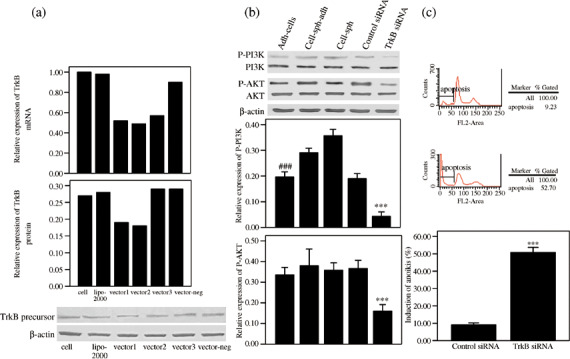
The phosphatidylinositol 3′‐kinase (PI3K) pathway was activated in anoikis‐survived OVCAR‐3 cells, but was inhibited and anoikis was increased when tyrosine kinase receptor (Trk) B was silenced by small interfering RNA (siRNA). (a) The expression of TrkB 48 h after transfection with siRNA plasmids was analyzed by real‐time polymerase chain reaction and western blotting. Three siRNA plasmids against different target sequences of TrkB (vector‐1, vector‐2, and vector‐3) were transfected into OVCAR‐3 cells. For the negative control, cells were transfected with scrambled siRNA. Transfection efficiency was determined by the green fluorescent protein gene (data not shown). Vector‐2 was selected for its best effect of suppressing TrkB expression. (b) P‐PI3K and P‐Ser‐AKT were analyzed by western blotting. The relative expression of P‐PI3K and P‐Ser‐AKT (normalized to β‐actin) from three independent experiments are expressed as mean ± SE. ***P < 0.001 versus adhesive cells (adh‐cells), cell spheroids replaced (cell‐sph‐adh), cell spheroids (cell‐sph), and the control siRNA group; ###P < 0.001 versus cell‐sph‐adh and cell‐sph. (c) Specific suppression of anoikis‐associated apoptosis by TrkB. OVCAR‐3 cells with scrambled siRNA (as control) or with silenced TrkB were seeded in hydrogel‐coated plates for 2 days, and then apoptosis in OVCAR3 cell‐sph was detected with fluorescence‐activated cell sorting by measuring the percentage of sub‐G1 cells stained with propidium iodide. Data were expressed as the mean ± SE. ***P < 0.001 versus the control group.
Expression of Bcl‐Xl and Bad was increased in anoikis‐survived OVCAR‐3 cells, but decreased when chemotherapy agents were combined with TrkB silencing. It has been reported that the PI3K–AKT pathway is associated with anoikis suppression by regulating the expression of Bcl‐2 family proteins. Our data had confirmed higher activation of the PI3K–AKT pathway in OVCAR‐3 multicellular spheroids,( 32 , 33 ) compared to adh‐cells. We then detected the expression of the anti‐apoptosis protein Bcl‐Xl and pro‐apoptosis protein Bad, which are downstream proteins in the PI3K pathway, in OVCAR‐3 cells by western blotting. We found that the expression of both Bcl‐Xl and Bad was higher in cell‐sph than in adh‐cells (P < 0.001). When the cells were treated with paclitaxel, the expression of Bcl‐Xl and Bad in cell‐sph was higher than that in adh‐cells, but the results were reversed when the cells were treated with cisplatin (P < 0.001). Moreover, when TrkB silencing was combined with chemotherapy agents, the expression of Bcl‐Xl and Bad in adh‐cells was significantly reduced compared to those cells treated with chemotherapy agents only (P < 0.001) (Fig. 5).
Figure 5.
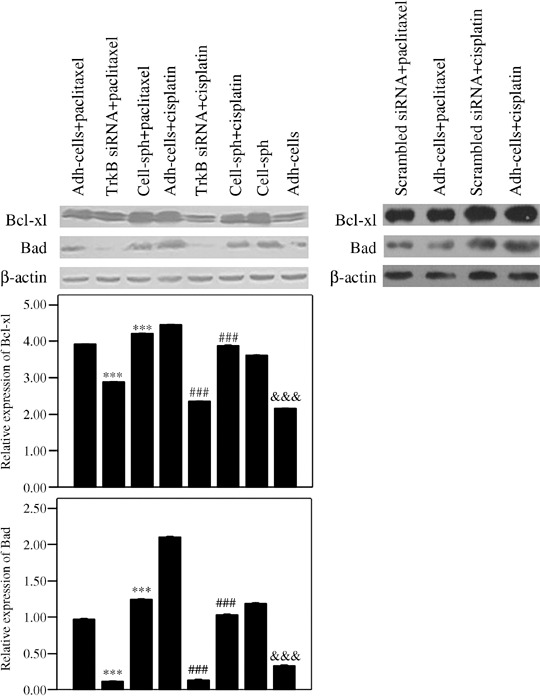
The expression of Bcl‐Xl and Bad was increased in anoikis‐survived OVCAR‐3 cells. Expression of Bcl‐Xl and Bad was detected in OVCAR‐3 cells under different culture conditions when treated with chemotherapeutic agents in combination with tyrosine kinase receptor (Trk) B silencing. OVCAR‐3 adhesive cells (adh‐cells) with or without TrkB silencing and OVCAR‐3 cell spheroids (cell‐sph) were treated with paclitaxel (10 µM) or cisplatin (40 µM) for 18 h, then Bcl‐Xl (upper) and Bad (lower) were detected by western blotting. The relative expression of Bcl‐Xl and Bad (normalized to β‐actin) from three independent experiments is expressed as the mean ± SE. ***P < 0.001 versus adh‐cells treated with paclitaxel; ###P < 0.001 versus adh‐cells treated with cisplatin; &&&P < 0.001 versus cell‐sph.
Anoikis‐survived OVCAR‐3 cells showed high chemotherapy resistance: Inhibition of the PI3K–AKT pathway by LY294002 impaired survival and enhanced apoptosis of OVCAR‐3 cells treated with paclitaxel. Our data have shown that TrkB overexpression could activate the PI3K–AKT pathway in OVCAR‐3 cells and such activation could be inhibited by TrkB silencing. Considering that anoikis suppression due to activation of the PI3K–AKT pathway may play an important role in the mechanism of chemotherapy resistance in OVCAR‐3 cells, we studied the chemosensitivity of OVCAR‐3 adh‐cells and cell‐sph‐adh treated with LY294002 (a highly selective inhibitor of PI3K) and chemotherapeutic agents by MTT and FACS. The cells were seeded and cultured in replaced culture medium with or without 50 µM LY294002 for 1 h, then treated with paclitaxel (10 µM) or cisplatin (40 µM) for 18 h (for MTT) or 6 h (for FACS). The results of the MTT assay showed that the survival rate of adh‐cells and cell‐sph‐adh treated with LY294002 and paclitaxel was significantly lower than that without LY294002 treatment (P < 0.01). Also, when the cells were treated with LY294002 and paclitaxel, the survival rate of adh‐cells was lower than that of cell‐sph‐adh (P < 0.01). However, when cells were treated with cisplatin instead of paclitaxel, cell‐sph‐adh showed a lower survival rate than adh‐cells (P < 0.05) (Fig. 6a). By FACS, the apoptotic rate of LY294002 and paclitaxel‐treated adh‐cells and cell‐shp‐adh was significantly higher than that of cells treated with paclitaxel only (P < 0.001). Taken together, OVCAR‐3 adh‐cells showed higher chemosensitivity to paclitaxel and cisplatin than cell‐sph‐adh (P < 0.001) (Fig. 6b,c).
Figure 6.
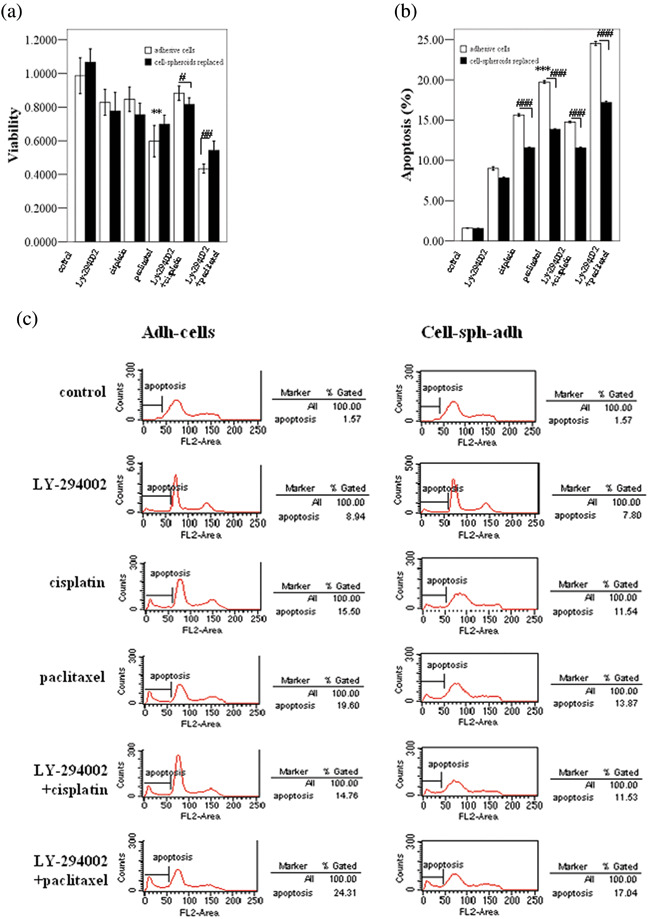
Anoikis‐survived OVCAR‐3 cells showed high chemotherapy resistance. The effects of phosphatidylinositol 3′‐kinase (PI3K)–protein kinase B (AKT) pathway inhibition by LY‐294002 in the survival and apoptosis of OVCAR‐3 cells was determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium and fluorescence‐activated cell sorting. (a) Adhesive cells (adh‐cells) and cell spheroids replaced (cell‐sph‐adh) with or without LY294002 (50 µM) treatment for 1 h followed by paclitaxel (10 µM) or cisplatin (40 µM) treatment for 18 h. The survival rate of cells is expressed as the relative density at 490 nm versus the untreated control of three independent experiments. (b,c) Adh‐cells and cell‐sph‐adh with or without LY294002 (50 µM) treatment for 1 h followed by paclitaxel (10 µM) or cisplatin (40 µM) treatment for 6 h. The percentage of apoptotic cells was calculated by determining the fraction of cells with sub‐G1 content in three independent experiments. Data are expressed as the mean ± SE. **P < 0.01 versus LY294002 + paclitaxel treatment group; #P < 0.05; ##P < 0.01; ###P < 0.001 adh‐cells versus cell‐sph‐adh.
Anoikis‐survived OVCAR‐3 cells showed high metastatic ability: Invasion ability of OVCAR‐3 cells was reduced when TrkB was silenced by siRNA in vitro and in vivo. Considering that anoikis suppression mediated by TrkB may play an important role in the mechanism of metastasis in OVCAR‐3 cells, invasion assays were carried out in vitro and in vivo. The Matrigel invasion assay in vitro using Boyden chambers showed that the anoikis‐survived cells (cell‐sph and cell‐sph‐adh) possessed the highest invasion capability and adh‐cells with silenced TrkB displayed the weakest invasion ability (P < 0.001) (Fig. 7a). The in vivo invasion assay in nude mice revealed that the cell‐sph formed distant and extensive metastatic lesions on the diaphragm, liver surface, and calcaneal region of the mesenterium. Cell‐sph‐adh developed larger tumors, whereas the adh‐cells with silenced TrkB developed smaller tumors (P < 0.03) (Fig. 7b; Table 1).
Figure 7.
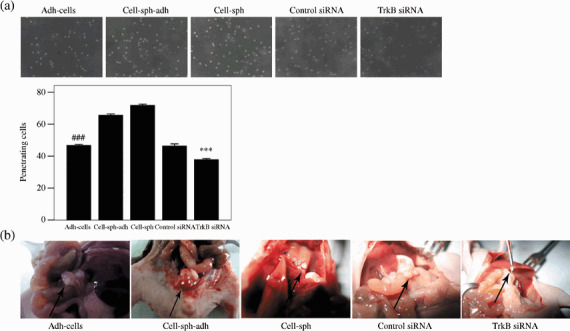
Anoikis‐survived OVCAR‐3 cells showed high metastasis ability. (a) Invasion analysis achieved by Matrigel invasion assay in vitro. Penetrating cells are expressed as the mean ± SE. ***P < 0.001 versus adhesive cells (adh‐cells), cell spheroids replaced (cell‐sph‐adh), cell spheroids (cell‐sph), and adh‐cells transfected with scrambled small interfering RNA; ###P < 0.001 versus cell‐sph‐adh and cell‐sph. Photographed at 400× magnification. (b) Invasion studies in vivo demonstrated that adh‐cells formed tumors in the mesenterium, cell‐sph‐adh generated a big tumor, cell‐sph formed distant metastatic lesions on the diaphragm, and adh‐cells with silenced tyrosine kinase receptor (Trk) B made a small tumor.
Table 1.
Tumorigenicity and metastatic potential of OVCAR‐3 cells under different culture conditions and tyrosine kinase receptor (Trk) B silenced by small interfering RNA (siRNA) in nude mice after intraperitoneal inoculation
| Clone | Tumorigenicity † | Tumor volume ‡ | Metastases § | |||
|---|---|---|---|---|---|---|
| Mesenterium | Peritoneum | Diaphragm | Others | |||
| Adh‐cells | 24 (n = 6) | 16.3 ± 4.7 | 5/5 | 1/5 | 0/5 | 0/5 |
| Cell‐sph‐adh | 37 (n = 6) | 50.7 ± 10.3 | 6/6 | 2/6 | 0/6 | 0/6 |
| Cell‐sph | 52 (n = 6) | 30.33 ± 10.2 | 6/6 | 4/6 | 2/6 | 2/6 |
| Adh‐cells with control siRNA | 22 (n = 6) | 15.73 ± 5.2 | 5/5 | 1/5 | 0/5 | 0/5 |
| Adh‐cells with TrkB silenced | 6 (n = 6) | 6.00 ± 1.4 ¶ | 3/3 | 0/3 | 0/3 | 0/3 |
Number of total tumors.
Mean ± SE mm3.
§ Number of mice with metastasis/number of mice with tumor.
Statistically significant when compared to the other four groups (P < 0.03). Adh‐cells, adhesive cells; cell‐sph, cell spheroids; cell‐sph‐adh, cell spheroids replaced.
Discussion
Peritoneal dissemination of tumor cells followed by ascites formation is very common in ovarian carcinoma. During this process, tumor cells must detach from their primary location and survive in the peritoneal cavity where the cells can not properly contact the extracellular matrix. This process must be controlled, at least in part, by some potent etiological factors that are responsible for anoikis suppression. Douma et al. indicated that TrkB generates a specific and potent prosurvival signal that renders epithelial cells resistant to anoikis, and acquires potent tumorigenic, invasive, and metastatic abilities.( 6 )
In the present study we found that TrkB and BDNF were overexpressed in epithelial ovarian cancer tissues, especially in greater omentum metastatic lesions, multicellular spheroids in ascites, high‐grade serous papillary cystadenocarcinomas, and OVCAR‐3 ovarian cancer cells. The higher the expression of TrkB, the more aggressive the behavior of cancer cells. Moreover, by means of the RNAi technique, we demonstrated that TrkB is associated with anoikis resistance, which conferred lower chemosensitivity and a higher invasion capability to OVCAR‐3 ovarian cancer cells via activation of the PI3K–AKT pathway.
Davidson et al. reported that the expression of both the mature glycosylated receptor of TrkA and its non‐glycosylated precursor were found in serous ovarian carcinoma tissues and malignant effusions.( 28 , 29 ) Similarly, we hypothesized that there were two forms of TrkB: the full‐length TrkB (145 kDa) expressed on cytomembranes, which is the mature and glycosylated receptor with full function, and the non‐glycosylated precursor (approximately 115 kDa) expressed in the cytoplasm, which is non‐functional or does not have fully functional kinase activity. Our data seem to support this hypothesis. We found that the 145 kDa full‐length TrkB, but not the 115 kDa precursor, was expressed more highly in anoikis‐survived OVCAR‐3 cells. In addition, more cytomembrane TrkB was found in multicellular spheroids in ascites and poorly differentiated carcinomas. TrkB has been investigated extensively in neuroblastoma and many other cancers, but the TrkB precursor has not been reported to date. We first mentioned the existence of the TrkB precursor in ovarian cancer cells here, but more evidence and regulation of the expression of the two forms TrkB should be investigated further.
It has been reported that TrkB is localized in both somatodendritic and axonal compartments in neurons.( 7 ) In unstimulated cells, the majority of TrkB is located intracellularly, and only a small amount of full‐length TrkB can be detected on the neuronal surface. It has been shown that when activated, the cell‐surface expression of full‐length TrkB is increased in both retinal ganglion cells and hippocampal neurons. Basal TrkA and TrkB receptor activation was indicated by low autophosphorylation in neuroblastomas, and physiological overexpression of the receptors may lead to dimerization, phosphorylation, and basal activation, contributing to the ability of mature neurons to survive in the absence of neurotrophin.( 34 ) We also observed that the TrkB precursor was overexpressed extensively in ovarian cancer tissues and OVCAR‐3 cancer cells under three culture conditions (adh‐cells, cell‐sph‐adh, and cell‐sph). Based on the overexpression of the precursor, TrkB can be auto‐activated through its overexpression of full‐length receptor.
In the present study we chose the ovarian cancer cell line OVCAR‐3 because it has a constitutively active PI3K–AKT pathway.( 20 , 23 , 35 ) Based on our data, the expression of TrkB was not obviously increased in SKOV3 cells, and this might be partially explained by the fact that the MAPK pathway has a central role in the induction and modulation of apoptosis in SKOV3 cells.( 36 )
Dodier et al. predicted that the overexpression of Bcl‐Xl‐mediated anoikis suppression should be a common feature of ovarian cancer cells, which would allow the cancer cells to survive in ascites.( 4 ) Consistent with this hypothesis, it was observed that there was a relatively high level of Bcl‐Xl expression in OVCAR‐3 cell‐sph in our study.
Our data also showed that the activation of PI3K and AKT was inhibited, and anoikis was increased in OVCAR‐3 cells when TrkB was silenced by siRNA, which demonstrated that TrkB mediated anoikis through the PI3K–AKT pathway in ovarian cancer cells. When TrkB silencing was combined with chemotherapeutic agents, the expression of Bcl‐Xl and Bad in OVCAR‐3 adh‐cells was reduced significantly compared with the cells treated only with chemotherapeutic agents. The expression of Bcl‐Xl was decreased when the PI3K pathway was inhibited, which is consistent with the findings of others.( 5 , 18 , 37 ) However, we found that the expression of Bad was also reduced, which presented apparent discrepancies with others,( 37 , 38 ) which concluded that PI3K–AKT inhibited apoptosis as it inhibited the release of cytochrome c and expression of Bad. Harnois et al. reported that inhibition of the PI3K–Akt‐1 pathway downregulated Bcl‐Xl and Bad.( 19 ) It was reported that Bcl‐Xl exists mainly on the outer membrane of mitochondria and inhibits the release of cytochrome c induced by chemotherapeutic agents,( 39 ) and when Bcl‐xl is combined with BAD on the outer membrane of mitochondria, the function of Bcl‐Xl is inhibited. Elevated levels of activated PKB/AKT can protect cells from undergoing apoptosis induced by cytotoxic drugs and contribute to drug resistance by phosphorylating BAD, resulting in BAD binding to 14‐3‐3 protein in the cytoplasm and sequestrating BAD away from Bcl‐Xl on the outer membrane of the mitochondria. We could investigate this further by localizing the different expression of Bcl‐Xl and Bad on the mitochondrial outer membrane or in the cytoplasm when the PI3K–AKT pathway is activated or inhibited.
In the present study, it was found that paclitaxel resistance is significantly attenuated when the PI3K pathway is inhibited, and the expression of Bcl‐Xl is reduced in OVCAR‐3 adh‐cells with silenced TrkB treated with paclitaxel. However, the inhibition of PI3K–AKT and the reduction of Bcl‐Xl expression did not provide chemosensitivity to cisplatin in OVCAR‐3 cells. Yan et al. suggested that the activation of p53 is a key determinant of sensitivity to cisplatin‐induced apoptosis, and PTEN expression and CDDP treatment to induce apoptosis is mediated through the upregulation of p53 and the activation of caspase‐3, which is p‐Akt/FKHR independent.( 40 ) However, p53 was not detected in OVCAR‐3 cells,( 20 ) and this may explain why the inhibition of PI3K–AKT activation and reduction of Bcl‐Xl expression could not increase the sensitivity of OVCAR‐3 cells to cisplatin.
In summary, the present study demonstrated that the expression of full‐length TrkB was increased in anoikis‐survived cells, and overexpression of TrkB plays a role in anoikis suppression via the PI3K–AKT pathway in ovarian cancer cells. These results may provide a new method of treatment for ovarian cancer involving blocking the expression of TrkB, which would improve the outcome of ovarian cancer by reducing metastasis and increasing chemosensitivity.
Acknowledgments
This work was supported by the Key Project of the Shanghai Health Bureau, grant 2005ZD002, and the Chinese National Natural Science Foundation, grant 30371482.
References
- 1. Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K‐ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med 2005; 11: 63–70. [DOI] [PubMed] [Google Scholar]
- 2. Westfall SD, Skinner MK. Inhibition of phosphatidylinositol3‐kinase sensitizes ovarian cancer cells to carboplatin and allows adjunct chemotherapy treatment. Mol Cancer Ther 2005; 4: 1764–71. [DOI] [PubMed] [Google Scholar]
- 3. Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer 2006; 6: 449–58. [DOI] [PubMed] [Google Scholar]
- 4. Dodier P, Piche A. Bcl‐XL is functionally non‐equivalent for the regulation of growth and survival in human ovarian cancer cells. Gynecol Oncol 2006; 100: 254–63. [DOI] [PubMed] [Google Scholar]
- 5. Liu Z, Li H, Derouet M et al . Oncogenic ras inhibits anoikis of intestinal epithelial cells by preventing the release of a mitochondrial pro‐apoptotic protein Omi/HtrA2 into the cytoplasm. J Biol Chem 2006; 281: 14 738–47. [DOI] [PubMed] [Google Scholar]
- 6. Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 2004; 430: 1034–9. [DOI] [PubMed] [Google Scholar]
- 7. Haapasalo A, Sipola I, Larsson K et al . Regulation of TRKB surface expression by brain‐derived neurotrophic factor and truncated TRKB isoforms. J Biol Chem 2002; 277: 43 160–7. [DOI] [PubMed] [Google Scholar]
- 8. Schramm A, Schulte JH, Astrahantseff K et al . Biological effects of TrkA and TrkB receptor signaling in neuroblastoma. Cancer Lett 2005; 228: 143–53. [DOI] [PubMed] [Google Scholar]
- 9. Satoh F, Mimata H, Nomura T et al . Autocrine expression of neurotrophins and their receptors in prostate cancer. Int J Urol 2001; 8: S28–34. [DOI] [PubMed] [Google Scholar]
- 10. Sclabas GM, Fujioka S, Schmidt C et al . Overexpression of tropomysin‐related kinase B in metastatic human pancreatic cancer cells. Clin Cancer Res 2005; 11: 440–9. [PubMed] [Google Scholar]
- 11. Yang ZF, Ho DW, Lam CT et al . Identification of brain‐derived neurotrophic factor as a novel functional protein in hepatocellular carcinoma. Cancer Res 2005; 65: 219–25. [PubMed] [Google Scholar]
- 12. Ricci A, Graziano P, Mariotta S et al . Neurotrophin system expression in human pulmonary carcinoid tumors. Growth Factors 2005; 23: 303–12. [DOI] [PubMed] [Google Scholar]
- 13. Bardelli A, Parsons DW, Silliman N et al . Mutational analysis of the tyrosine kinome in colorectal cancers. Science 2003; 300: 949. [DOI] [PubMed] [Google Scholar]
- 14. Desmet CJ, Peeper DS. The neurotrophic receptor TrkB: a drug target in anti‐cancer therapy? Cell Mol Life Sci 2006; 63: 755–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Z, Jaboin J, Dennis PA, Thiele CJ. Genetic and pharmacologic identification of Akt as a mediator of brain‐derived neurotrophic factor/TrkB rescue of neuroblastoma cells from chemotherapy‐induced cell death. Cancer Res 2005; 65: 2070–5. [DOI] [PubMed] [Google Scholar]
- 16. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006; 6: 184–92. [DOI] [PubMed] [Google Scholar]
- 17. Schmidt M, Hovelmann S, Beckers TL. A novel form of constitutively active farnesylated Akt1 prevents mammary epithelial cells from anoikis and suppresses chemotherapy‐induced apoptosis. Br J Cancer 2002; 87: 924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee S, Choi EJ, Jin C, Kim DH. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol 2005; 97: 26–34. [DOI] [PubMed] [Google Scholar]
- 19. Harnois C, Demers MJ, Bouchard V et al . Human intestinal epithelial crypt cell survival and death: Complex modulations of Bcl‐2 homologs by Fak, PI3‐K/Akt‐1, MEK/Erk, and p38 signaling pathways. J Cell Physiol 2004; 198: 209–22. [DOI] [PubMed] [Google Scholar]
- 20. Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol3′‐kinase increases the efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 2002; 15: 1087–92. [PubMed] [Google Scholar]
- 21. Mabuchi S, Ohmichi M, Nishio Y et al . Inhibition of inhibitor of nuclear factor‐κB phosphorylation increases the efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Clin Cancer Res 2004; 10: 7645–54. [DOI] [PubMed] [Google Scholar]
- 22. Sain N, Krishnan B, Ormerod MG et al . Potentiation of paclitaxel activity by the HSP90 inhibitor 17‐allylamino‐17‐demethoxygeldanamycin in human ovarian carcinoma cell lines with high levels of activated AKT. Mol Cancer Ther 2006; 5: 1197–208. [DOI] [PubMed] [Google Scholar]
- 23. Treeck O, Wackwitz B, Haus U, Ortmann O. Effects of a combined treatment with mTOR inhibitor RAD001 and tamoxifen in vitro on growth and apoptosis of human cancer cells. Gynecol Oncol 2006; 102: 292–9. [DOI] [PubMed] [Google Scholar]
- 24. Frankel A, Rosen K, Filmus J, Kerbel RS. Induction of anoikis and suppression of human ovarian tumor growth in vivo by down‐regulation of Bcl‐XL . Cancer Res 2001; 61: 4837–41. [PubMed] [Google Scholar]
- 25. Cheng KW, Lahad JP, Kuo WL et al . The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat Med 2004; 10: 1251–6. [DOI] [PubMed] [Google Scholar]
- 26. Li X, Cao X, Li X, Zhang W, Feng Y. Expression level of insulin‐like growth factor binding protein 5 mRNA is a prognostic factor for breast cancer. Cancer Sci 2007; 98: 1592–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sui G, Soohoo C, Affarel B et al . DNA vector‐based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 2002; 99: 5515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Davidson B, Lazarovici P, Ezersky A et al . Expression levels of the nerve growth factor receptors TrkA and p75 in effusions and solid tumors of serous ovarian carcinoma patients. Clin Cancer Res 2001; 7: 3457–64. [PubMed] [Google Scholar]
- 29. Davidson B, Reich R, Lazarovici P et al . Expression and activation of the nerve growth factor receptor TrkA in serous ovarian carcinoma. Clin Cancer Res 2003; 9: 2248–59. [PubMed] [Google Scholar]
- 30. Spears N, Molinek MD, Robinson LL et al . The role of neurotrophin receptors in female germ‐cell survival in mouse and human. Development 2003; 130: 5481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paredes A, Romero C, Dissen GA et al . TrkB receptors are required for follicular growth and oocyte survival in the mammalian ovary. Dev Biol 2004; 267: 430–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Uttamsingh S, Zong CS, Wang LH. Matrix‐independent activation of phosphatidylinositol3‐kinase, Stat3, and cyclin A‐associated Cdk2 is essential for anchorage‐independent growth of v‐Ros‐transformed chicken embryo fibroblasts. J Biol Chem 2003; 278: 18 798–810. [DOI] [PubMed] [Google Scholar]
- 33. Rytomaa M, Lehmann K, Downward J. Matrix detachment induces caspase‐dependent cytochrome c release from mitochondria: inhibition by PKB/Akt, but not Raf signaling. Oncogene 2000; 19: 4461–8. [DOI] [PubMed] [Google Scholar]
- 34. Schulte JH, Schramm A, Klein‐Hitpass L et al . Microarray analysis reveals differential gene expression patterns and regulation of single target genes contributing to the opposing phenotype of TrkA‐ and TrkB‐expressing neuroblastomas. Oncogene 2005; 24: 165–77. [DOI] [PubMed] [Google Scholar]
- 35. Burgering BM, Coffer PJ. Protein kinase B (c‐AKT) in phosphatidylinositol‐3‐OH kinase signal transduction. Nature 1995; 376: 599–602. [DOI] [PubMed] [Google Scholar]
- 36. Coltella N, Rasola A, Nano E et al . p38 MAPK turns hepatocyte growth factor to a death signal that commits ovarian cancer cells to chemotherapy‐induced apoptosis. Int J Cancer 2006; 118: 2981–90. [DOI] [PubMed] [Google Scholar]
- 37. Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol 2000; 151: 484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsuruta F, Masuyanam N, Gotoh Y. The phosphatidylinositol3‐kinase (PI3K)–Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem 2000; 277: 14 040–7. [DOI] [PubMed] [Google Scholar]
- 39. Gao N, Flynn DC, Zhang Z et al . G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am J Physiol Cell Physiol 2004; 287: C281–91. [DOI] [PubMed] [Google Scholar]
- 40. Yan X, Fraser M, Qiu Q, Tsang BK. Over‐expression of PTEN sensitizes human ovarian cancer cells to cisplatin‐induced apoptosis in a p53‐dependent manner. Gynecol Oncol 2006; 102: 348–55. [DOI] [PubMed] [Google Scholar]