

Abstract
Medullary thyroid carcinoma (MTC) is a rare endocrine tumor that frequently metastasizes, and treatment with irinotecan (CPT‐11) is limited because of side effects. Mutations in the Rearranged during transfection (RET) proto‐oncogene are considered the causative event of MTC. The objective of this study was to examine whether small interfering RNA (siRNA) and its combined treatment with CPT‐11 could inhibit MTC cell growth in vitro and in vivo. The transfection of RET siRNA suppressed RET expression, reduced proliferation, and increased caspase‐3/7 activity via the down‐regulation of Bcl‐2 expression. Combined treatments with CPT‐11 or SN‐38 significantly increased caspase 3/7 activity compared with RET siRNA, CPT‐11 or SN‐38 treatment alone. Importantly, intratumoral injection of RET siRNA along with intravenous injection of CPT‐11 significantly inhibited the tumor growth of MTC xenografts via an increased apoptotic effect. These findings that RET siRNA enhanced sensitivity for CPT‐11 will provide a novel strategy for the treatment of MTC with RET mutation.
(Cancer Sci 2010; 101: 941–947)
Medullary thyroid carcinoma (MTC) is a rare endocrine tumor originating from calcitonin‐secreting C cells. MTC may be sporadic or hereditary, including multiple endocrine neoplasia (MEN) type 2A, MEN type 2B and familial medullary thyroid carcinoma (FMTC).( 1 ) MTC has an overall 10‐year cause‐specific survival of 60–70%, with a particularly poor prognosis for patients with MEN2B and sporadic MTC, who have a 5‐year mortality of 30–50%.( 2 ) Hereditary MTC is caused by a germline mutation in the Rearranged during transfection (RET) gene. About 40% of sporadic MTCs harbor a somatic RET mutation.( 1 ) The RET gene encodes a receptor tyrosine kinase with an extracellular ligand‐binding domain and a cysteine‐rich domain, a signal transmembrane domain and an intracellular domain containing the catalytic tyrosine kinase domain.( 3 ) RET has a glial cell line‐derived neurotrophic factor as a ligand,( 4 ) but mutated RET leads to a constitutive active RET tyrosine kinase, causing malignant behavior of C cells.( 5 )
Surgery is currently the only effective treatment for primary MTC.( 6 ) Radioactive iodine has no benefit since C cells do not take up iodine. The role of chemotherapeutic regimens is also limited.( 7 ) Imatinib specifically inhibits the tyrosine kinase activity of Abl, Kit and platelet‐derived growth factor receptor (PDGFR).( 8 ) However, imatinib therapy for MTC yielded no objective responses and induced considerable toxicity in patients.( 9 ) CPT‐11 injected intraperioneally was relatively effective for TT tumor xenografts.( 10 ) There is, therefore, no curative therapy for patients with metastatic MTC, which is responsible for many of the deaths caused by MTC. Gene therapy is a potentially useful alternative treatment for MTC. Specific inhibition of RET signaling using the dominant‐negative RET mutant and ribozyme resulted in both growth suppression and subsequently the death of MTC cells.( 11 , 12 , 13 ) Expressions of thymidine kinase, interleukin (IL‐2) and IL‐12 by adenoviral vectors have resulted in tumor suppression in MTC animal models;( 14 ) however, the observed antitumor effect in vivo was mostly transient.
New technology based on specific inhibition of the RET gene may be useful for MTC treatment to improve safety. RNA interference (RNAi) is a powerful gene‐silencing process that holds great promise in the field of cancer therapy.( 15 ) Small interfering RNAs (siRNAs) are expected to have a medicinal application in human gene therapy as drugs with high specificity for molecular targeting. RET siRNA can inhibit the expression of RET tyrosine kinase receptor, and it is expected to inhibit all signals following RET tyrosine kinase; however, gene therapy with RET siRNA has not been reported in vivo for thyroid cells endogenously expressing mutated RET.
It is rational to select tyrosine kinase inhibitors for the treatment of MTC, although a selective drug that targets RET has not yet been found. Therefore, in this study, we selected RET siRNA as potentially therapeutic molecules. Human MTC (TT) cells endogenously expressed RET bearing the C634W mutation associated with MEN2A mutations, which have been identified mainly in one of six cysteine residues (codon 609, 611, 618, 620 in exon 10, and codon 630, 634 in exon 11) in the RET extracellular domain;( 1 , 16 ) therefore, TT cells are useful as a model for studying MTC.( 16 )
In this study, we investigated whether the transfection of RET siRNA alone and in combination with CPT‐11 could induce the growth inhibition of TT cells and tumor xenografts. Combination therapy with RET siRNA and CPT‐11 may be effective for MTC.
Materials and Methods
Cell culture. TT cells were obtained from the European Collection of Cell Cultures (ECACC, Wiltshire, UK). The cells were grown in Ham’s F‐12 culture medium (Wako, Osaka, Japan) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) (Gibco BRL) at 37°C in a 5% CO2 humidified atmosphere.
siRNA. The stealth RNA interference duplex‐targeting nucleotides of RET (RET‐1 siRNA, RET 2‐siRNA and RET‐3 siRNA) and stealth RNAi Negative Control Low GC Duplex and Medium GC Duplex as control siRNAs (Cont‐L siRNA and Cont‐M siRNA, respectively) were synthesized by Invitrogen (Carlsbad, CA, USA). The stealth siRNA sequences of RET were RET‐1 sense: 5′‐AAAUCCGAAAUCUUCAUCUUCCGCC‐3′ and antisense strand 5′‐GGCGGAAGAUGAAGAUUUCGGAUUU‐3′, RET‐2: sense 5′‐AAUCCUCUUCAUAAACAU‐CUCGGGA‐3′ and antisense 5′‐UCCCGAGAUGUUUAUGAAGAGGAUU‐3′, RET‐3: sense 5′‐AAAUCAGGGAGUCAGAUGGAGUGGA‐3′ and antisense 5′‐UCCACUCCAUC‐UGACUCCCUGAUUU‐3′. RET siRNA cocktail is used as a cocktail of three duplexes (100 nmol of RET stealth siRNA cocktail containing 33.3 nmol of each individual siRNA).
Western blotting. TT cells were transfected at a concentration of 100 nm siRNA by Lipofectamine 2000 (Invitrogen Corp.), and then incubated for 72 h. Cell protein extracts were prepared with sampling buffer containing 1% Triton X‐100 in phosphate‐buffered saline pH 7.4 (PBS), and then Western blotting was performed as previously reported.( 17 ) Expression of RET protein was identified using rabbit anti‐RET polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and goat anti‐rabbit IgG peroxidase conjugate (Santa Cruz Biotechnology) as the secondary antibody. Expression of Bcl‐2 and β‐actin protein was identified as previously described.( 17 )
Antiproliferative activity. TT cells at 50% confluence in 96‐well plates were transfected with 100 nm siRNA by Lipofectamine 2000, and then incubated for 72 h. In combination with CPT‐11 or SN‐38, the culture medium was exchanged for medium containing CPT‐11 (Irinotecan; Yakuruto, Tokyo, Japan) ranging from 0.1 to 1000 μg/mL or SN‐38 (ethyl‐10‐hydroxycamptothecin; Wako, Osaka, Japan) ranging from 0.001 to 10 μg/mL 48 h after transfection of siRNA, and then incubated for another 48 h. After incubation, cell viability was measured by a cell proliferation assay kit (Dojindo, Kumamoto, Japan) as previously reported.( 17 )
The numbers of dead cells after treatment with RET siRNA, CPT‐11, SN‐38 or their combination were measured by propidium iodide (PI) staining. The cells were transfected with 100 nm siRNA and then incubated for 48 h. The culture medium was exchanged for medium containing 10 μg/mL CPT‐11 or 0.2 μg/mL SN‐38, and then incubated for another 48 h. The percentage of PI‐positive cells was determined by examining fluorescence intensity on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA) as previously described.( 17 )
Real‐time RT‐PCR for quantification of RET and Bcl‐2 mRNA. TT cells at 50% confluence in 6‐well plates were transfected with 100 nM siRNA with Lipofectamine 2000 and then incubated for 72 h. Total RNA was isolated from transfected cells using the NucleoSpin RNA II (Macherey‐Nagel, Dueren, Germany). SYBR green quantitative RT‐PCR amplification for Bcl‐2 and β‐actin cDNA was carried out by MyiQ™ Single Color Real‐Time PCR Detection System (Bio‐Rad Laboratories, Hercules, CA, USA) as previously reported.( 17 ) For the amplification of human RET cDNA, primers RET‐FW, 5′‐GGCAGCCAGAAACATCCT‐3′, and RET‐RW, 5′‐ACTTTGCGTGGTG‐TAGAT‐3′, were used.
Caspase 3/7 assay. TT cells at 50% confluence in 12‐well plates were transfected with 100 nm siRNA using Lipofectamine 2000 and then incubated for 48 h. The culture medium was then exchanged for medium containing 10 μg/mL CPT‐11 or 0.2 μg/mL SN‐38 and then incubated for another 48 h. After the incubation, caspase 3/7 activity was measured by Caspase3/7 assay system (Caspase‐Glo® 3/7 assay; Promega, Madison, WI, USA) as previously reported.( 18 )
Assessment of TT tumor growth. Female ICR nu/nu mice (6 weeks of age) were purchased from Oriental Yeast Co., Ltd. (Tokyo, Japan). To generate TT tumor xenografts, 1 × 107 TT cells suspended in 50 μL PBS containing 50% Matrigel (Collaborative Research, Bedford, MA, USA) were inoculated subcutaneously into the flank region of mice. When the average volume of xenograft tumors reached about 100 mm3, therapy was started (day 0). For siRNA transfection in vivo, we used cationic nanoparticles (NP) consisting of cholesteryl‐3β‐carboxyamidoethylene‐N‐hydroxyethylamine as a cationic lipid, and 5 mol% Tween‐80, as previously reported.( 19 ) For transfection into tumors, 10 μg siRNA was mixed with NP at a charge ratio (+/−) of cationic lipid/siRNA of 1/1 with gentle shaking and standing for 15 min at room temperature. The complex of NP and siRNA was directly injected into the xenografts on days 0, 3, and 6, and CPT‐11 was intravenously injected at a dose of 30 mg/kg on day 2, 5, and 8. On 24 days, blood was collected into tubes and centrifuged to separate plasma from blood cells. Serum concentrations of calcitonin were measured using a human calcitonin ELISA kit (Biosource International, Inc., CA, USA). Animal experiments were conducted with ethics approval from our institutional animal care and use committee.
Preparation of probes for in situ hybridization. The RET cDNA fragment (873 bp) was amplified from TT cells cDNA. The RET primers used for RET cDNA amplification were: forward primer, 5′‐TCCTGGGAGAAGCTCAGTGT‐3′ and reverse primer, 5′‐CACGTTGAAGTGGAGCAAGA‐3′. The fragments were cloned into pGEM‐T vector (Promega), and 35S‐labeled cRNA probes were prepared as previously reported.( 20 )
In situ hybridization. For transfection into tumors, the complex of NP and 10 μg siRNA per tumor was directly injected into TT tumor xenografts on days 0, 3, and 6. At 48 h after final injection the tumors were frozen in powdered dry ice, and coronal sections were cut 16 μm thick with a cryostat, thaw‐mounted onto gelatin and poly‐lysine‐coated slides. In situ hybridization was carried out as previously described.( 20 ) All sections were counterstained with hematoxylin and eosin.
In vivo detection of apoptosis. For siRNA transfection into tumors, 10 μg siRNA per tumor was directly injected by NP into TT tumor xenografts on days 0, 3, and 6. CPT‐11 was intravenously injected at a dose of 30 mg/kg on day 2, 5, and 8. On day 11, the Red in vivo FLIVO™ apoptosis Kit (Immunochemistry Technologies, Bloomington, MN, USA) was injected i.v. and allowed to circulate for 30 min. The tumors were frozen in powdered dry ice, and coronal sections were cut 20 μm thick with a cryostat, thaw‐mounted onto poly‐lysine‐coated slides. The specimens were examined microscopically using an Eclipse TS100‐F microscope (Nikon, Tokyo, Japan).
Statistical analysis. The statistical significance of the data was evaluated with Student’s t‐test. P < 0.05 was considered significant.
Results
In vitro growth inhibitory effect of RET siRNA. We initially characterized the expression of RET protein in TT cells after transfection of RET siRNA with Lipofectamine 2000. Here, we used RET‐1, RET‐2, RET‐3 and their cocktail siRNAs as RET siRNAs (40%, 44%, 48%, and 44% in GC contents of the siRNA sequence, respectively), and Cont‐L and ‐M siRNAs as negative control siRNAs (36% and 48% in GC contents of the siRNA sequence, respectively). Expression of RET protein was strongly suppressed in TT cells 48 h after transfection of RET‐1, RET‐2, RET‐3 siRNA or their siRNA cocktail, but unaffected by the transfection of Cont‐L or ‐M siRNA (Fig. 1A). Furthermore, the transfection of RET siRNA suppressed the expression of RET mRNA to about 40–50% of untransfected cells (Fig. 1B).
Figure 1.
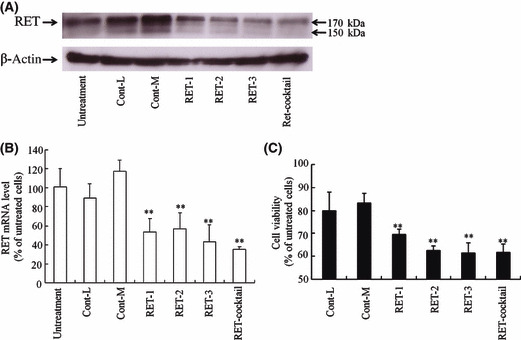
Inhibition of RET expression by transfection of RET siRNA with Lipofectamine 2000 into TT cells. Expressions of RET protein (170 KDa) and mRNA were detected by Western blot (A) and quantitative RT‐PCR (B) analyses, respectively, 72 h after transfection with 100 nm Cont‐L and ‐M siRNA, RET‐1, ‐2 and ‐3 siRNA and their cocktail. The number of viable cells was determined by WST‐8 assay 72 h after transfection (C). n = 4 for each sample. **P < 0.01, compared with Cont‐L siRNA transfected cells.
To investigate whether the suppression of RET expression induced the inhibition of cell growth, we examined cell viability 72 h after transfection of RET siRNA at 100 nm. After transfection, the cell viabilities of all RET siRNAs were 60–70%, but those of Cont‐L and ‐M were 80 and 83%, respectively (Fig. 1C). When transfected at a concentration of 12.5 nM siRNA, the RET siRNA cocktail showed a stronger decrease of cell viability (67%) than RET‐1, RET‐2 and RET‐3 siRNA (76, 72, and 75%, respectively) (data not shown). The slight inhibition of cell growth by transfection with Cont‐L and ‐M siRNA may be due to the toxicity of Lipofectamine 2000. In subsequent studies, we used the RET siRNA cocktail as a RET siRNA, and Cont‐L siRNA as a control siRNA.
Bcl‐2 expression and caspase 3/7 activity after transfection with RET siRNA into TT cells. It has been reported that the expression of a dominant‐negative RET down‐regulated the expression of Bcl‐2.( 21 ) Therefore, we investigated whether transfection of RET siRNA affected Bcl‐2 expression in TT cells (Fig. 2A,B). Transfection of RET siRNA decreased the expression of Bcl‐2 mRNA and protein (Fig. 2A,B) and furthermore, significantly increased caspase 3/7 activity in cells compared with Cont siRNA (Fig. 2C). These findings suggested that the suppression of RET expression may increase caspase 3/7 activity via the down‐regulation of Bcl‐2 expression.
Figure 2.
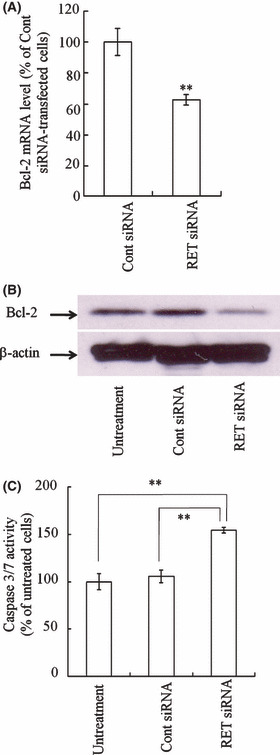
Effect of transfection with RET siRNA with Lipofectamine 2000 on Bcl‐2 expression and caspase 3/7 activity in TT cells. Cells were incubated for 72 h after transfection with 100 nm RET siRNA or Cont siRNA. Expressions of Bcl‐2 mRNA and protein were detected by quantitative RT‐PCR (A) and Western blot (B). Caspase 3/7 activity was measured by caspase 3/7 luminometric assay (C). n = 4 for each sample. **P < 0.01.
In vitro sensitivity of CPT‐11 or SN‐38. Here, we used CPT‐11 as a potent antitumor drug for TT tumor xenografts, because it has been reported that MTC cell lines were sensitive to a topoisomerase I inhibitor, camptothecin (CPT).( 22 ) To investigate whether the transfection of RET siRNA enhanced sensitivity for chemotherapy, we incubated CPT‐11 or its active metabolite SN‐38 for 48 h in the cells 48 h after transfection of RET siRNA. In cells transfected with RET siRNA, cytotoxicities by CPT‐11 and SN‐38 were similar with those in cells transfected with Cont siRNA (Fig. 3A,B), indicating that their activities were at least additive for tumor suppression by RET siRNA.
Figure 3.
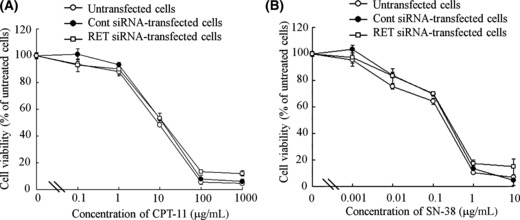
Concentration‐dependent effect of CPT‐11 or SN‐38 on cytotoxicity after transfection with RET siRNA. Cells were transfected with 100 nm RET siRNA or Cont siRNA with Lipofectamine 2000 and incubated for 48 h. After incubation, the cells were treated with various concentrations of CPT‐11 (A) or SN‐38 (B) and incubated for another 48 h. The number of viable cells was determined by WST‐8 assay. n = 4 for each sample.
We next investigated whether RET siRNA transfection affected the number of cell deaths and caspase 3/7 activity by CPT‐11 or SN‐38 treatment. The cells were transfected with RET or Cont siRNA for 48 h, and then treated with 10 μg/mL CPT‐11 or 0.2 μg/mL SN‐38 for 48 h, the concentrations of which were around the IC50 of CPT‐11 and SN‐38 in untransfected cells, respectively. To measure the number of cell deaths, PI staining of the cellular nucleus was used as a marker. RET siRNA transfection increased the number of PI‐staining cells and caspase 3/7 activity compared with Cont siRNA transfection (Fig. 4A,B). Among treatments, the combination of RET siRNA plus SN‐38 significantly increased activity and cells compared to RET siRNA or SN‐38 alone (Fig. 4A,B) (P < 0.01).
Figure 4.
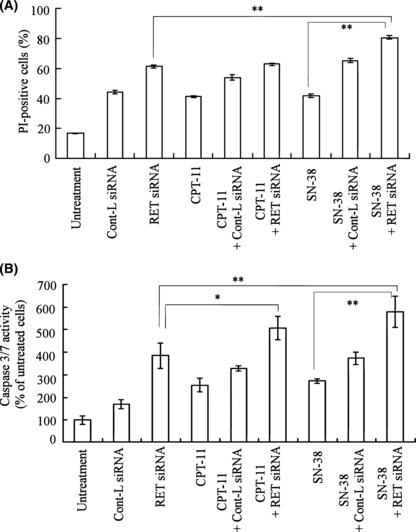
Dead cells (A) and caspase‐3/7 activity (B) by combined treatment with RET siRNA plus CPT‐11 or SN‐38 in TT cells. The number of dead cells was measured by a FACSCalibur flow cytometer after staining with PI (A). Caspase‐3/7 activity was measured by caspase‐3/7 luminometric assay (B). n = 3 for each sample. *P < 0.05, **P < 0.01; compared with RET siRNA transfected cells.
Suppression of RET expression in TT tumor xenografts. Next, we investigated whether the expression of RET mRNA was decreased by intratumoral injection of RET siRNA. Here, we used NP as a siRNA transfection vector for in vivo experiments.( 19 ) Cont or RET siRNA was directly injected with NP into TT tumor xenografs 3 times at 3‐day intervals and then tumors were excised 2 days after the final injection. The sense probe for the RET gene was used as a negative control probe. Silver grain distribution in the dark field and black grain in the bright field following in situ hybridization exhibiting RET mRNA expression (Fig. 5A,B, supplemental Figs S1 and S2). Positive cells for RET mRNA were strongly detected in the tumor cells, but not stroma cells with the antisense probe for the RET gene (Fig. 5B). Most of the tumor cells were positive for the expression of RET mRNA in saline‐injected and Cont siRNA‐transfected cells. In contrast, RET siRNA transfection decreased the number of silver grains in the dark field (Fig. 5A) and black grains in the bright field (Fig. 5B) in some tumor sections, indicated that the expression of RET mRNA was suppressed by RET siRNA injection.
Figure 5.
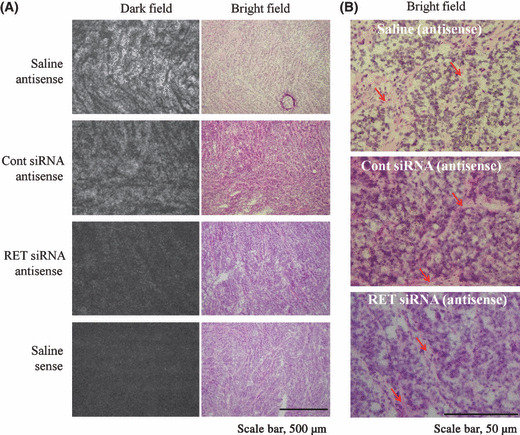
In situ hybridization of a 35S‐labeled antisense or sense probe generated against RET mRNA. An antisense probe was used to detect RET mRNA. The sense probe for the RET gene was used as a negative control probe. In (A), silver grains in a dark field indicate RET mRNA expression. Scale bar: 500 μm. In (B), microautoradiographs in bright fields in Fig. 5(A) were enlarged. Black grains indicate RET mRNA expression. Red arrows indicate stroma cells. Scale bar: 50 μm.
Synergistic inhibition of the growth of TT tumor xenografts. We evaluated the efficacy of combination therapy of RET siRNA and CPT‐11 in inhibiting the growth of subcutaneous TT tumors. The anti‐tumor effect was evaluated by direct injection of Cont or RET siRNA with NP into xenografts once a day three times (day 0, 3, and 6) following three i.v. injections of CPT‐11 (day 2, 5, and 8). No significant growth inhibitory effect was observed in mice treated with RET siRNA, CPT‐11 or CPT‐11 plus Cont siRNA compared with mice injected with saline (Fig. 6A). CPT‐11 plus Cont siRNA exhibited a similar tumor suppressive effect with CPT‐11 alone. A significant growth inhibitory effect was observed in combination therapy with CPT‐11 and RET siRNA compared with CPT‐11 alone. All treatments, the transfection of RET siRNA, the injection of CPT‐11 alone, and their combination did not significantly change body weight during 3 weeks of treatment (data not shown). In this study, the effect of the combination on tumor growth was significantly higher than that of CPT‐11 alone at lower doses that did not exhibit serve adverse effects.
Figure 6.
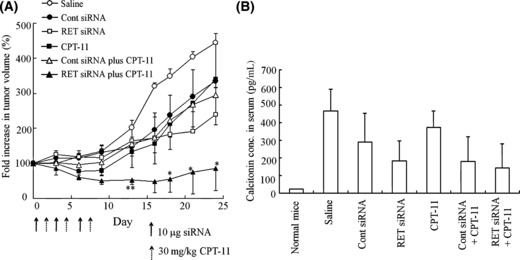
In vivo combination therapy of RET siRNA and CPT‐11 for TT tumor xenografts. Ten microgram of RET or Cont siRNA per tumor was directly injected with NP into the tumor on days 0, 3, and 6. CPT‐11 at a dose of 30 mg/kg was injected i.v. on days 2, 5, and 8. Tumor volume was measured for 24 days (A). *P < 0.05, **P < 0.01; compared with the group injected with CPT‐11. All mice were sacrificed on day 24 and the concentration of calcitonin in blood was measured (B). Data in A and B are shown as the mean ± SE. n = 3 for each group.
RET siRNA transfection alone could not significantly suppress tumor growth. The results of in situ hybridization showed that the suppression of RET expression in the tumors was restricted to the tumor in the vicinity of the injection site (data not shown). Strong inhibition of in vivo tumor growth by RET siRNA depends on the development of a gene vector having the ability to introduce siRNA widely into tumors.
Serum calcitonin concentration and in vivo apoptosis. C cells of the thyroid gland secrete calcitonin, the level of which is elevated in patients with C cell hyperplasia and MTC; therefore, calcitonin is a very sensitive and reliable marker for the presence of MTC, and the level of calcitonin correlates with tumor burden and the response to therapy.( 23 , 24 ) On day 24 in Fig. 6A, all mice were sacrificed, and serum calcitonin concentration was measured. Serum calcitonin level in TT tumor xenografts showed a 21‐fold increase compared with that in normal mice (Fig. 6B). RET siRNA or CPT‐11 alone decreased serum calcitonin levels to 39% and 80% compared with saline (Fig. 6B). Corresponding to the highest growth inhibition, the combination of CPT‐11 plus RET siRNA markedly decreased the serum calcitonin level to 30%.
To confirm the effectiveness in vivo, we detected apoptotic cells 48 h after combination therapy with RET siRNA and CPT‐11. In tumor sections after injections of RET siRNA or CPT‐11 alone, red, indicating apoptotic cells, was slightly observed, but not in saline or Cont siRNA (Fig. 7). On the other hand, combination treatment with RET siRNA plus CPT‐11 strongly induced apoptotic cells. These data also indicated that combination therapy was effective for MTC.
Figure 7.
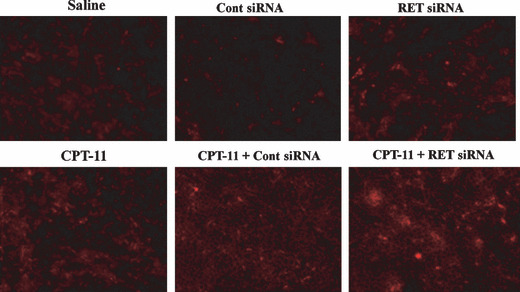
Distribution of apoptotic cells in TT tumor xenografts after injection of RET siRNA and CPT‐11. Experimental conditions were the same as in Fig 6. On day 11, all mice were sacrificed and apoptosis cells were detected by the Red in vivo FLIVO™ apoptosis Kit. Red signals indicate apoptotic cells. Magnification ×100.
Discussion
In this study, we demonstrated that the combination with RET siRNA and CPT‐11 could induce the growth inhibition of TT cells and tumor xenografts. Recently, RET siRNA delivered by chitosan‐coated poly(isobutylcyanoacrylate) nanoparticles led to tumor growth inhibition after intratumoral administration in mice transplanted with NIH3T3 cells transformed with the ret/PTC1 gene;( 25 ) however, it has been reported that the ret/PTC‐1 signaling pathway was different between thyroid and fibroblast cells;( 26 ) therefore, this is the first report that RET siRNA transfected with NP inhibited tumor growth in mice bearing TT cells.
The combination therapy in vivo revealed greater than additive effects in cytotoxicity in vitro. CPT‐11 is known as an effective inhibitor of angiogenesis for endothelial cells in tumor as well as cytotoxic drug.( 27 ) The mechanism of increasing growth inhibition for TT tumor xenografts by RET siRNA and CPT‐11 was not clear, although inhibition of receptor tyrosine kinase in tumor cells could sensitize cells to CPT‐11.( 28 , 29 ) Overexpression of Bcl‐2 has been observed in MTC,( 30 , 31 ) and is associated with the poor response of MTC to chemotherapy. The expression of dominant‐negative RET in TT cells decreased the expression of Bcl‐2 and could reduce cell viability,( 21 ) and the antitumor effect by CPT‐11 was enhanced by the combination with RET inhibitor, CEP‐751, for TT tumor xenografts.( 10 ) As another possibility, RET has been shown to activate nuclear factor κB (NFκB) via the phosphatidylinositol‐3‐kinase/Akt pathway.( 1 ) Akt mediates multiple cellular responses, such as survival signaling by NFκB activation and BAD inactivation, and cell cycle progression; therefore, inhibition of RET expression might modulate cellular sensitivity through the survival signaling pathway.( 32 , 33 ) From these findings, the combination of RET siRNA and CPT‐11 will have great potential for gene therapy in MTC.
In conclusion, we demonstrated that combining RET siRNA with CPT‐11 resulted in significant greater growth suppression of TT cells and tumor xenografts via increased apoptosis. As the thyroid gland is anatomically accessible, future treatment of primary MTC tumors by direct intratumoral injection of siRNA/NP complex expressing mutated genes is possible. Thus, the combination of RET siRNA and CPT‐11 may serve as a novel tool for gene therapy.
Supporting information
Fig. S1. In situ hybridization of a 35S‐labeled antisense or sense probe generated against RET mRNA. Microautoradiographs in bright fields of Fig. 5A were enlarged. Black grains indicate RET mRNA expression. Scale bar: 500 μm.
Fig. S2. In situ hybridization of a 35S‐labeled antisense or sense probe generated against RET mRNA. Microautoradiographs in bright fields of Fig. 5B were enlarged. Black grains indicate RET mRNA expression. Scale bar: 50 μm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Acknowledgments
We thank Ms. Manami Kubo for assistance with the experimental work. This study was supported in part by the Japan Health Sciences Foundation, by the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by the Open Research Center Project.
References
- 1. Kodama Y, Asai N, Kawai K et al. The RET proto‐oncogene: a molecular therapeutic target in thyroid cancer. Cancer Sci 2005; 96: 143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moley JF. Medullary thyroid carcinoma. Curr Treat Options Oncol 2003; 4: 339–47. [DOI] [PubMed] [Google Scholar]
- 3. Hansford JR, Mulligan LM. Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. J Med Genet 2000; 37: 817–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Treanor JJ, Goodman L, de SF et al. Characterization of a multicomponent receptor for GDNF. Nature 1996; 382: 80–3. [DOI] [PubMed] [Google Scholar]
- 5. Santoro M, Melillo RM, Carlomagno F, Vecchio G, Fusco A. Minireview: RET: normal and abnormal functions. Endocrinology 2004; 145: 5448–51. [DOI] [PubMed] [Google Scholar]
- 6. Cohen MS, Moley JF. Surgical treatment of medullary thyroid carcinoma. J Intern Med 2003; 253: 616–26. [DOI] [PubMed] [Google Scholar]
- 7. Gimm O. Thyroid cancer. Cancer Lett 2001; 163: 143–56. [DOI] [PubMed] [Google Scholar]
- 8. Jones RL, Judson IR. The development and application of imatinib. Expert Opin Drug Saf 2005; 4: 183–91. [DOI] [PubMed] [Google Scholar]
- 9. de Groot JW, Zonnenberg BA, van Ufford‐Mannesse PQ et al. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J Clin Endocrinol Metab 2007; 92: 3466–9. [DOI] [PubMed] [Google Scholar]
- 10. Strock CJ, Park JI, Rosen DM et al. Activity of irinotecan and the tyrosine kinase inhibitor CEP‐751 in medullary thyroid cancer. J Clin Endocrinol Metab 2006; 91: 79–84. [DOI] [PubMed] [Google Scholar]
- 11. Drosten M, Frilling A, Stiewe T, Putzer BM. A new therapeutic approach in medullary thyroid cancer treatment: inhibition of oncogenic RET signaling by adenoviral vector‐mediated expression of a dominant‐negative RET mutant. Surgery 2002; 132: 991–7. [DOI] [PubMed] [Google Scholar]
- 12. Drosten M, Stiewe T, Putzer BM. Antitumor capacity of a dominant‐negative RET proto‐oncogene mutant in a medullary thyroid carcinoma model. Hum Gene Ther 2003; 14: 971–82. [DOI] [PubMed] [Google Scholar]
- 13. Parthasarathy R, Cote GJ, Gagel RF. Hammerhead ribozyme‐mediated inactivation of mutant RET in medullary thyroid carcinoma. Cancer Res 1999; 59: 3911–4. [PubMed] [Google Scholar]
- 14. Messina M, Robinson BG. Technology insight: gene therapy and its potential role in the treatment of medullary thyroid carcinoma. Nat Clin Pract Endocrinol Metab 2007; 3: 290–301. [DOI] [PubMed] [Google Scholar]
- 15. Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF, Wu TC. Prospects of RNA interference therapy for cancer. Gene Ther 2006; 13: 464–77. [DOI] [PubMed] [Google Scholar]
- 16. Marsh DJ, Theodosopoulos G, Martin‐Schulte K et al. Genome‐wide copy number imbalances identified in familial and sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 2003; 88: 1866–72. [DOI] [PubMed] [Google Scholar]
- 17. Hattori Y, Yoshizawa T, Koga K, Maitani Y. NaCl induced high cationic hydroxyethylated cholesterol‐based nanoparticle‐mediated synthetic small interfering RNA transfer into prostate carcinoma PC‐3 cells. Biol Pharm Bull 2008; 31: 2294–301. [DOI] [PubMed] [Google Scholar]
- 18. Hattori Y, Fukushima M, Maitani Y. Non‐viral delivery of the connexin 43 gene with histone deacetylase inhibitor to human nasopharyngeal tumor cells enhances gene expression and inhibits in vivo tumor growth. Int J Oncol 2007; 30: 1427–39. [PubMed] [Google Scholar]
- 19. Yoshizawa T, Hattori Y, Hakoshima M, Koga K, Maitani Y. Folate‐linked lipid‐based nanoparticles for synthetic siRNA delivery in KB tumor xenografts. Eur J Pharm Biopharm 2008; 70: 718–25. [DOI] [PubMed] [Google Scholar]
- 20. Hattori Y, Koga K, Izumisawa T et al. The distribution of mRNA expression and protein after hydrodynamic injection of transgene in mice. Biol Pharm Bull 2009; 32: 755–9. [DOI] [PubMed] [Google Scholar]
- 21. Drosten M, Hilken G, Bockmann M et al. Role of MEN2A‐derived RET in maintenance and proliferation of medullary thyroid carcinoma. J Natl Cancer Inst 2004; 96: 1231–9. [DOI] [PubMed] [Google Scholar]
- 22. Kaczirek K, Schindl M, Weinhausel A et al. Cytotoxic activity of camptothecin and paclitaxel in newly established continuous human medullary thyroid carcinoma cell lines. J Clin Endocrinol Metab 2004; 89: 2397–401. [DOI] [PubMed] [Google Scholar]
- 23. Moley JF, Wells SA, Dilley WG, Tisell LE. Reoperation for recurrent or persistent medullary thyroid cancer. Surgery 1993; 114: 1090–5. [PubMed] [Google Scholar]
- 24. Tisell LE, Dilley WG, Wells SA Jr. Progression of postoperative residual medullary thyroid carcinoma as monitored by plasma calcitonin levels. Surgery 1996; 119: 34–9. [DOI] [PubMed] [Google Scholar]
- 25. de MH, Bertrand JR, Fusco A et al. siRNA nanoformulation against the ret/PTC1 junction oncogene is efficient in an in vivo model of papillary thyroid carcinoma. Nucleic Acids Res 2008; 36: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barone MV, Sepe L, Melillo RM et al. RET/PTC1 oncogene signaling in PC Cl 3 thyroid cells requires the small GTP‐binding protein Rho. Oncogene 2001; 20: 6973–82. [DOI] [PubMed] [Google Scholar]
- 27. Ji Y, Hayashi K, Amoh Y et al. The camptothecin derivative CPT‐11 inhibits angiogenesis in a dual‐color imageable orthotopic metastatic nude mouse model of human colon cancer. Anticancer Res 2007; 27: 713–8. [PubMed] [Google Scholar]
- 28. Koizumi F, Kanzawa F, Ueda Y et al. Synergistic interaction between the EGFR tyrosine kinase inhibitor gefitinib (“Iressa”) and the DNA topoisomerase I inhibitor CPT‐11 (irinotecan) in human colorectal cancer cells. Int J Cancer 2004; 108: 464–72. [DOI] [PubMed] [Google Scholar]
- 29. Shao RG, Cao CX, Shimizu T, O’Connor PM, Kohn KW, Pommier Y. Abrogation of an S‐phase checkpoint and potentiation of camptothecin cytotoxicity by 7‐hydroxystaurosporine (UCN‐01) in human cancer cell lines, possibly influenced by p53 function. Cancer Res 1997; 57: 4029–35. [PubMed] [Google Scholar]
- 30. Wang HG, Reed JC. Mechanisms of Bcl‐2 protein function. Histol Histopathol 1998; 13: 521–30. [DOI] [PubMed] [Google Scholar]
- 31. Hinze R, Gimm O, Taubert H et al. Regulation of proliferation and apoptosis in sporadic and hereditary medullary thyroid carcinomas and their putative precursor lesions. Virchows Arch 2000; 437: 256–63. [DOI] [PubMed] [Google Scholar]
- 32. Ludwig L, Kessler H, Wagner M et al. Nuclear factor‐kappaB is constitutively active in C‐cell carcinoma and required for RET‐induced transformation. Cancer Res 2001; 61: 4526–35. [PubMed] [Google Scholar]
- 33. Segouffin‐Cariou C, Billaud M. Transforming ability of MEN2A‐RET requires activation of the phosphatidylinositol 3‐kinase/AKT signaling pathway. J Biol Chem 2000; 275: 3568–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. In situ hybridization of a 35S‐labeled antisense or sense probe generated against RET mRNA. Microautoradiographs in bright fields of Fig. 5A were enlarged. Black grains indicate RET mRNA expression. Scale bar: 500 μm.
Fig. S2. In situ hybridization of a 35S‐labeled antisense or sense probe generated against RET mRNA. Microautoradiographs in bright fields of Fig. 5B were enlarged. Black grains indicate RET mRNA expression. Scale bar: 50 μm.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item