

Abstract
Osteosarcoma is a highly vascular and extremely destructive malignancy, and the survival of patients with osteosarcoma has not improved significantly in recent years. Antiangiogenic therapy currently holds great potential in conjunction with conventional treatment modalities for osteosarcoma. However, there are examples of gradual loss of response, and perhaps acquired resistance to antiangiogenic drugs. The acquired resistance of antiangiogenesis may be associated with a lot of hypoxia‐response genes. The human apurinic/apyrimidinic endonuclease (Ape1) protein, a bifunctional redox factor and apurinic/apyrimidinic (AP) endonuclease, plays a crucial role in protecting against cell death due to hypoxia. We therefore hypothesized that Ape1 may contribute to the resistance of antiangiogenic therapy. To investigate the effect of Ape1 on the sensitivity of human osteosarcoma cells to endostatin, we constructed an Ape1 small interfering RNA expression vector, pSilenceApe1. Transfection of human osteosarcoma 9901 and HOS cells with pSilenceApe1 resulted in a dose‐dependent loss of Ape1 protein. pSilenceApe1 also significantly suppressed the expression of vascular endothelial growth factor (VEGF) protein in the 9901 cells. Combined treatment with pSilenceApe1 and recombinant human endostatin (rhES) showed potent antiangiogenic effects in the transwell chamber invasion assay. Then, 20 nude mice bearing 9901 xenografts were divided into four groups: the phosphate‐buffered saline treatment control group; the rhES treatment group (1.5 mg/kg, daily); the pSilenceApe1 treatment group (20 µg, once every 3 days); and the combination of rhES and pSilenceApe1 treatment group. pSilenceApe1 significantly suppressed the expression of Ape1 and VEGF protein in the 9901 xenografts. The tumor‐inhibition rate of the pSilenceApe1, rhES, and combination of rhES and pSilenceApe1 treatment groups was 38.23, 35.29, and 62.18%, respectively. Furthermore, a significant decrease in microvessel density with an increase in apoptosis was observed following combined treatment with pSilenceApe1 and rhES, compared with control and either agent alone in 9901 xenografts. These results indicate that Ape1 small interfering RNA could enhance the sensitivity of osteosarcoma cells to endostatin. (Cancer Sci 2007; 98: 1993–2001)
Osteosarcoma is the most common malignant bone tumor in children and young people. The peak incidence of osteosarcoma usually occurs in the second and third decade of life, and approximately 1000 new cases a year are diagnosed in the USA. Over the past three decades, advances in treatment have been responsible for improved limb salvage, reduced metastases, and overall higher survival rates.( 1 ) Multiagent dose‐intensive chemotherapy regimens have resulted in long‐term disease‐free survival rates of ~60–76% in patients with localized disease.( 2 ) Osteosarcoma patients whose tumors respond poorly to chemotherapy are at a higher risk of relapse and adverse outcome due to the genetic instability and intrinsic concomitant tumor resistance.( 3 ) Angiogenesis inhibitors are new anticancer drugs considered potentially capable of circumventing or significantly delaying acquired drug resistance, because they target the normal – and hence genetically stable – host endothelial cells of a tumor's growing vasculature.( 4 , 5 ) However, there are examples of gradual loss of response, and perhaps acquired resistance to antiangiogenic drugs or treatment strategies, especially when the drugs are administered as monotherapies.( 6 ) These experiments demonstrate that the genetic background of a tumor cell, in this case the presence or absence of wild‐type p53, may be an important determinant of response to antiangiogenic therapy.( 7 ) The acquired resistance to antiangiogenic therapy may be associated with a lot of hypoxia‐response genes.( 8 , 9 ) The human apurinic/apyrimidinic endonuclease (Ape1), a bifunctional redox factor/apurinic/apyrimidinic (AP) endonuclease, plays a crucial role in protecting against cell death due to hypoxia.( 10 ) We therefore hypothesized that Ape1 may contribute to the resistance to antiangiogenic therapy, and the knockdown of Ape1 may enhance tumor sensitivity to antiangiogenic therapy.
In the present study, we constructed the specific Ape1 small interfering RNA (siRNA) stable expression vector pSilenceApe1 to knockdown Ape1 expression in the osteosarcoma cells, and to determine the knockdown cell response to antiangiogenic treatment in the nude mice experiment using growth‐curve observation, immunohistochemistry, and microvascular count. One strategy for tumor gene therapy is to inhibit expression of the DNA damage and repair gene Ape1, and to selectively enhance the sensitivity of antiangiogenesis in osteosarcoma cells with the purpose of eliminating tumor cells rapidly and effectively.
Materials and Methods
Cell lines and reagents. The human osteogenic sarcoma HOS cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). The human osteogenic sarcoma 9901 cells were a kind gift from Professor Qingyu Fan (Fourth Military Medical University, Xian, China). Both cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mmol/L l‐glutamine, 0.1 mmol/L non‐essential amino acids, 1.0 mmol/L pyruvate, 100 units/mL penicillin, and 100 µg/mL streptomycin. The human umbilical cord endothelial cell line ECV304 was grown in DMEM supplemented with 10% fetal bovine serum. The cells were maintained in a humidified incubator at 37°C in an atmosphere of 5% CO2 and 95% air. The endostatin was provided by Medejin (Yantai, China); it was a modified recombinant human endostatin (rhES, YH‐16; Endostar, Yantai, Shandong, China).( 11 ) Rat antimouse CD31 PECAM‐1 antibody was obtained from BD Pharmingen (San Diego, CA, USA) and secondary donkey antirat antibody was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Ape1 monoclonal antibody was purchased from Novos Biologicals (Littleton, CO, USA). Mouse antihuman β‐actin monoclonal antibody was purchased from Sigma (St Louis, MO, USA). Mouse antihuman vascular endothelial growth factor (VEGF) monoclonal antibody was purchased from Santa Cruz (Santa Cruz, CA, USA).
Construction of the Ape1 siRNA expression vector. The vector‐based short hairpin RNA expression system pSilencer 2.0‐U6 (Ambion, Austin, TX) was used to express short hairpin RNA endogenously in mammalian cells. We selected the target region according to Kelley Laboratory. (12) The two pairs of complementary oligonucleotides (5′‐GATCCGCTGGTACGACTGGAGTACCTTCAAGAGAGGTACTCCAGTCGTACCAGACTTTTTTGGAAA‐3′; 5′‐AGCTTTTCCAAAAAAGTCTGGTACGACTGGAGTACCTCTCTTGAAGGTACTCCAGTCGTACCAG CG‐3′) were annealed and inserted into pSilencer 2.0‐U6 via the BamHI and HindIII sites. The recombinants were identified with double digests and verified by sequencing.( 10 ) Transient transfection was carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer's instructions.
Immigration of endothelial cells measured using the transwell chamber invasion assay. The in vitro immigration properties of ECV304 cells were evaluated using the transwell chamber invasion assay (Costar, Cambridge, MA, USA). Each transwell filter (8‐µm pore size) was coated with 50 µg/filter of the solubilized tissue basement membrane Matrigel (Collaborative Research, Bedford, MA, USA). Culture medium (150 µL) that contained 7.5 × 104 ECV304 cells was applied to the upper compartments of the transwells. The lower compartment was filled with culture medium that contained 5 × 104 9901 cells treated with rhES, pSilenceApe1, or a combination of both. After 48 h incubation, the cells on the top of the filters were fixed and stained with hematoxylin–eosin. The determination of immigration of endothelial cells was counted with microscopy observations in five random areas at ×200 magnification.
Western blot analysis. For whole‐cell extracts, cells were washed with ice‐cold phosphate‐buffered saline and collected by scraping. Cell pellets were homogenized in extraction buffer (50 mM Tris‐HCl, 0.1% sodium dodecylsulfate [SDS], 150 mM NaCl, 100 µg/mL phenylmethylsulfonyl fluoride, 1 µg/mL aprotinin, 1% nonidet P‐40, and 0.5% sodium orthovanadate), then incubated at 4°C for 20 min and centrifuged for 20 min at 12000 g . The protein levels in the extracts were quantified using the Bio‐Rad DC protein assay. For western blotting, whole‐cell extracts (20 µg/lane) were resolved in 8–12% SDS‐polyacrylamide gels, and transferred onto nitrocellulose membrane (0.45 µm; Millipore, Bedford, MA, USA) in 25 mM Tris‐base, 190 mM glycine, and 20% methanol using a semidry blotter. Membranes were blocked with 8% fat‐free milk and 0.1% Tween 20 in Tris‐buffered saline. The Ape1 and VEGF monoclonal antibodies were diluted 1:1000 and the β‐actin monoclonal antibody was used at the 1:2000 dilution recommended by the supplier. The membranes were then incubated with a horseradish peroxidase‐conjugated secondary antibody (1:2000) (Pierce, Rockford, IL, USA). The proteins were detected using an enhanced chemiluminescence detection system (Pierce), and light emission was captured on X‐ray films (Kodak, Rochester, NY, USA).
Tumor model and treatment. All surgical procedures and care given to the animals were carried out in accordance with institutional and regulatory guidelines. Twenty BALB/C nude mice (4–5 weeks old, comprising five in each group) were inoculated subcutaneously on their oxters of right‐anterior limbs with 9901 cells (1 × 107 per 0.2 mL). These nude mice bearing 20 grafted tumors were divided into four groups, including a phosphate‐buffered saline‐treatment control, rhES treatment only (1.5 mg/kg, intratumoral injection, daily), pSilenceApe1 treatment only (20 µg, intratumoral injection, once every 3 days), and rhES combined with pSilenceApe1. After continuous monitoring for 14 days, all nude mice were killed, and each tumor was isolated and weighed. The maximum diameters (Dmax) and minimum diameters (Dmin) of each tumor were measured daily. The gross tumor volume of each tumor was calculated according to the formula: tumor size (mm3) = (Dmax × Dmin2)/2, and the growth curve and tumor inhibition rate was measured.
Immunohistochemical analysis of tumors for Ape1, VEGF, and CD31 expression. Sections from paraffin‐embedded tumors were incubated overnight with rat antimouse CD31 polyclonal antibody. The sections were then incubated with donkey antirat secondary antibody. Also, the sections were incubated with mouse antihuman Ape1 and VEGF monoclonal antibodies, then incubated with rabbit antimouse secondary antibody. Antigen–antibody complexes were visualized by incubation with 3,3′‐diaminobenzidine substrate and counterstained with diluted Harris hematoxylin. Ape1, VEGF, and CD31 staining were quantified using computer‐assisted image analysis with Image Pro Plus software (Media Cybernetics, Atlanta, GA, USA). The image analysis was done on four random fields per slide (magnification, ×100) from a total of five slides per group. For the quantification of microvessel density (MVD), 10 random fields at ×100 magnification were captured for each tumor, and CD31‐positive microvessels were quantified as relative vessel density.
In situ apoptosis detection by TUNEL staining. The formalin‐fixed and paraffin‐embedded 5 µm‐thick sections of all tumor samples were studied by terminal dUTP nick end labeling (TUNEL) staining using the Apoptag Kit (Intergen, Purchase, NY, USA). The extent of apoptosis was evaluated by counting the positive brown‐stained cells as well as the total number of cells in 10 arbitrarily selected fields in a blinded manner. The apoptotic index was calculated as the number of apoptotic cells per ×100 microscope field.
Statistical analysis. All error terms are expressed as the standard deviation of the mean. Significance levels for comparison of differences between groups in the in vitro experiments were analyzed using Student's t‐test. All reported P‐values were two‐sided. In the animal model tumor therapy studies, the treatment groups were compared with respect to tumor size and percentage of original tumor size over time. To test for significant differences in tumor size between treatment groups, a one‐way analysis of variance (ANOVA) test was conducted. The differences were considered significant when the P‐value was <0.05.
Results
Knockdown of Ape1 protein in osteosarcoma cells with pSilenceApe1. After evaluation and sequencing, the Ape1 siRNA expression vector pSilenceApe1 was constructed successfully( 13 ) and transfected into 9901 and HOS cells using Lipofectamine 2000. Immunoblot analysis demonstrated that Ape1 expression was significantly downregulated in both cell lines transfected with pSilenceApe1 compared to the Lipofectamine group. The inhibition rate of Ape1 gene expression reached 78–95% with 2.0 µg and 3.0 µg of pSilenceApe1 at 48 h (Fig. 1).
Figure 1.
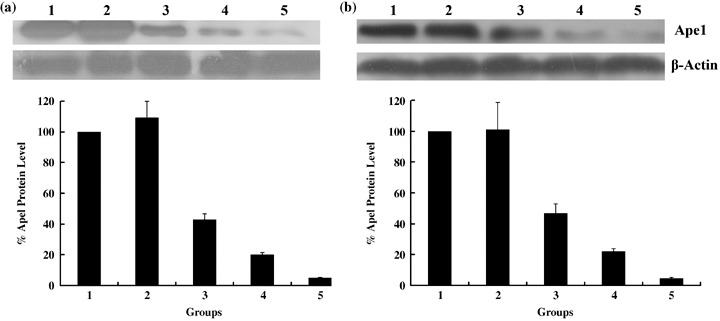
Knockdown of apurinic/apyrimidinic endonuclease (Ape1) protein using the constructed small interfering RNA vector pSilenceApe1. Western blotting of cell lysates for the protein expression of Ape1 in (a) 9901 and (b) HOS cells. Cells were treated with different doses of pSilenceApe1. Samples were collected at 48 h after pSilenceApe1 treatment, and western blot analysis was done with Ape1 monoclonal antibody and reprobed with β‐actin antibody as a loading control. B, normalized Ape1 protein levels after adjusting for loading; 1, normal 9901 cells; 2, Lipofectamine; 3, 1 µg pSilenceApe1; 4, 2 µg pSilenceApe1; and 5, 3 µg pSilenceApe1.
Inhibition of endothelial cell immigration following treatment with pSilenceApe1 and rhES. The number of endothelial cells that underwent immigration was much less in both the low‐dose rhES (350 ng/mL) and high‐dose rhES groups (700 ng/mL) compared with the normal‐cell control group (P < 0.01), much less in the latter than the former. Also, the inhibition of endothelial cell immigration was found in groups treated with 2 or 3 µg pSilenceApe1, which was much higher in the latter than the former. The results indicated that both rhES and pSilenceApe1 could inhibit the immigration of endothelial cells in a dose‐dependent manner. When we added 2 µg pSilenceApe1 with low‐dose rhES (350 ng/mL), the level of endothelial cell immigration was much less in this combined group than the single‐reagent treatment group (P < 0.01) (Table 1; Fig. 2).
Table 1.
Inhibition of endothelial cell immigration following treatment with pSilenceApe1 and recombinant human endostatin (rhES)
| Group | No. immigrated endothelial cells |
|---|---|
| Normal‐cell control | 67.33 ± 2.08 |
| Lipofectamine control | 62.33 ± 2.52 |
| rhES | |
| Low dose (350 ng/mL) | 30.67 ± 1.53* |
| High dose (700 ng/mL) | 12.67 ± 0.57* |
| pSilenceApe1 | |
| 2 µg | 25.67 ± 2.52*,** |
| 3 µg | 8.67 ± 1.15*,** |
| 350 ng/mL rhES + 2 µg pSilenceApe1 | 7.33 ± 0.57*,**,*** |
Versus normal‐cell control, *P < 0.01; versus Lipofactamine control, **P < 0.01; versus rhES low‐dose and pSilenceApe1 2 µg groups: ***P < 0.01.
Figure 2.
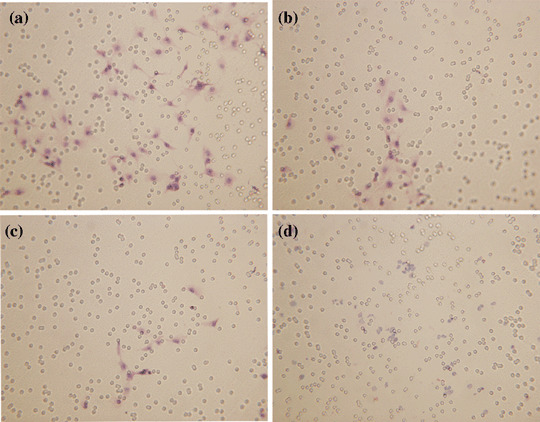
In vitro immigration of endothelial cells was detected using a transwell chamber model (hematoxylin–eosin, ×200). A significant decrease in the level of endothelial cell immigration was observed following combined treatment with pSilenceApe1 and recombinant human endostatin (rhES) compared with treatment with pSilenceApe1 and rhES alone in 9901 cells. (a) Lipofectamine; (b) 350 ng/mL rhES; (c) 2 µg pSilenceApe1; and (d) 350 ng/mL rhES + 2 µg pSilenceApe1.
Combined treatment with pSilenceApe1 and rhES inhibits tumor growth in human osteosarcoma xenografts. To investigate the effects of the combination of pSilenceApe1 and rhES treatment on tumor growth in vivo, we injected 9901 cells subcutaneously into the athymic nude mice. Before treatment, the mean tumor sizes in the groups of 20 mice at baseline were not significantly different between treatment groups (P > 0.05), and the within‐treatment variances were not significantly different (P > 0.05). The baseline mean and standard deviation for tumor sizes was 26 ± 4.3 mm3. We initiated in vivo tumor therapy on day 1, which corresponded to 4 days after tumor‐cell injection. We noted an inhibition of tumor growth in groups of mice treated with pSilenceApe1 plus rhES, pSilenceApe1 and rhES alone versus the pSilenceControl‐injected control group (Fig. 3). Comparisons of mean tumor volumes of the pSilenceApe1 in combination with rhES treatment group versus pSilenceApe1 and rhES alone showed significant differences (P < 0.05). This difference in tumor size was evident at day 14 and persisted for the duration of the experiment (Fig. 3). On day 14, the average tumor weight was 0.9 ± 0.03 g in mice treated with pSilenceApe1 in combination with rhES, 1.47 ± 0.04 g in mice injected with pSilenceApe1 alone, 1.54 ± 0.04 g in mice treated with rhES, and 2.38 ± 0.03 g in control mice (P < 0.01). The tumor‐inhibition rates of the pSilenceApe1, rhES, and combined groups were 38.23, 35.29, and 62.18%, respectively.
Figure 3.
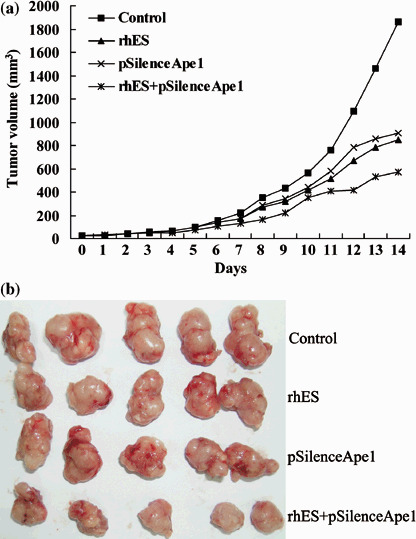
In vivo evaluation of tumor growth in 9901 xenografts. Combined treatment with pSilenceApe1 and recombinant human endostatin (rhES) produced significant tumor regression compared with either agent alone. (a) The effect of combined treatment with pSilenceApe1 and rhES was evaluated by measuring the volume of xenografts. (b) Photo of 9901 xenografts.
In vivo knockdown of Ape1 protein by pSilenceApe1 in human 9901 xenografts. The in vivo knockdown of Ape1 protein by pSilenceApe1 in 9901 cells was evaluated. We investigated the expression of Ape1 protein in 9901 xenografts by immunohistochemistry. In the control group, the Ape1 protein was predominantly localized to the nucleus of tumor cells (Fig. 4a). Ape1 expression in the rhES‐treated group was not changed compared with the control group (Fig. 4b). However, the tumors in both the pSilenceApe1 and combined groups showed significant decreases in Ape1 expression of approximately 78 and 74%, respectively, compared with the control group (P < 0.01) (Fig. 4c–e).
Figure 4.
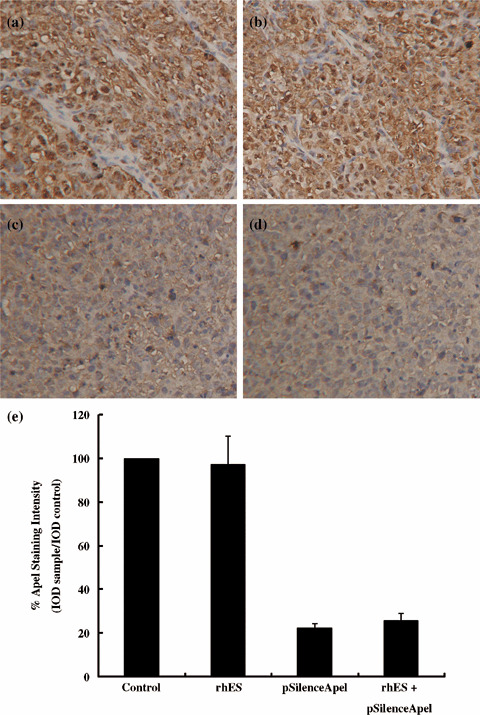
In vivo knockdown of apurinic/apyrimidinic endonuclease (Ape1) protein by pSilenceApe1 in 9901 xenografts. The expression of Ape1 protein in xenografts was evaluated by immunohistochemistry with ×200 magnification. Ape1 protein was predominantly localized in the nucleus of tumor cells. A significant decrease in Ape1 protein was observed following treatment with pSilenceApe1 or combined treatment with pSilenceApe1 and recombinant human endostatin (rhES), compared with control or treatment with rhES alone in 9901 xenografts. (a) Control; (b) rhES; (c) pSilenceApe1; (d) rhES + pSilenceApe1; and (e) quantitative analysis of Ape1 staining.
Antiangiogenesis in human 9901 xenografts following treatment with pSilenceApe1 and rhES. The results of the immunohistochemical detection of CD31 (Fig. 5a) showed that there were several blood vessels with intact walls and high CD31 expression in the control group, and its MVD was 27.66 ± 3.00. The MVD was 16.10 ± 0.91 and 15.04 ± 0.47 in the rhES‐ and pSilenceApe1‐treated groups, respectively. In the combined group, blood vessels were rare and the frames of vessel walls collapsed. CD31 expression was low and the MVD was only 8.74 ± 0.69, which was three times lower than that of the control group, as well as nearly two times lower than that of the pSilenceApe1 alone and rhES alone groups with significant difference (P < 0.01). Furthermore, the image analysis of mean CD31 staining intensity showed a similar change in each treated group compared with the control group (Fig. 5b).
Figure 5.
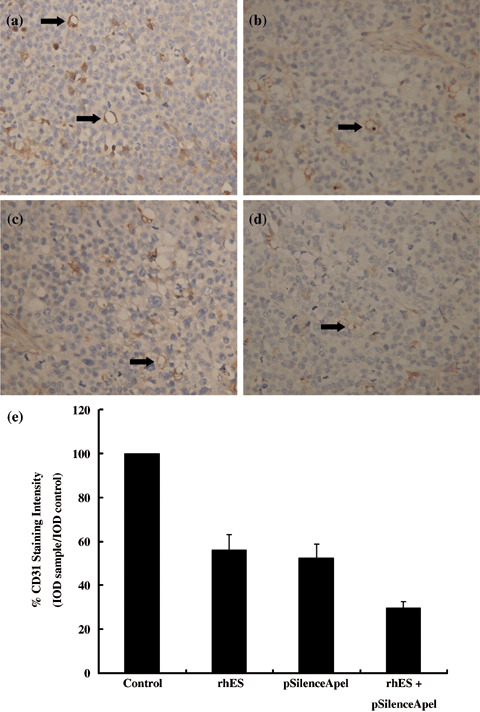
Effect of combined treatment with pSilenceApe1 and recombinant human endostatin (rhES) on angiogenesis in vivo. Paraffin‐embedded sections were made and microvessels were stained with antimouse CD31 monocolonal antibodies by immunohistochemistry. Microvessel density (MVD) was determined by microscopic counting of the CD31‐stained microvessels at ×200 magnification. Representative vessels are shown (arrow). A significant decrease in MVD was observed following combined treatment with pSilenceApe1 and rhES compared with control or either agent alone in 9901 xenografts. (a) Control; (b) rhES; (c) pSilenceApe1; (d) rhES + pSilenceApe1; and (e) quantitative analysis of CD31 staining.
Histological observation and detection of apoptosis. The xenograft tumor showed sarcoma‐like pattern with diffuse tumor cells. There was much more necrosis in all of the groups, including pSilenceApe1 treatment alone, rhES treatment alone, and the combined‐treatment group, compared with the control group (P < 0.01). The semiqualitative results of necrosis in xenografts is shown in Table 2. Moreover, the apoptosis index was calculated using an in situ apoptosis detection assay (Fig. 6). A much higher apoptosis index was observed in the combined‐treatment, pSilenceApe1 alone, and rhES alone groups than in the control group (P < 0.01).
Table 2.
Percentage of necrosis and apoptosis index in osteosarcoma xenografts
| Group | n | Necrosis (%) | Apoptosis index (%) |
|---|---|---|---|
| Normal‐cell control | 5 | 2.10 ± 0.17 | 2.52 ± 0.24 |
| pSilenceApe1 | 5 | 9.04 ± 0.062* | 7.96 ± 0.58* |
| rhES | 5 | 9.14 ± 1.03* | 8.10 ± 0.34* |
| rhES + pSilenceApe1 | 5 | 21.98 ± 1.82*,** | 18.66 ± 0.69*,** |
Versus normal‐cell control, *P < 0.01; versus pSilenceApe1 and rhES groups, **P < 0.01. rhES, recombinant human endostatin.
Figure 6.
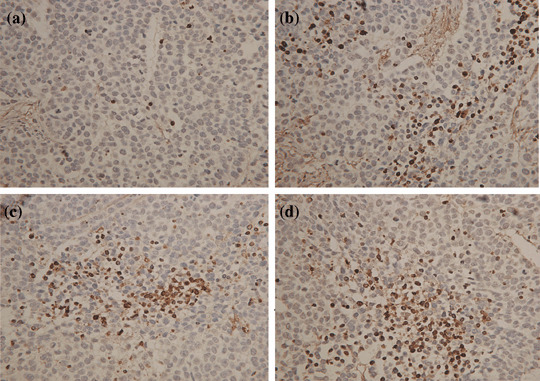
Combined treatment with pSilenceApe1 and recombinant human endostatin (rhES) induces apoptosis in vivo. Paraffin‐embedded sections were made and apoptosis was determined by terminal dUTP nick end labeling (TUNEL) staining at ×100 magnification. A significant increase in apoptotic cells was observed following combined treatment with pSilenceApe1 and rhES compared with control and either agent alone in 9901 xenografts. (a) Control; (b) rhES; (c) pSilenceApe1; and (d) rhES + pSilenceApe1.
Suppression of VEGF protein expression by pSilenceApe1 in vitro and in vivo. In vitro, western blotting analysis demonstrated that VEGF protein expression was significantly downregulated in 9901 cells transfected with pSilenceApe1 compared to the lipofectamine group, and the inhibition rate of VEGF expression reached 63–93% with 2.0 µg and 3.0 µg of pSilenceApe1 after 48 h (Fig. 7). In vivo, VEGF protein expression in the rhES‐treated group was not changed compared with the control group. However, the tumors in both the pSilenceApe1 and combined‐treatment groups showed significant decreases in VEGF expression, approximately 61 and 63%, respectively, compared with the control group (P < 0.01) (Fig. 8).
Figure 7.
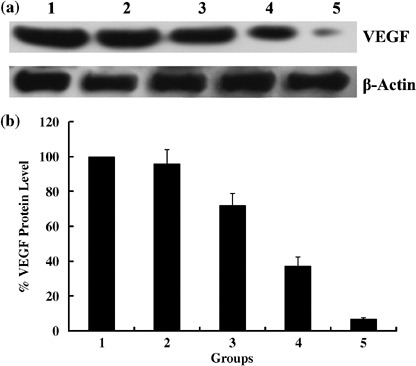
Suppression of the vascular endothelial growth factor (VEGF) protein expression by pSilenceApe1 in vitro. (a) Western blotting of cell lysates for the protein expression of VEGF. 9901 Cells treated with different dose of pSilenceApe1. Sample were collected at 48 h after pSilenceApe1 treatment and Western blot analysis was done with VEGF monoclonal antibody and reprobed with β‐actin antibody as a loading control. (b) Normalized VEGF protein levels after adjusting for loading. 1, normal 9901 cells; 2, lipofectamine; 3, 1 µg pSilenceApe1; 4, 2 µg pSilenceApe1; 5, 3 µg pSilenceApe1.
Figure 8.
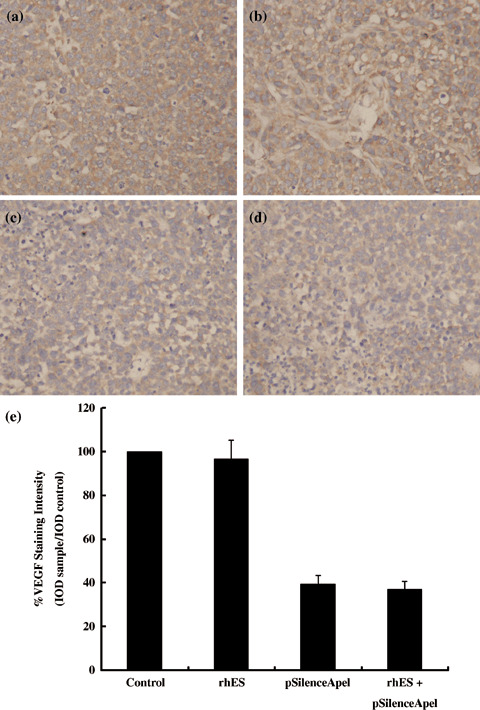
Suppression of vascular endothelial growth factor (VEGF) protein expression by pSilenceApe1 in vivo. The expression of VEGF protein in 9901 xenografts was evaluated by immunohistochemistry with 200× magnification. A significant decrease in VEGF protein was observed following treatment with pSilenceApe1 or combined treatment with pSilenceApe1 and recombinant human endostatin (rhES), compared with control or treatment with rhES alone in 9901 xenografts. (a) Control; (b) rhES; (c) pSilenceApe1; (d) rhES + pSilenceApe1; and (e) quantitative analysis of VEGF staining.
Discussion
Ape1 is a ubiquitous bifunctional protein. In addition to its DNA repair functions, Ape1 is also a multifunctional protein that participates in other crucial cellular processes, including the response to oxidative stress, regulation of transcription factors, cell cycle control, and apoptosis.( 10 ) As a reduction–oxidation factor, Ape1 can reduce a conserved cysteine residue in members of the Jun/Fos and related activating transcription factor/cAMP‐responsive element binding protein families of proteins, facilitating the formation of heterodimers and homodimers that bind to transcriptional regulatory elements containing activator protein 1 (AP‐1).( 14 ) In addition, Ape1 stimulates the DNA binding of other transcription factors such as nuclear factor κB (NF‐κB). Ape1 has also been implicated in regulating the transactivation and pro‐apoptotic activity of p53.( 15 ) Overexpression of Ape1 has been shown to enhance the transcriptional activity of the key transcription factor governing cellular adaptation to hypoxia, hypoxia‐inducible factor (HIF)‐1.( 16 ) Ape1 seems to promote redox‐dependent interactions between HIF‐1 and transcriptional coactivators, including p300 and cAMP response element binding protein (CREB), which form a multiprotein complex binding to adjacent and sometimes non‐adjacent sequences in the hypoxic response element of hypoxia‐inducible genes.( 17 ) Ape1 is a critical component of the hypoxia‐inducible transcriptional complex formed on the hypoxic response element of VEGF, which modulates VEGF expression.( 18 ) As VEGF is known to be a crucial factor for angiogenesis and the above transcription factors, such as Ap‐1, NF‐κB, p53, and HIF‐1, play an important role in promoting angiogenesis, it is reasonable to postulate that Ape1 is associated with angiogenesis, although there was no direct evidence of this before the present study.
The tumor hypoxic environment results in DNA‐damaging free radicals and DNA repair capacity. In both in vivo and in vitro studies, oxidative agents, including ischemia, hypoxia, and reactive oxygen species (ROS), induce Ape1 expression, which is characterized by a transient increase in Ape1 protein and mRNA.( 19 , 20 ) The antiangiogenic therapy can aggravate hypoxia in the tumor, leading to serious DNA damage. Cells repair DNA damage via four main mechanisms: direct reversal; base excision repair; nucleotide excision repair; and mismatch repair. The damage induced by oxidative damage is effectively repaired by base excision repair, and Ape1 is one of the main enzymes within the base excision repair pathway. Ape1 is abundant in human cells and accounts for nearly all of the abasic site cleavage activity observed in cellular extracts.( 21 ) Ape1 possesses a strong Mg2+‐dependent endonuclease activity that hydrolyzes the phosphodiester bond 5′ of potentially cytotoxic abasic sites, leaving a 3′‐OH and 5′‐deoxyribose phosphate.( 22 ) In addition, Ape1 has a 3′‐phosphodiesterase activity that excises deoxyribose fragments and phosphate groups at the 3′ terminus of strand breaks caused by ionizing radiation, yielding a 3′‐OH substrate for polymerase β repair synthesis.( 23 ) Ape1 has recently been shown to possess a 3′ mismatch exonuclease activity as well.( 24 , 25 )
Combining the redox and DNA repair functions, Ape1 may play an important role in angiogenesis, and may contribute to the molecular mechanism of resistance to antiangiogenic treatment. Our previous study showed that Ape1 was overexpressed in human osteosarcoma, and its knockdown made the tumor cells sensitive to alkylators and ionizing irradiation with synthesized Ape1 siRNA.( 12 ) In the present study, we successfully constructed the Ape1 siRNA vector pSilenceApe1, which could inhibit Ape1 expression from 80 to 95% at doses of 2–3 µg. First, pSilenceApe1 was shown to inhibit endothelial cell immigration in vitro both alone and synergistically with rhES by a transwell model. Furthermore, with the nude mice tumor experiment, the tumor‐inhibition rate of combined treatment with pSilenceApe1 and rhES reached 62.18%, which was significantly higher than the rhES and pSilenceApe1 groups alone, which had lower MVD and much more necrosis, as well as cell apoptosis. The results confirmed that pSilenceApe1 had a synergistic role with rhES in antiangiogenic therapy in vivo. There have been a small number of previous studies using DNA antisense methodology that implicates Ape1 in cellular resistance to a variety of agents. For example, studies using antisense Ape1 in human HeLa, rat glioma, or human lung carcinoma cells indicated that cells could be made hypersensitive to alkylating and oxidative agents, as well as ionizing radiation but not ultraviolet radiation.( 26 , 27 ) Although much more information has been published implicating Ape1 in tumor‐cell growth, proliferation, and drug resistance,( 10 ) little has been done to determine its role in angiogenesis. Our results suggest that Ape1 plays an important role in tumor‐cell resistance to antiangiogenic therapy.
Osteosarcoma is a hypervascular tumor. However, the role of angiogenesis in osteosarcoma remains a matter of debate. Our previous report showed evidence for decreased overall survival in osteosarcoma patients with high MVD.( 12 ) Kreuter et al. reported that high MVD correlated with good response to neoadjuvant chemotherapy and favorable prognosis in systemically treated osteosarcoma.( 28 ) The apparent contrary findings might in part be explained by different treatments, study populations, and size. However, antiangiogenic therapy for malignancy has been the focus of extensive laboratory and clinical research in recent times. Moreover, the use of antiangiogenic agents in combination with chemotherapy and ionizing irradiation for solid tumors has been supported in numerous in vivo animal studies.( 29 , 30 ) The role of angiogenesis inhibitors as adjuvants to chemotherapy and irradiation in osteosarcoma has not been investigated. Our previous study showed that synthesized Ape1 siRNA suppressed Ape1 endonuclease activity and made the tumor cell sensitive to alkylators and irradiation.( 12 ) The data presented in the present paper demonstrated that pSilenceApe1 suppressed the expression of VEGF protein and enhanced the sensitivity of tumor cells to endostatin in vitro and in vivo. Downregulation of VEGF protein might contribute to augmented antiangiogenic activity of endostatin. Therefore, targeting of Ape1 holds great potential in conjunction with conventional treatment modalities for osteosarcoma.
Endostatin is a 20‐kDa internal fragment of the carboxyterminus of collagen XVIII. It was discovered by Michael O’Reilly of the Folkman laboratory (Laboratory of Surgical Research, Children's Hospital, Boston, MA, USA), based on Folkman's hypothesis of a mechanism to explain the phenomenon that surgical removal of certain tumors leads to rapid growth of remote metastases.( 31 ) Endostatin inhibits 65 different tumor types and modifies 12% of the human genome to downregulate pathological angiogenesis without side‐effects.( 32 ) The first angiogenesis inhibitor, recombinant human endostatin (rhES, YH‐16, Endostar) has now been approved by China, and showed antitumor activity with high clinical benefit rate. It was also well tolerated in pretreated advanced non‐small cell lung cancer patients.( 11 ) However, mutant tumor cells and some factors may, over time, produce redundant angiogenic factors. Therefore, Dr Folkman emphasized that a combination of angiogenesis inhibitors or broad‐spectrum angiogenesis inhibitors will be needed for long‐term use in cancer.( 32 )
The data presented in this paper suggest that Ape1 may be an important therapeutic target in tumor angiogenesis. Comparing antisense and ribozyme therapy, siRNA was likely to be more effective in knocking down gene expression. siRNA are short oligonucleotides of 21–23 nucleotides in length that can be used in vitro to produce sequence‐specific gene silencing of mammalian cells.( 33 , 34 ) It has been shown that siRNA can be used effectively in vivo to suppress gene expression in adult mice.( 35 ) The present study implies that Ape1 may be a crucial factor in the regulation of angiogeneis and supports the notion that Ape1 siRNA could be an effective therapeutic agent to sensitize osteosarcoma cells to antiangiogenic treatment.
Acknowledgments
This study was supported by Grants from the National Natural Science Foundation of China (nos 30340044, 30472004, and 30670628).
References
- 1. Ferguson WS, Goorin AM. Current treatment of osteosarcoma. Cancer Invest 2001; 19: 292–315. [DOI] [PubMed] [Google Scholar]
- 2. Ferrari S, Bertoni F, Mercuri M et al . Predictive factors of disease‐free survival for non‐metastatic osteosarcoma of the extremity: an analysis of 300 patients treated at the Rizzoli Institute. Ann Oncol 2001; 12: 1145–50. [DOI] [PubMed] [Google Scholar]
- 3. Ragland BD, Bell WC, Lopez RR et al . Cytogenetics and molecular biology of osteosarcoma. Laboratory Invest 2002; 82: 365–73. [DOI] [PubMed] [Google Scholar]
- 4. Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti‐cancer therapeutic agents. Bioessays 1991; 13: 31–6. [DOI] [PubMed] [Google Scholar]
- 5. Folkman J, Hahnfeldt P, Hlatky L. Cancer: looking outside the genome. Nat Rev Mol Cell Biol 2000; 1: 76–9. [DOI] [PubMed] [Google Scholar]
- 6. Kerbel RS, Yu J, Tran J, Man S et al . Possible mechanisms of acquired resistance to anti‐angiogenic drugs: implications for the use of combination therapy approaches. Cancer Metastasis Rev 2001; 20: 79–86. [DOI] [PubMed] [Google Scholar]
- 7. Yu JL, Rak JW, Coomber BL et al . Effect of p53 status on tumor response to antiangiogenic therapy. Science 2002; 295: 1526–8. [DOI] [PubMed] [Google Scholar]
- 8. Longo R, Sarmiento R, Fanelli M et al . Anti‐angiogenic therapy: rationale, challenges and clinical studies. Angiogenesis 2002; 5: 237–56. [DOI] [PubMed] [Google Scholar]
- 9. Carmeliet P, Dor Y, Herbert JM et al . Role of HIF‐1α in hypoxic‐mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998; 394: 485–90. [DOI] [PubMed] [Google Scholar]
- 10. Evans AR, Limp‐Foster M, Kelley MR. Going APE over ref‐1. Mutat Res 2000; 461: 83–108. [DOI] [PubMed] [Google Scholar]
- 11. Yang L, Wang JW, Sun Y et al . Randomized phase II trial on escalated doses of Rh‐endostatin (YH‐16) for advanced non‐small cell lung cancer. Zhonghua Zhong Liu Za Zhi 2006; 28: 138–41. [PubMed] [Google Scholar]
- 12. Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther 2004; 3: 679–86. [PubMed] [Google Scholar]
- 13. Wang D, Zhong ZY, Li ZP et al . Construction of Ape1 siRNA expression vector and its inhibition to Ape1 in osteosarcoma cells. Acta Academiae Med Militaris Tertiae 2006; 28: 97–100. [Google Scholar]
- 14. Xanthoudakis S, Miao G, Wang F et al . Redox activation of Fos‐Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J 1992; 11: 3323–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jayaraman L, Murthy KG, Zhu C et al . Identification of redox/repair protein Ref‐1 as a potent activator of p53. Genes Dev 1997; 11: 558–70. [DOI] [PubMed] [Google Scholar]
- 16. Huang LE, Arany Z, Livingston DM et al . Activation of hypoxia inducible transcription factor depends primarily upon redox‐sensitive stabilization of its alpha subunit. J Biol Chem 1996; 271: 32 253–9. [DOI] [PubMed] [Google Scholar]
- 17. Carrero P, Okamoto K, Coumailleau P et al . Redox‐regulated recruitment of the transcriptional coactivators CREB‐binding protein and SRC‐1 to hypoxia‐inducible factor 1α. Mol Cell Biol 2000; 20: 402–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ziel KA, Campbell CC, Wilson GL et al . Ref‐1/Ape is critical for formation of the hypoxia‐inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. FASEB J 2004; 18: 986–8. [DOI] [PubMed] [Google Scholar]
- 19. Yao KS, Xanthoudakis S, Curran T et al . Activation of AP‐1 and of a nuclear redox factor, Ref‐1, in the response of HT29 colon cancer cells to hypoxia. Mol Cell Biol 1994; 14: 5997–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gillardon F, Bottiger B, Hossmann KA. Expression of nuclear redox factor Ref‐1 in the rat hippocampus following global ischemia induced by cardiac arrest. Brain Res Mol Brain Res 1997; 52: 194–200. [DOI] [PubMed] [Google Scholar]
- 21. Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3′‐blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res 1991; 19: 5907–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Ann Rev Biochem 1994; 63: 915–48. [DOI] [PubMed] [Google Scholar]
- 23. Izumi T, Hazra TK, Boldogh I et al . Requirement for human AP endonuclease 1 for repair of 3′‐blocking damage at DNA single‐strand breaks induced by reactive oxygen species. Carcinogenesis 2000; 21: 1329–34. [PubMed] [Google Scholar]
- 24. Chou KM, Cheng YC. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3′ mispaired DNA. Nature 2002; 415: 655–9. [DOI] [PubMed] [Google Scholar]
- 25. Chou KM, Kukhanova M, Cheng YC. A novel action of human apurinic/apyrimidinic endonuclease: excision of 1‐configuration deoxyribonucleoside analogs from the 3′ termini of DNA. J Biol Chem 2000; 275: 31 009–15. [DOI] [PubMed] [Google Scholar]
- 26. Walker LJ, Craig RB, Harris AL et al . A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res 1994; 22: 4884–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ono Y, Furuta T, Ohmoto T et al . Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutat Res 1994; 315: 55–63. [DOI] [PubMed] [Google Scholar]
- 28. Kreuter M, Bieker R, Bielack SS et al . Prognostic relevance of increased angiogenesis in osteosarcoma. Clin Cancer Res 2004; 10: 8531–7. [DOI] [PubMed] [Google Scholar]
- 29. Klement G, Baruchel S, Rak J et al . Continuous low‐dose therapy with vinblastine and VEGF receptor‐2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 2000; 105: R15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wachsberger P, Burd R, Dicker AP. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin Cancer Res 2003; 9: 1957–71. [PubMed] [Google Scholar]
- 31. O’Reilly MS, Boehm T, Shing Y et al . Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997; 88: 277–85. [DOI] [PubMed] [Google Scholar]
- 32. Folkman J. Antiangiogenesis in cancer therapy – endostatin and its mechanisms of action. Exp Cell Res 2006; 312: 594–607. [DOI] [PubMed] [Google Scholar]
- 33. Shuey DJ, McCallus DE, Giordano T. RNAi: gene‐silencing in therapeutic intervention. Drug Discov Today 2002; 7: 1040–6. [DOI] [PubMed] [Google Scholar]
- 34. Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nat Rev Mol Cell Biol 2003; 4: 457–67. [DOI] [PubMed] [Google Scholar]
- 35. Takei Y, Kadomatsu K, Yuzawa Y et al . A small interfering RNA targeting vascular endothelial growth factor as cancer therapeutics. Cancer Res 2004; 64: 3365–70. [DOI] [PubMed] [Google Scholar]