

Abstract
Adult T‐cell leukemia (ATL) is an aggressive malignancy of activated CD4+ T cells associated with human T‐cell leukemia virus type I (HTLV‐I) infection. No conventional chemotherapy regimen has appeared successful in patients with ATL, thus establishing effective therapy is urgently required. In some cases, ATL tumor cells express CD30 on the cell surface, therefore, a therapy with mAb against CD30 would be beneficial. To investigate the effect of CD30‐mediated therapy on ATL, we assessed SGN‐30, a chimeric anti‐CD30 mAb, and SGN‐35, a monomethyl auristatin E‐conjugated anti‐CD30 mAb, in vitro and in vivo. Three HTLV‐I‐infected cell lines were co‐cultured with SGN‐30 or SGN‐35, and the growth‐inhibitory effects on the HTLV‐I‐infected cells were evaluated using an in vitro cell proliferation assay and cell cycle analysis. SGN‐30 and SGN‐35 showed growth‐inhibitory activity against the HTLV‐I‐infected cell lines by apoptosis and/or cell growth arrest in vitro. To further investigate the effects of SGN‐30 and SGN‐35 on HTLV‐I‐infected cells in vivo, we used NOD/SCID mice subcutaneously engrafted with HTLV‐I‐infected cells. Both mAbs significantly inhibited the growth of HTLV‐I‐infected cell tumors in the NOD/SCID murine xenograft models. These data suggest that CD30‐mediated therapy with SGN‐30 or SGN‐35 would be useful for patients with ATL. (Cancer Sci 2009)
Adult T‐cell leukemia is an aggressive malignancy of activated CD4+ T cells associated with HTLV‐I infection.( 1 ) Although the mechanism of ATL tumorigenesis by HTLV‐I has been intensively studied during the past 25 years, the prognosis of ATL patients remains poor due to the innate resistance of the disease to conventional chemotherapy regimens. Therefore, establishing convincing therapy to prolong life in patients with ATL is urgently required.
Receptors expressed by activated lymphocytes but not by other normal tissues would be attractive candidates for therapeutic targeting to maximize targeting specificity and minimize toxicity of experimental reagents. As ATL cell lines highly express CD2, CD25, and CD52 on the cell surface, humanized mAbs MEDI‐507, HAT, and Campath‐1H directed toward CD2, CD25, and CD52, respectively, have been evaluated for their therapeutic efficacy on ATL cells in vivo.( 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ) ATL cells also express the CC chemokine receptor 4 on the cell surface, and chimeric anti‐CC chemokine receptor 4 mAb, KM2760, the Fc region of which is defucosylated, was reported to augment antitumor activity on ATL cells in a SCID mice model.( 14 ) Thus, it is believed that anti‐ATL chemotherapy with agonistic mAbs would be a promising strategy. As well as these cell surface antigens, ATL tumor cells have in some cases been shown to express CD30.( 15 , 16 , 17 , 18 , 19 , 20 , 21 ) CD30 is a 120‐kDa type I cell surface glycoprotein belonging to the TNFR superfamily, including TNFR1, TNFR2, CD40, Fas, or tumor necrosis factor‐related apoptosis‐inducing ligand receptor.( 22 ) CD30 is expressed on a variety of malignant cells of hematopoietic origin, but is rarely found on non‐malignant cells, making it a good target for cancer therapy.( 23 ) Although CD30 does not contain a death domain in the cytoplasmic tail, its stimulation with CD30L leads to activate cellular pathways for apoptosis or cell growth arrest.( 24 ) Since an agonistic mAb to CD30 mimics CD30L depending on the epitope, several CD30 mAbs have been evaluated for therapy against hematopoietic malignancies, including ALCL and HL.( 25 , 26 , 27 , 28 , 29 ) We have reported an mAb against CD30, AC10, which was originally produced by immunizing mice with a CD30‐positive large granular lymphoma cell line.( 30 ) The variable regions from AC10 were subsequently cloned into an expression vector containing the human γ1 heavy chain and κ light chain constant regions to humanize the Ab.( 31 ) The resulting chimeric Ab, SGN‐30, retained the binding and in vitro growth‐inhibitory activities of the parental Ab, and was able to inhibit the growth of ALCL and HL cell lines in vitro and in vivo.( 31 , 32 , 33 , 34 , 35 ) To enhance the potency of SGN‐30, we also generated an Ab‐drug conjugate termed SGN‐35.( 36 , 37 , 38 , 39 , 40 , 41 ) SGN‐35 consists of the AC10 chemically conjugated to a cytotoxic drug MMAE, made up synthetic antimitotic agents related to marine natural product dolastin 10 which act by inhibiting polymerization of tubulin and thus prevent formation of the mitotic apparatus followed by apoptosis.( 36 ) The resulting Ab, SGN‐35, showed more potent antitumor activity against ALCL cell lines in vitro.( 36 , 37 , 38 ) Anti‐CD20 mAb conjugated with MMAE effectively and specifically killed CD20+ B‐lymphoma cell lines.( 42 ) In addition, anti‐CD70 ADC, consisting of auristatin phenylalanine phenylenediamine or monomethyl auristatin phenylalanine, two derivatives of the antitubulin agent auristatin, mediated potent antigen‐dependent cytotoxicity in CD70‐expressing cells.( 43 ) Therefore, the use of an auristatin derivative would be a promising strategy for cancer therapy.
In this study we investigated the effects of SGN‐30 and SGN‐35 on HTLV‐I‐infected cells in vitro and in vivo, as a CD30‐mediated therapeutic approach for ATL has never been reported. We found that both mAbs significantly inhibited the growth of HTLV‐I‐infected cells in vitro and in vivo, suggesting that CD30‐mediated therapy with SGN‐30 or SGN‐35 would be useful for patients with ATL.
Materials and Methods
Binding of SGN‐30 to CD30+ cell lines. HTLV‐I‐infected cell lines, HUT‐102,( 44 ) MT‐2,( 45 ) S1T,( 46 ) and an ALCL cell line Karpas 299( 47 ) were grown in RPMI‐1640 supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL). Anti‐CD30 mAbs, SGN‐30 and SGN‐35, were prepared and provided by Seattle Genetics (Seattle, WA, USA). Cells were incubated with biotin‐conjugated SGN‐30 or isotype‐matched control IgG for 30 min at 4°C followed by incubation with phycoerythrin‐conjugated streptavidin for 30 min at 4°C. The binding of SGN‐30 was evaluated by using a FACSCalibur flow cytometer (BD Biosciences, San Diego, CA, USA). Background‐corrected MFI was determined for each cell type.
In vitro growth inhibition assay. Cells were cultured in the absence or presence of various concentrations of SGN‐30 with 10‐fold excess of goat antihuman IgG, or SGN‐35, for 48 h at 37°C. The cells were further incubated with Cell Proliferation Reagent WST‐1 (Roche, Mannheim, Germany) for 4 h at 37°C. The absorbance of the samples was measured at 450 and 550 nm (reference wavelength) with a microplate reader.
Cell cycle analysis. Cells were cultured in the absence or presence of various concentrations of SGN‐30 with 10‐fold excess of goat antihuman IgG, or SGN‐35, for 48 h at 37°C. The cell cycle was analyzed with Bromodeoxyuridine Flow Kit (BD Biosciences) according to the manufacturer’s instructions. The population of cells in each cell cycle phase was evaluated by a FACSCalibur flow cytometer.
Xenograft model of ATL. Non‐obese diabetic/severe combined immunodeficient mice were purchased from Charles River Laboratories (Kanagawa, Japan). Mice were engrafted s.c. with HUT‐102( 48 , 49 , 50 ) or Karpas 299 cells.( 36 , 37 , 38 ) One day after injection of cells, mice received i.p. SGN‐30 (2 mg/kg) or SGN‐35 (1 mg/kg) diluted in 200 μL of PBS every 3 days for a total of five injections. The tumor size was measured at 11, 14 and 17 days and determined using the formula (L × W2)/2, in which L is the length and W is the width. Statistically significant differences from mAb‐treated and control mice were calculated by Student’s t‐test and shown with P‐values. Differences of P < 0.05 were considered statistically significant. All experiments were approved and done in accordance with the guidelines of the Committee of Ethics on Animal Experiments (Kyushu University, Fukuoka, Japan).
Results
Specific binding of SGN‐30 to HTLV‐I‐infected cells. We first examined if SGN‐30 specifically binds to HTLV‐I‐infected cell lines. In this study we used three HTLV‐I‐infected cell lines, HUT‐102, MT‐2 and S1T, and an ALCL cell line, Karpas 299. As shown in Figure 1, SGN‐30 could bind to Karpas 299 cells (MFI = 4466.9). It also bound to S1T cells (MIF = 1894.3), MT‐2 cells (MFI = 967.5), and HUT‐102 cells (MFI = 601.7), but at a lower intensity than Karpas 299 cells.
Figure 1.
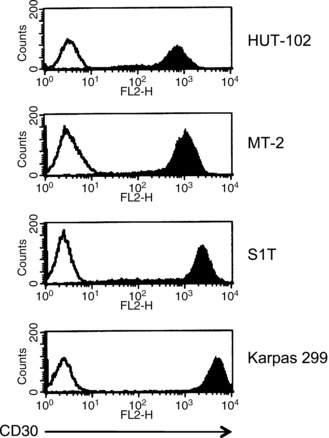
Binding of SGN‐30 to CD30+ cell lines. Cells (1 × 106) were incubated with biotin‐conjugated SGN‐30 (shaded) or isotype‐matched control IgG (unshaded) for 30 min at 4°C, followed by incubation with phycoerythrin‐conjugated streptavidin for 30 min at 4°C. The binding of SGN‐30 was evaluated using a flow cytometer. The histogram shows expression of CD30. Data of a representative experiment are shown from three independent experiments.
Growth inhibitory effects of SGN‐30 and SGN‐35 on HTLV‐I‐infected cells in vitro. To evaluate the effects of SGN‐30 on cell growth in vitro, we carried out a cell proliferation assay. Cross‐linking with human IgG was carried out to augment the antiproliferative activity of SGN‐30 in vitro as described by Wahl et al.,( 31 ) as it could be possible that cross‐linking could reflect the situation in vivo in which Fc receptor‐mediated clustering of SGN‐30 bound to CD30 could induce a similar effect. In this manner, CD30 clustering by SGN‐30 could provide transduction of growth arrest or apoptotic signals that would promote antitumor activity. Upon cross‐linking, SGN‐30 showed growth‐inhibitory activity against S1T and Karpas 299 cells with an IC50 of 80 and 5 ng/mL, respectively (Fig. 2a). The growth of HUT‐102 and MT‐2 cells was also inhibited in a dose‐dependent manner, but they were relatively resistant to SGN‐30 (Fig. 2a).
Figure 2.
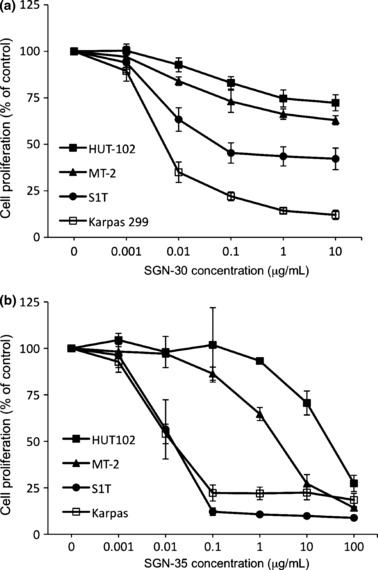
Growth inhibitory effects of SGN‐30/SGN‐35 on HTLV‐I‐infected cells in vitro. HUT‐102, MT‐2, S1T, and Karpas 299 cells (1 × 104 cells/each cell line) were cultured in the absence or presence of various concentrations of (a) SGN‐30 with 10‐fold excess of goat antihuman IgG or (b) SGN‐35 for 48 h at 37°C in flat‐bottomed 96‐well plates. The cells were further incubated with Cell Proliferation Reagent WST‐1 for 4 h at 37°C. The absorbance of the samples was measured at 450 and 550 nm (reference wavelength) with a microplate reader. Data shown represent mean value ± SD obtained from a representative experiment in three independent experiments.
We next examined the effects of SGN‐35 on cell growth of these cell lines in vitro. In the cell proliferation assay, SGN‐35 inhibited the growth of S1T as well as Karpas 299 cells with an IC50 of approximately 10 ng/mL, whereas the growth of HUT‐102 and MT‐2 cells was inhibited in a dose‐dependent manner with IC50 values greater than 1 μg/mL (Fig. 2b). As previously confirmed by Sutherland et al.,( 40 ) SGN‐35 could be internalized by CD30 and transported to the lysosomes, followed by MMAE release, therefore the effect of SGN‐35 on cell growth could be, at least in part, due to the MMAE activity. SGN‐35 is effective for all cell lines tested, but HUT‐102 and MT‐2 were less susceptible to SGN‐35 than S1T or Karpas 299.
Effects of SGN‐30 and SGN‐35 on cell cycle of HTLV‐I‐infected cells. One of the mechanisms of the inhibitory effects by mAbs is thought to be due to cell cycle arrest.( 31 , 37 ) As previously reported,( 31 ) in Karpas 299 cells, co‐cultivation with SGN‐30 drastically increased the population of the cells in the G1 phase (from 36% in the untreated cells to 79%) with a reduction of the cells in the S phase (from 42% in the untreated cells to 4%) even at 0.1 μg/mL (Fig. 3a). In S1T cells, SGN‐30 upon cross‐linking also increased the population of the cells in the G1 phase (from 38% in the untreated cells to 61%) with a reduction of the cells in the S phase (from 42% in the untreated cells to 26%) at 0.1 μg/mL, and the effect was dose‐dependent (Fig. 3b). In contrast, in HUT‐102 cells, SGN‐30 only increased the population of cells in the G1 phase (from 44% in the untreated cells to 60%) with a reduction of the cells in the S phase (from 45% in the untreated cells to 31%) at the highest concentration (10 μg/mL), thus SGN‐30 was less effective on the cell cycle of HUT‐102 cells (Fig. 3c). These results were consistent with the in vitro cell proliferation assay shown in Figure 2a.
Figure 3.
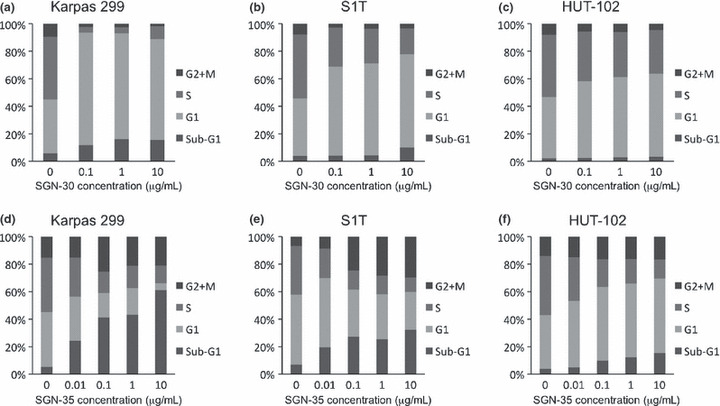
Growth inhibitory effects of SGN‐30 and SGN‐35 on cell cycle. HUT‐102, S1T, and Karpas 299 cells (5 × 105 cells/each cell line) were cultured in the absence or presence of various concentrations of SGN‐30 with 10‐fold excess of goat antihuman IgG (a–c) or SGN‐35 (d–f) for 48 h at 37°C in flat‐bottomed 24‐well plates. The cell cycle was analyzed with a bromodeoxyuridine flow kit according to the manufacturer’s instructions. Briefly, the cells were fixed and permeabilized with Perm buffer, and treated with 30 μg of DNase for 1 h at 37°C, and stained with FITC‐conjugated anti‐bromodeoxyuridine Ab and 7‐amino‐actinomycin D. The percentage of cells in each cell cycle phase (sub‐G1, G1, S, and G2/M) was evaluated using a flow cytometer. Data of a representative experiment are shown from two independent experiments.
We next examined the effects of SGN‐35 on the cell cycle of three HTLV‐I‐infected cell lines. As previously reported,( 37 ) in Karpas 299 cells, co‐cultivation with SGN‐35 decreased the population of cells in the S phase (from 39% in the untreated cells to 12%) with an increase in the percentage of apoptotic cells in the sub‐G1 phase (from 5% in the untreated cells to 61%), due to the effect of MMAE, in a dose‐dependent manner (Fig. 3d). In S1T cells, SGN‐35 also decreased the population of cells in the S phase (from 35% in the untreated cells to 10%) with an increase in the percentage of apoptotic cells in the sub‐G1 phase (from 7% in the untreated cells to 32%) in a dose‐dependent manner (Fig. 3e). In contrast, the effect of SGN‐35 on the cell cycle of HUT‐102 cells was lower than on S1T cells (Fig. 3f).
We also observed that treatment with SGN‐35 increased the G2/M phase of S1T cells (from 6% in the untreated cells to 29%) as well as Karpas 299 cells (from 15% in the untreated cells to 25%) but had little effect on HUT‐102 cells (Fig. 3d–f).
Growth inhibitory effects of SGN‐30 and SGN‐35 on HTLV‐I‐infected cells in vivo. To further investigate the effects of SGN‐30 and SGN‐35 on HTLV‐I‐infected cells, we subsequently used NOD/SCID mice to subcutaneously engraft with HUT‐102 or Karpas 299 cells. HUT‐102 and Karpas 299 cells grew rapidly in the mice (Fig. 4). We also used S1T cells in these experiments but they did not grow in the mice (data not shown). In several published reports, treatment of SCID mice with reagents for evaluation of anti‐ATL effects was begun at one day after HUT‐102 cell inoculation,( 49 , 50 ) so we applied the identical protocol to evaluate the anti‐ATL activities of the CD30 mAbs in NOD/SCID mice. SGN‐30 (2 mg/kg) was given every 3 days for a total of five injections. SGN‐30 significantly inhibited the tumor growth of Karpas 299 cells compared with the control (**P < 0.01; Fig. 4a). On day 17, SGN‐30 also induced a significant delay in tumor growth of HUT‐102 cells compared with the control (**P < 0.01; Fig. 4b).
Figure 4.
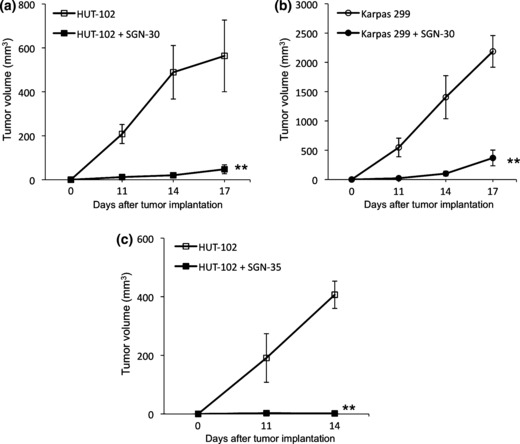
Effect of SGN‐30 and SGN‐35 on growth of HTLV‐I‐infected cells in NOD/SCID mice. Seven‐week‐old female mice were subcutaneously engrafted with HUT‐102 or Karpas 299 cells (1 × 107 and 2 × 107 cells, respectively). One day after injection of cells, mice received i.p. SGN‐30 (2 mg/kg) (a,b) or SGN‐35 (1 mg/kg) (c) diluted in 200 μL PBS every 3 days for a total of five injections. The tumor size was measured at 11, 14 and 17 days. Data shown represent mean value ± SD for four mice of each group obtained from a representative experiment in two independent experiments. Statistically significant differences between mAb‐treated and control mice are shown (**P < 0.01).
As previously described, SGN‐35 (1 mg/kg) induced complete tumor regression in subcutaneous xenograft models of ALCL tumors in SCID mice.( 37 ) In this study SGN‐35 (1 mg/kg) was given every 3 days for a total of five injections. On day 14, SGN‐35 also inhibited the tumor growth of HUT‐102 cells more effectively than SGN‐30 (**P < 0.01; Fig. 4c). We carried out the experiments with the appropriate doses as previously described; doses of SGN‐30 and SGN‐35 up to 100 and 30 mg/kg, respectively, were well tolerated with no apparent toxicity.( 31 , 37 )
Discussion
CD30 is an attractive target for therapeutic intervention because of its restricted pattern of expression.( 23 ) In this study we showed for the first time that SGN‐30, a chimeric anti‐CD30 mAb, and SGN‐35, an MMAE‐conjugated anti‐CD30 mAb, had growth‐inhibitory activity against the HTLV‐I‐infected cell lines in vitro and had a curative potential in the NOD/SCID murine model.
As expected, all three HTLV‐I‐infected cell lines tested express CD30 on the cell surface. However, the MFI of the HTLV‐I‐infected cell lines was quite different and lower than that of Karpas 299 cells. The antiproliferative effect on each cell line in vitro was closely related to the binding activity. These data suggest that SGN‐30 is definitely effective on HTLV‐I‐infected cells, but the growth inhibitory effect of SGN‐30 could be influenced by the binding activity to CD30. It is notable that co‐cultivation with SGN‐30 drastically increased the population of HTLV‐I‐infected cells in the G1 phase with a reduction of cells in the S phase depending on the MFI. SGN‐30 had little effect on an increase of the sub‐G1 phase on the cell cycle of HTLV‐I‐infected cells, suggesting that the effects of SGN‐30 on HTLV‐I‐infected cells were mainly cell cycle arrest but not cell death including apoptosis or necrosis. Thus, molecules associated with cell cycle arrest should be involved. Hübinger et al. reported that anti‐CD30 mAbs such as M44 and M67 can inhibit the growth of ALCL cell lines through the induction of cell cycle arrest and without the induction of apoptosis, and also showed that cyclin‐dependent kinase inhibitor p21waf1 was not expressed in ALCL cells but induced by anti‐CD30 mAb HeFi‐1 in a p53‐independent mechanism,( 51 ) suggesting that the molecule p21waf1 might be involved in the CD30‐induced cell cycle arrest of Karpas 299 cells. In addition, Wright et al. reported that CD30 stimulation elicited p21waf1‐mediated cell cycle arrest through the canonical but not the alternate NF‐κB pathway in Karpas 299 cells.( 24 ) A possible explanation for the differing effects of CD30 mAbs on ALCL cells and HL cells was suggested by Mir et al., who demonstrated that constitutive NF‐κB activation via a specific defect in inhibitor κBα (IκBα) in HL cell line L428 rendered them resistant to CD30‐induced growth inhibition,( 25 ) and Horie et al. reported that ligand‐independent signaling by overexpressed CD30 drives constitutive NF‐κB activation in HL cells.( 52 ) However, the expression of a chimeric protein nucleophosmin–anaplastic lymphoma kinase by a chromosomal translocation t(2;5)(p23;q35) that fuses nucleophosmin sequences on chromosome 5 to anaplastic lymphoma kinase sequences on chromosome 2, abrogates recruitment and aggregation of TNFR‐associated factor proteins by competitive binding to CD30, resulting in abrogation of NF‐κB activation in ALCL cells.( 23 ) In HTLV‐I‐infected cells, Tax, a trans‐regulatory protein encoded in a pX region of the HTLV‐I genome, is well known to, at least in part, influence constitutive NF‐κB activation and cell cycle progression followed by dysregulating cell growth.( 53 ) The constitutive NF‐κB activation is in part induced by a proteolytic degradation of inhibitor κBα in HTLV‐I‐infected cells.( 54 ) In this regard the responsiveness to the CD30 mAb in HTLV‐I‐infected cells might be similar to that in HL cells rather than ALCL cells. It would be definitely interesting to investigate the precise function of CD30‐mediated p21waf1 or NF‐κB activation on the cell cycle of HTLV‐I‐infected cells.
An effective approach to augment the antitumor activity of mAb is linkage to cytotoxic drugs.( 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 ) We originally reported that SGN‐35, a chemically modified version of SGN‐30 with MMAE, was designed for optimal stability in human plasma and efficient cleavage and release of multiple drug types by the human lysosomal protease cathepsin B,( 39 , 40 ) and induced antitumor effect on ALCL cells and HL cells.( 36 , 37 , 38 ) In this study, we observed that SGN‐35 showed a more antiproliferative effect on HUT‐102 and MT‐2 cells than SGN‐30, which was also correlated with the MFI as observed in the experiments with SGN‐30 in vitro. In contrast, it is interesting that the antiproliferative effect of SGN‐35 on S1T cells was fully effective, the same as that on Karpas 299 cells. Francisco et al. described that there was no direct correlation between CD30 expression level and sensitivity to SGN‐35.( 37 ) The HL cell line HDLM‐2 with a relatively high level of CD30 was only moderately sensitive to SGN‐35 with an IC50 of 302 ng/mL, whereas the ALCL cell line SU‐DHL‐1 had a lower binding ratio but was approximately 1000‐fold more sensitive, suggesting that the role of other possible factors, including the rate of internalization, intracellular trafficking of ADC, and enzymatic cleavage and release of MMAE from the lysosome after internalization, should be involved in the action of ADC.( 37 ) In addition, we found that SGN‐35 in S1T and Karpas 299 cells increased not only the sub‐G1 population (apoptotic cells) but also G2/M arrest and onset of apoptosis due to the effects of MMAE as described previously.( 37 , 42 , 43 ) The molecular mechanism of G2/M growth arrest by SGN‐35 on CD30 signaling remains to be determined. Nevertheless, we would expect that SGN‐35 could be used as a highly potent and selective agent against not only ALCL/HL but also ATL by inducing both cell growth arrest and apoptosis.
It is intriguing that SGN‐30 given in vivo was apparently effective without overt toxicities on the HUT‐102 tumor xenograft model in NOD/SCID mice, whereas the HUT‐102 cells in vitro were quite resistant to SGN‐30 in a proliferation assay. It is possible for the action of mAbs that antibody‐dependent cell cytotoxicity activity functions against HUT‐102 cells in vivo. Oflazoglu et al. ( 33 ) reported that macrophages contribute significantly to the activity of SGN‐30 in vitro by inducing cellular cytotoxicity through antibody‐dependent cellular phagocytosis and complement–dependent cytotoxicity activity, and depletion of macrophages reduced survival of HL tumor‐bearing mice treated with SGN‐30 in vivo, suggesting that Fc‐dependent effector cell functions contribute significantly to the antitumor activity of SGN‐30 in HL cells. Furthermore, we found that SGN‐35 showed a greater antitumor effect on HUT‐102 without overt toxicities in NOD/SCID mice. It is very important that the effective dose of 1 mg/kg SGN‐35 contains 0.036 mg/kg of MMAE, a dose equivalent to 5% of the maximum tolerated dose of the free drug alone, supporting the usefulness of targeted drug delivery by SGN‐35, as the maximum tolerated dose in mice of dolastatin 10 is approximately 0.45 mg/kg.( 37 ) Taken together, these results in vivo strongly suggest that SGN‐30 and SGN‐35 could be expected to be clinically applied for ATL chemotherapy.
Currently both SGN‐30 and SGN‐35 are in clinical trials for patients with ALCL and HL.( 34 , 35 , 55 ) Preliminary results from phase I/II trials using SGN‐30 in 20 patients with recurrent/relapse systemic ALCL showed an overall response rate of 20%.( 55 ) Current trials are investigating SGN‐30 as a single agent for relapsed/refractory HL and cutaneous ALCL, and in combination with gemcitabine, vinorelbine, and pegylated liposomal doxorubicin for patients with relapsed/refractory HL.( 55 ) These data indicated that both SGN‐30 and SGN‐35 would also be applicable for ATL. Several groups independently examined CD30 expression in cells from patients with ATL; it was observed in 7 out of 36 ATL cases (19.4%)( 15 ) and in 8 out of 66 ATL cases (12.1%).( 19 ) The expression level of CD30 in primary ATL cells was variable among patients and lower than that in HTLV‐I‐infected cell lines.( 19 , 20 ) They suggested that the CD30 expression level might be correlated with development of ATL. The reason for the difference in CD30 expression levels between HTLV‐I‐infected cell lines and primary ATL cells is unknown. It would be possible that the expression level of CD30 might also be correlated with NF‐κB activation.( 19 ) Tax induces the constitutive activation of transcription factor activator protein‐1,( 56 , 57 ) which might result in an autocrine loop of CD30‐mediated activation of NF‐κB in HTLV‐I‐infected cells, as previously observed in HL and ALCL cells.( 58 ) In addition to the membrane‐associated form of CD30, sCD30 in the sera of patients with ATL( 20 , 59 , 60 , 61 ) would be a useful marker to evaluate the clinical progression of ATL as well as HL or ALCL.( 59 , 62 , 63 , 64 , 65 ) The sCD30 binds to CD30L with high affinity and blocks transmembrane signaling by CD30,( 66 ) suggesting that the elevated level of sCD30 in sera might also decrease antitumor effects of the CD30 mAbs by blocking the antiproliferative signaling through cell cycle arrest and apoptosis. It will be of great interest to determine whether or not SGN‐30 and SGN‐35 show antitumor activity on primary ATL cells followed by clinical trials.
In conclusion, we have evaluated the effects of SGN‐30 and SGN‐35 on HTLV‐I‐infected cells. Our results indicate for the first time that SGN‐30 and SGN‐35 could induce antiproliferative activity on HTLV‐I‐infected cells through apoptosis and/or cell growth arrest in vitro and in vivo. Based on our new findings, we propose the use of SGN‐30 and SGN‐35 as novel therapeutic agents for the treatment of ATL.
Abbreviations
- ADC
antibody–drug conjugates
- ALCL
anaplastic large cell lymphoma
- ATL
adult T‐cell leukemia
- CD30L
CD30 ligand
- HL
Hodgkin’s lymphoma
- HTLV‐I
human T‐cell leukemia virus type I
- MFI
mean fluorescence intensity
- MMAE
monomethyl auristatin E
- NF‐κB
nuclear factor‐κB
- sCD30
soluble form of CD30
- TNFR
tumor necrosis factor receptor
Acknowledgments
We thank Dr. M. Shiratsuchi for critical suggestions. This work was supported by Grants‐in‐Aid for Young Scientists (B) (to N. Maeda), for Scientific Research (C) (to H. Muta), and for Scientific Research on Priority Areas (to Y. Yoshikai) by the Japan Society for the Promotion of Science, and the Ministry of Education, Culture, Sports, Science and Technology, Japan; and by the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases, which was launched as a project commissioned by the Ministry of Education, Culture, Sports, Science and Technology, Japan (to Y. Yoshikai).
References
- 1. Matsuoka M, Jeang KT. Human T‐cell leukaemia virus type 1 (HTLV‐1) infectivity and cellular transformation. Nat Rev Cancer 2007; 7: 270–80. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Z, Zhang M, Ravetch JV, Goldman C, Waldmann TA. Effective therapy for a murine model of adult T‐cell leukemia with the humanized anti‐CD2 monoclonal antibody, MEDI‐507. Blood 2003; 102: 284–8. [DOI] [PubMed] [Google Scholar]
- 3. Waldmann TA, Goldman CK, Bongiovanni KF et al. Therapy of patients with human T‐cell lymphotrophic virus I‐induced adult T‐cell leukemia with anti‐Tac, a monoclonal antibody to the receptor for interleukin‐2. Blood 1988; 72: 1805–16. [PubMed] [Google Scholar]
- 4. Waldmann TA, White JD, Goldman CK et al. The interleukin‐2 receptor: a target for monoclonal antibody treatment of human T‐cell lymphotrophic virus I‐induced adult T‐cell leukemia. Blood 1993; 82: 1701–12. [PubMed] [Google Scholar]
- 5. Waldmann TA, White JD, Carrasquillo JA et al. Radioimmunotherapy of interleukin‐2R alpha‐expressing adult T‐cell leukemia with Yttrium‐90‐labeled anti‐Tac. Blood 1995; 86: 4063–75. [PubMed] [Google Scholar]
- 6. Phillips KE, Herring B, Wilson LA et al. IL‐2Ralpha‐Directed monoclonal antibodies provide effective therapy in a murine model of adult T‐cell leukemia by a mechanism other than blockade of IL‐2/IL‐2Ralpha interaction. Cancer Res 2000; 60: 6977–84. [PubMed] [Google Scholar]
- 7. Tan C, Waldmann TA. Proteasome inhibitor PS‐341, a potential therapeutic agent for adult T‐cell leukemia. Cancer Res 2002; 62: 1083–6. [PubMed] [Google Scholar]
- 8. Zhang M, Yao Z, Garmestani K et al. Pretargeting radioimmunotherapy of a murine model of adult T‐cell leukemia with the α‐emitting radionuclide, bismuth 213. Blood 2002; 100: 208–16. [DOI] [PubMed] [Google Scholar]
- 9. Zhang M, Zhang Z, Garmestani K et al. Activating Fc receptors are required for antitumor efficacy of the antibodies directed toward CD25 in a murine model of adult T‐cell leukemia. Cancer Res 2004; 64: 5825–9. [DOI] [PubMed] [Google Scholar]
- 10. Zhang M, Zhang Z, Goldman CK, Janik J, Waldmann TA. Combination therapy for adult T‐cell leukemia‐xenografted mice: flavopiridol and anti‐CD25 monoclonal antibody. Blood 2005; 105: 1231–6. [DOI] [PubMed] [Google Scholar]
- 11. Zhang Z, Zhang M, Garmestani K et al. Effective treatment of a murine model of adult T‐cell leukemia using 211At‐7G7/B6 and its combination with unmodified anti‐Tac (daclizumab) directed toward CD25. Blood 2006; 108: 1007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Z, Zhang M, Goldman CK, Ravetch JV, Waldmann TA. Effective therapy for a murine model of adult T‐cell leukemia with the humanized anti‐CD52 monoclonal antibody, Campath‐1H. Cancer Res 2003; 63: 6453–7. [PubMed] [Google Scholar]
- 13. Hakimi J, Ha VC, Lin P et al. Humanized Mikβ1, a humanized antibody to the IL‐2 receptor β‐chain that acts synergistically with humanized anti‐TAC. J Immunol 1993; 151: 1075–85. [PubMed] [Google Scholar]
- 14. Ishida T, Iida S, Akatsuka Y et al. The CC chemokine receptor 4 as a novel specific molecular target for immunotherapy in adult T‐Cell leukemia/lymphoma. Clin Cancer Res 2004; 10: 7529–39. [DOI] [PubMed] [Google Scholar]
- 15. Ohtsuka E, Kikuchi H, Nasu M, Takita‐Sonoda Y, Fujii H, Yokoyama S. Clinicopathological features of adult T‐cell leukemia with CD30 antigen expression. Leuk Lymphoma 1994; 15: 303–10. [DOI] [PubMed] [Google Scholar]
- 16. Takeshita M, Akamatsu M, Ohshima K et al. CD30 (Ki‐1) expression in adult T‐cell leukaemia/lymphoma is associated with distinctive immunohistological and clinical characteristics. Histopathology 1995; 26: 539–46. [DOI] [PubMed] [Google Scholar]
- 17. Takahara T, Masutani K, Kajiwara E et al. Adult T‐cell leukemia/lymphoma in which the pathohistological diagnosis was identical to that of Ki‐1 positive anaplastic large cell lymphoma. Intern Med 1999; 38: 824–8. [DOI] [PubMed] [Google Scholar]
- 18. Fujiwara H, Arima N, Ohtsubo H et al. Clinical significance of serum neuron‐specific enolase in patients with adult T‐cell leukemia. Am J Hematol 2002; 71: 80–4. [DOI] [PubMed] [Google Scholar]
- 19. Higuchi M, Matsuda T, Mori N et al. Elevated expression of CD30 in adult T‐cell leukemia cell lines: possible role in constitutive NF‐kappaB activation. Retrovirology 2005; 2: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nishioka C, Takemoto S, Kataoka S et al. Serum level of soluble CD30 correlates with the aggressiveness of adult T‐cell leukemia/lymphoma. Cancer Sci 2005; 96: 810–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bittencourt AL, Barbosa HS, Vieira MD, Farré L. Adult T‐cell leukemia/lymphoma (ATL) presenting in the skin: Clinical, histological and immunohistochemical features of 52 cases. Acta Oncol 2009; 48: 598–604. [DOI] [PubMed] [Google Scholar]
- 22. Watts TH. TNF/TNFR family members in costimulation of T cell response. Annu Rev Immunol 2005; 23: 23–68. [DOI] [PubMed] [Google Scholar]
- 23. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008; 8: 11–23. [DOI] [PubMed] [Google Scholar]
- 24. Wright CW, Rumble JM, Duckett CS. CD30 activates both the canonical and alternative NF‐κB pathways in anaplastic large cell lymphoma cells. J Biol Chem 2007; 282: 10252–62. [DOI] [PubMed] [Google Scholar]
- 25. Mir SS, Richter BW, Duckett CS. Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood 2000; 96: 4307–12. [PubMed] [Google Scholar]
- 26. Borchmann P, Treml JF, Hansen H et al. The human anti‐CD30 antibody 5F11 shows in vitro and in vivo activity against malignant lymphoma. Blood 2003; 102: 3737–42. [DOI] [PubMed] [Google Scholar]
- 27. Zhang M, Yao Z, Zhang Z et al. Effective therapy for a murine model of human anaplastic large‐cell lymphoma with the anti‐CD30 monoclonal antibody, HeFi‐1, does not require activating Fc receptors. Blood 2006; 108: 705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang M, Yao Z, Patel H et al. Effective therapy of murine models of human leukemia and lymphoma with radiolabeled anti‐CD30 antibody, HeFi‐1. Proc Natl Acad Sci U S A 2007; 104: 8444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ansell SM, Horwitz SM, Engert A et al. Phase I/II study of an anti‐CD30 monoclonal antibody (MDX‐060) in Hodgkin’s lymphoma and anaplastic large‐cell lymphoma. J Clin Oncol 2007; 25: 2764–9. [DOI] [PubMed] [Google Scholar]
- 30. Bowen MA, Olsen KJ, Cheng L, Avila D, Podack ER. Functional effects of CD30 on a large granular lymphoma cell line, YT. Inhibition of cytotoxicity, regulation of CD28 and IL‐2R, and induction of homotypic aggregation. J Immunol 1993; 151: 5896–906. [PubMed] [Google Scholar]
- 31. Wahl AF, Klussman K, Thompson JD et al. The anti‐CD30 monoclonal antibody SGN‐30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin’s disease. Cancer Res 2002; 62: 3736–42. [PubMed] [Google Scholar]
- 32. Cerveny CG, Law CL, McCormick RS et al. Signaling via the anti‐CD30 mAb SGN‐30 sensitizes Hodgkin’s disease cells to conventional chemotherapeutics. Leukemia 2005; 19: 1648–55. [DOI] [PubMed] [Google Scholar]
- 33. Oflazoglu E, Stone IJ, Gordon KA et al. Macrophages contribute to the antitumor activity of the anti‐CD30 antibody SGN‐30. Blood 2007; 110: 4370–2. [DOI] [PubMed] [Google Scholar]
- 34. Bartlett NL, Younes A, Carabasi MH et al. A phase 1 multidose study of SGN‐30 immunotherapy in patients with refractory or recurrent CD30+ hematologic malignancies. Blood 2008; 111: 1848–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Forero‐Torres A, Leonard JP, Younes A et al. A phase II study of SGN‐30 (anti‐CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br J Haematol 2009; 146: 171–9. [DOI] [PubMed] [Google Scholar]
- 36. Doronina SO, Toki BE, Torgov MY et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 2003; 21: 778–84. [DOI] [PubMed] [Google Scholar]
- 37. Francisco JA, Cerveny CG, Meyer DL et al. cAC10‐vcMMAE, an anti‐CD30‐monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003; 102: 1458–65. [DOI] [PubMed] [Google Scholar]
- 38. Hamblett KJ, Senter PD, Chace DF et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10: 7063–70. [DOI] [PubMed] [Google Scholar]
- 39. Sanderson RJ, Hering MA, James SF et al. In vivo drug‐linker stability of an anti‐CD30 dipeptide‐linked auristatin immunoconjugate. Clin Cancer Res 2005; 11: 843–52. [PubMed] [Google Scholar]
- 40. Sutherland MS, Sanderson RJ, Gordon KA et al. Lysosomal trafficking and cysteine protease metabolism confer target‐specific cytotoxicity by peptide‐linked anti‐CD30‐auristatin conjugates. J Biol Chem 2006; 281: 10540–7. [DOI] [PubMed] [Google Scholar]
- 41. Oflazoglu E, Kissler KM, Sievers EL, Grewal IS, Gerber HP. Combination of the anti‐CD30‐auristatin‐E antibody‐drug conjugate (SGN‐35) with chemotherapy improves antitumour activity in Hodgkin lymphoma. Br J Haematol 2008; 142: 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Law CL, Cerveny CG, Gordon KA et al. Efficient elimination of B‐lineage lymphomas by anti‐CD20‐auristatin conjugates. Clin Cancer Res 2004; 10: 7842–51. [DOI] [PubMed] [Google Scholar]
- 43. Law CL, Gordon KA, Toki BE et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potent therapeutic target for anti‐CD70 antibody‐drug conjugates. Cancer Res 2006; 66: 2328–37. [DOI] [PubMed] [Google Scholar]
- 44. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T‐cell lymphoma. Proc Natl Acad Sci U S A 1980; 77: 7415–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miyoshi I, Kubonishi I, Yoshimoto S et al. Type C virus particles in a cord T‐cell line derived by co‐cultivating normal human cord leukocytes and human leukaemic T cells. Nature 1981; 294: 770–1. [DOI] [PubMed] [Google Scholar]
- 46. Arima N, Molitor JA, Smith MR, Kim JH, Daitoku Y, Greene WC. Human T‐cell leukemia virus type I Tax induces expression of the Rel‐related family of kappa B enhancer‐binding proteins: evidence for a pretranslational component of regulation. J Virol 1991; 65: 6892–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischer P, Nacheva E, Mason DY et al. A Ki‐1 (CD30)‐positive human cell line (Karpas 299) established from a high‐grade non‐Hodgkin’s lymphoma, showing a 2;5 translocation and rearrangement of the T‐cell receptor β‐chain gene. Blood 1988; 72: 234–40. [PubMed] [Google Scholar]
- 48. Mori N, Matsuda T, Tadano M et al. Apoptosis induced by the histone deacetylase inhibitor FR901228 in human T‐cell leukemia virus type 1‐infected T‐cell lines and primary adult T‐cell leukemia cells. J Virol 2004; 78: 4582–90. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49. Ishikawa C, Matsuda T, Okudaira T et al. Bisphosphonate incadronate inhibits growth of human T‐cell leukaemia virus type I‐infected T‐cell lines and primary adult T‐cell leukaemia cells by interfering with the mevalonate pathway. Br J Haematol 2007; 136: 424–32. [DOI] [PubMed] [Google Scholar]
- 50. Nakazato T, Okudaira T, Ishikawa C et al. Anti‐adult T‐cell leukemia effects of a novel synthetic retinoid, Am80 (Tamibarotene). Cancer Sci 2008; 99: 2286–94. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51. Hübinger G, Müller E, Scheffrahn I et al. CD30‐mediated cell cycle arrest associated with induced expression of p21CIP1/WAF1 in the anaplastic large cell lymphoma cell line Karpas 299. Oncogene 2001; 20: 590–8. [DOI] [PubMed] [Google Scholar]
- 52. Horie R, Watanabe T, Morishita Y et al. Ligand‐independent signaling by overexpressed CD30 drives NF‐κB activation in Hodgkin‐Reed‐Sternberg cells. Oncogene 2002; 21: 2493–503. [DOI] [PubMed] [Google Scholar]
- 53. Marriott SJ, Semmes OJ. Impact of HTLV‐I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 2005; 24: 5986–95. [DOI] [PubMed] [Google Scholar]
- 54. Mori N, Fujii M, Ikeda S et al. Constitutive activation of NF‐κB in primary adult T‐cell leukemia cells. Blood 1999; 93: 2360–8. [PubMed] [Google Scholar]
- 55. Castillo J, Winer E, Quesenberry P. Newer monoclonal antibodies for hematological malignancies. Exp Hematol 2008; 36: 755–68. [DOI] [PubMed] [Google Scholar]
- 56. Mori N, Fujii M, Iwai K et al. Constitutive activation of transcription factor AP‐1 in primary adult T‐cell leukemia cells. Blood 2000; 95: 3915–21. [PubMed] [Google Scholar]
- 57. Fujii M, Iwai K, Oie M et al. Activation of oncogenic transcription factor AP‐1 in T cells infected with human T cell leukemia virus type 1. AIDS Res Hum Retroviruses 2000; 16: 1603–6. [DOI] [PubMed] [Google Scholar]
- 58. Watanabe M, Sasaki M, Itoh K et al. JunB induced by constitutive CD30‐extracellular signal‐regulated kinase 1/2 mitogen‐activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and Reed‐Sternberg cells of Hodgkin lymphoma. Cancer Res 2005; 65: 7628–34. [DOI] [PubMed] [Google Scholar]
- 59. Pfreundschuh M, Pohl C, Berenbeck C et al. Detection of a soluble form of the CD30 antigen in sera of patients with lymphoma, adult T‐cell leukemia and infectious mononucleosis. Int J Cancer 1990; 45: 869–74. [DOI] [PubMed] [Google Scholar]
- 60. Birmann BM, Mueller NE, Okayama A et al. Patterns of serum type 1 and type 2 immune markers in healthy carriers of HTLV‐I. J Med Virol 2006; 78: 847–52. [DOI] [PubMed] [Google Scholar]
- 61. Birmann BM, Breen EC, Stuver S et al. Population differences in immune marker profiles associated with human T‐lymphotropic virus type I infection in Japan and Jamaica. Int J Cancer 2009; 124: 614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pizzolo G, Vinante F, Chilosi M et al. Serum levels of soluble CD30 molecule (Ki‐1 antigen) in Hodgkin’s disease: relationship with disease activity and clinical stage. Br J Haematol 1990; 75: 282–4. [DOI] [PubMed] [Google Scholar]
- 63. Gause A, Pohl C, Tschiersch A et al. Clinical significance of soluble CD30 antigen in the sera of patients with untreated Hodgkin’s disease. Blood 1991; 77: 1983–8. [PubMed] [Google Scholar]
- 64. Nadali G, Vinante F, Stein H et al. Serum levels of the soluble form of CD30 molecule as a tumor marker in CD30+ anaplastic large‐cell lymphoma. J Clin Oncol 1995; 13: 1355–60. [DOI] [PubMed] [Google Scholar]
- 65. Zinzani PL, Pileri S, Bendandi M et al. Clinical implications of serum levels of soluble CD30 in 70 adult anaplastic large‐cell lymphoma patients. J Clin Oncol 1998; 16: 1532–7. [DOI] [PubMed] [Google Scholar]
- 66. Hargreaves PG, Al‐Shamkhani A. Soluble CD30 binds to CD153 with high affinity and blocks transmembrane signaling by CD30. Eur J Immunol 2002; 32: 163–73. [DOI] [PubMed] [Google Scholar]