

Abstract
The recent clinical application of granulocyte macrophage colony‐stimulating factor (GM‐CSF)‐transduced autologous tumor vaccines revealed substantial antitumor activity and valuable clinical results. However, for these vaccines to be optimally effective, the antitumor efficacies must be improved. Recently, Sendai virus (SeV) vectors, which are cytoplasmic RNA vectors, have emerged as safe vectors with high gene transduction. In the current study, the in vivo therapeutic antitumor efficacies of irradiated GM‐CSF‐transduced mouse renal cell carcinoma (RENCA) vaccine cells mediated by either fusion gene‐deleted non‐transmissible SeV encoding mouse GM‐CSF (SeV/dF/G) or adenovirus (E1, E3 deleted serotype 5 adenovirus) encoding mouse GM‐CSF (AdV/G) (respectively described as irRC/SeV/GM or irRC/AdV/GM) were compared in RENCA‐bearing mice. The results showed that the antitumor effect was equivalent between irRC/SeV/GM and irRC/AdV/GM cells, even though the former produced less GM‐CSF in vitro. The cell numbers of activated (CD80+, CD86+, CD80+CD86+) dendritic cells in lymph nodes from mice treated with irRC/AdV/GM or irRC/SeV/GM cells were increased significantly compared with those of mice treated with the respective controls, at both the earlier and later phases. In an in vitro cytotoxicity assay, splenocytes harvested from mice treated with both irRC/SeV/GM and irRC/AdV/GM cells showed tumor‐specific responses against RENCA cells. The restimulated splenocytes harvested from mice treated with irRC/SeV/GM or irRC/AdV/GM cells produced significantly higher levels of interleukin‐2, interleukin‐4, and interferon‐γ compared with their respective controls (P < 0.05). Furthermore, vaccination with irRC/AdV/GM or irRC/SeV/GM cells induced significantly enhanced recruitment of the cytolytic effectors of CD107a+CD8+ T cells and CD107a+ natural killer cells into tumors compared with those induced by their respective controls (P < 0.05). Taken together, our results suggest that the SeV/dF/G vector is a potential candidate for the production of effective autologous GM‐CSF‐transduced tumor vaccines in clinical cancer immune gene therapy. (Cancer Sci 2008; 99: 2315–2326)
Abbreviations:
- APC
antigen‐presenting cell
- CM
complete medium
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- DMEM
Dulbecco's modified Eagle's medium
- DLN
draining lymph node
- F
fusion
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- GM‐CSF
granulocyte macrophage colony‐stimulating factor
- HBSS
Hank's buffered salt solution
- IL
interleukin
- IFN
interferon
- LLC
Lewis lung carcinoma
- MHC
major histocompatibility complex
- MOI
multiplicity of infection
- NK
natural killer
- PBS
phosphate‐buffered saline
- RCC
renal cell carcinoma
- SeV
Sendai virus
- TIL
tumor‐infiltrating leukocyte
- TNF
tumor necrosis factor
Several studies have evaluated the capacity to augment antitumor immunity using various mouse models and have shown that GM‐CSF is one of the most potent immunostimulatory cytokines.( 1 , 2 , 3 ) GM‐CSF is an important maturation and differentiation factor for DC,( 4 ) including Langerhans cells to APC,( 5 ) enhancing their capacity to present tumor‐associated antigens to activate CTL effectively.( 6 ) Moreover, the cytotoxic activity of NK cells or CTL can be enhanced or induced by GM‐CSF‐recruited DC. Therefore, GM‐CSF has been postulated to be a critical mediator of the initial antitumor immune response.( 7 ) In the past decade, clinical trials have shown that autologous GM‐CSF gene‐transduced tumor vaccine therapy is feasible, safe, and has effective antitumor immunomodulating activity against melanoma,( 8 , 9 ) RCC,( 10 , 11 ) prostate cancer,( 12 ) pancreatic cancer( 13 ) and non‐small‐cell lung cancer.( 14 ) To generate effective GM‐CSF‐transduced tumor vaccines, it is essential to efficiently transduce tumor cells and to obtain appropriate expression of the induced gene. Serotype 5 E1, E3 gene‐deleted adenovirus encoding human GM‐CSF is one of the most promising vectors for tumor vaccines. However, adenoviral gene transduction is limited because the receptors for adenovirus serotype 5, including Coxsackie adenovirus receptor, integrin αvβ3, and integrin αvβ5, are not expressed on many tumor cells. SeV, a member of the Paramyxoviridae family, has a non‐segmented negative‐strand RNA genome and infects via sialic acid residues on surface glycoproteins or asialoglycoproteins, which are present on most mammalian cells.( 15 , 16 ) Because of the ubiquitous expression of the SeV receptor and high gene transduction capacity, SeV is emerging as a promising gene therapy tool. As SeV possesses a cytoplasmic transcription system, it can transfer exogenous genes to the cytoplasm, where genomic replication and translation are carried out by virally encoded RNA polymerase. This replication system reduces the risk of malignant transformation due to genomic integration of the vector into the host‐cell chromosome and increases the safety of this viral vector.( 17 , 18 , 19 , 20 ) To further improve the safety of the SeV vector, we used a newly developed genetically modified temperature‐sensitive mutant recombinant vector of non‐transmissible SeV (SeV/dF). This vector can self‐replicate but can not be transmitted to adjacent cells due to the lack of the F, thereby increasing the clinical application of this system.( 21 , 22 )
In the present study, we successfully transduced GFP, mouse GM‐CSF, and human GM‐CSF cDNA using SeV/dF encoding GFP (SeV/dF/GFP), mouse GM‐CSF (SeV/dF/mGM), and human GM‐CSF (SeV/dF/hGM) cDNA, respectively, into various tumor‐cell lines in vitro. Subsequently, the antitumor efficacies of irradiated SeV‐mediated GM‐CSF‐transduced RENCA (irRC/SeV/GM) cells and irradiated AdV‐mediated GM‐CSF‐transduced RENCA (irRC/AdV/GM) cells were compared in a RENCA‐bearing mouse model, demonstrating that irRC/SeV/GM cells and irRC/AdV/GM cells had equivalent antitumor effects.
Materials and Methods
Mice. Six to eight week old female immunocompetent BALB/c and C57BL/6 mice were purchased from Clea Japan, (Tokyo, Japan) and housed in the Animal Maintenance Facility at Kyushu University under specific pathogen‐free conditions. All animal experiments were approved by the Committee of the Ethics on Animal Experiments in the Faculty of Medicine, Kyushu University and carried out following the Guidelines for Animal Experiments in the Faculty of Medicine, Kyushu University, Fukuoka, Japan and The Law and Notification of the Government. Mouse experiments were carried out at least twice to confirm results.
Tumor cell lines. WEHI‐3B, a mouse myelomonocytic leukemia cell line, was kindly provided by Dr D. Metcalf (University of Melbourne), and RENCA, a mouse renal cell carcinoma cell line, was kindly provided by Dr M. Azuma (Tokyo Medical and Dental University). Mouse LLC and EL4 (lymphoma) cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). The human non‐small‐cell carcinoma cell lines PC9, H1299, H460, and LK87, were kindly provided by Dr K. Takayama (Kyushu University). The human RCC cell lines Caki‐1, Caki‐2, and A498 were purchased from the American Type Culture Collection. OSRC‐2 and VMRC‐RCW cells were purchased from the Riken BioResource Center (Ibaraki, Japan) and the Japanese Collection of Research Bioresources (Osaka, Japan), respectively. All cell lines were cultured at 37°C in a humidified atmosphere containing 5% CO2. All tumor‐cell lines except for LLC cells (which were maintained in DMEM [Gibco, New York, NY, USA]) were cultured in tissue flasks or Petri dishes containing RPMI‐1640 (Gibco) supplemented with 10% heat‐inactivated FBS and penicillin (100 units/mL), streptomycin (0.1 mg/mL), and 2 mmol/L glutamine (CM).
Preparation of non‐transmissible recombinant Sendai virus vectors. Preparation and recovery of recombinant temperature‐sensitive non‐transmissible SeV vectors harboring GFP, mouse GM‐CSF, or human GM‐CSF (SeV/dF/GFP, SeV/dF/mGM, and SeV/dF/hGM, respectively) were constructed as described previously.( 21 , 22 , 23 ) A series of SeV/dF vectors were prepared using recombinant LLC‐MK2 cells carrying the F gene (LLC‐MK2/F7). An adenovirus vector, AxCANCre, expressing Cre recombinase was used to induce the F protein into LLC‐MK2/F7 cells (referred to as LLC‐MK2/F7/A). Recombinant vaccinia virus vTF7‐3 carrying a T7 RNA polymerase was inactivated with psoralen and long‐wave ultraviolet irradiation, and then used to recover the ribonucleoprotein complex. The viral vectors were further amplified by several rounds of propagation. The titers of the recovered viral vectors were expressed as cell infectious units. These vectors were kept frozen at –80°C until use (Fig. 1a).
Figure 1.
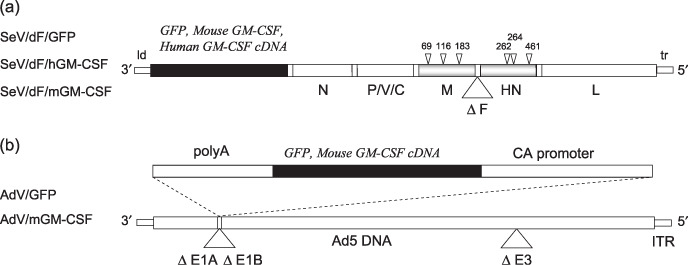
Schematic representation of the viral vectors used in the present study. (a) The three recombinant Sendai virus (SeV) vectors (SeV/dF/GFP, SeV/dF/mGM, and SeV/dF/hGM) were based on the Z‐strain of SeV. The SeV genome is delimited by two promoter regions: the leader (ld) and the trailer (tr) regions. The respective exogenous genes (green fluorescent protein [GFP], mouse granulocyte macrophage colony‐stimulating factor [GM‐CSF], and human GM‐CSF) were inserted between the ld and the open reading frame of the N gene. The SeV genes encode the envelope‐related proteins M, F, and HN, and the negative‐stranded genomic ribonucleotide‐protein complex (RNP) proteins N, P/V/C, and L. Temperature‐sensitive recombinant SeV/dF vectors lose expression of the envelope‐related M and HN genes, and have ribonucleotide substitutions in the M, HN, and L genes, as indicated by the arrowheads.( 23 ) (b) The recombinant adenovirus vectors containing the GFP or mouse GM‐CSF cDNA expression cassettes (AdV/GFP or AdV/mGM‐CSF) were constructed by homologous recombination between the expression cosmid cassette and the parental virus genome.( 24 ) The expression of these genes was driven by a CAG promoter. These replication‐defective adenovirus serotype 5 (Ad5)‐based vectors have deletions in the E1A, E1B, and E3 regions. ITR, internal terminal repeat.
Preparation of recombinant adenovirus vectors. The replication‐defective recombinant adenovirus serotype 5 vectors that lack the E1A, E1B, and E3 genes and harbor the GFP or mouse GM‐CSF genes (AdV/GFP and AdV/G, respectively) were constructed as described previously( 24 ) and kindly provided by the Riken BioResource Center. The recombinant virus vectors were propagated in 293 cells (American Type Culture Collection), and titers were determined by a plaque‐forming assay on 293 cells (TCID50 method). The adenovirus solution was stored at –80°C until use. The recombinant adenovirus vector was used as a control to compare its antitumor activity with that of the recombinant SeV vectors in the present study (Fig. 1b).
SeV/dF/GFP‐mediated green fluorescent protein transduction efficiency. One million cells of the various mouse (five) and human (nine) cell lines were seeded in six‐well plates and transduced with SeV/dF/GFP when monolayers reached 60–80% confluence. As the standard inoculation procedure for vaccination, monolayers were washed twice with PBS and overlaid with serum‐free medium containing SeV/dF/GFP at a MOI of 0, 1, 10, 50, 100, or 300. After a 90‐min incubation at 37°C, nonadsorbed virus was removed, medium containing 10% FBS was added, and the cells were incubated for over 48 h at 37°C. The transduction studies were carried out in triplicate for each MOI. Microscopy was used to detect transduced cells by GFP fluorescence. At 48 h after tranduction, the GFP‐transduced cells were analyzed for GFP expression using a FACS Calibur (BD Pharmingen, Franklin Lakes, NJ, USA).
Gene transduction and preparation for tumor vaccine cells.
Adenovirus‐mediated gene transduction. Tumor cells seeded in 10‐cm Petri dishes were washed with PBS, and 1000 µL viral solution containing 5–10% FBS was added to each dish. After a 60‐min incubation at 37°C, CM was added.
Sendai virus‐mediated gene transduction. Tumor cells seeded in 10‐cm Petri dishes were washed with PBS, and 1 mL FBS‐free viral solution was added to each dish. After a 90‐min incubation at 37°C, CM was added.
These adenovirus or SeV genetically modified tumor cells were incubated for an additional 24 h in CM, and then irradiated with 50 Gy (for mouse tumor cell lines) or 100 Gy (for human cell lines) using a 137Cs‐source γ cell 40 (Atomic Energy of Canada, Missisauga, ON, Canada). These irradiated cells were incubated for an additional 1–2 days (for RENCA vaccine cells, a 2‐day incubation), and trypsinized cells were subjected to the following tumor vaccine experiments.
Quantification of granulocyte macrophage colony‐stimulating factor production levels from granulocyte macrophage colony‐stimulating factor‐transduced tumor cells. The in vitro levels of mouse or human GM‐CSF produced from adenovirus‐ or SeV‐mediated GM‐CSF‐transduced tumor cells at the indicated MOI and times, with or without irradiation, were measured using mouse GM‐CSF and human GM‐CSF enzyme‐linked immunosorbent assay kits (BD Pharmingen), respectively.
In vitro viability of tumor cells after non‐transmissible Sendai virus transduction. Cell viability was determined by trypan blue exclusion. Two million parental RENCA or LLC cells were seeded onto 100‐mm Petri dishes, transduced with SeV/dF/GFP (MOI = 100) or SeV/dF/mGM (MOI = 100) for 90 min in serum‐free RPMI‐1640 or DMEM, cultured in CM for 48 h, and then trypsinized, diluted, and stained with 0.4% (w/v) trypan blue (Gibco). The number of trypan blue‐positive and ‐negative cells was counted under a light microscope. The percentage of cells excluding trypan blue was taken as an index of cell viability. Cell morphology was also visualized under the light microscope.
In vitro proliferation assay. For the proliferation assay, RENCA or LLC cells were cultured separately in 96‐well microplates at a concentration of 1 × 104 cells/well. After a 4–5‐h incubation with CM to promote cell adhesion, the tumor cells were washed with PBS, transduced with SeV/dF/GFP (MOI = 1, 10 or 100) or SeV/dF/mGM (MOI = 1, 10 or 100) for 90 min in serum‐free RPMI‐1640 or DMEM, and incubated for 1–4 days. At each time point (days 0, 1, 2, and 4 after SeV transduction), the number of viable cells was estimated spectrophotometrically by the incorporation of tetrazolium dye using Cell Count Reagent SF (Nacalai Tesque, Kyoto, Japan). The reagent was added, and the cells were incubated for an additional 1 h, after which an optical density value at 450 nm was determined using a microplate reader. All assays from three independent experiments were carried out in triplicate.
Experimental design of granulocyte macrophage colony‐stimulating factor‐transduced tumor vaccines. On the day of tumor challenge, RENCA cells that had been thawed from frozen stores and cultured in vitro for 1–2 weeks were trypsinized, washed twice in HBSS, and inoculated subcutaneously into the right flank of BALB/c mice (day 0, 1 × 106 cells/mouse, n = 9). Tumor volume was monitored two or three times per week. RENCA vaccine cells were inoculated subcutaneously into the left flank three times weekly, starting 7 days after tumor inoculation. The treatment groups included HBSS, irRC cells, irRC/AdV/GFP cells (MOI = 300), irRC/AdV/GM cells (MOI = 300), irRC/SeV/GFP cells (MOI = 100), or irRC/SeV/GM cells (MOI = 100).
In all tumor‐implantation experiments, each injection was diluted in 100 µL HBSS using a 1‐mL tuberculin syringe with a 27‐gauge needle. Two bisecting diameters of each tumor were measured with calipers. The tumor volumes were calculated using the formula: volume = 0.4ab 2, where a is the larger diameter and b is the smaller diameter. Changes in tumor growth were monitored two or three times per week.
Immunofluorescence analysis for costimulation‐related molecules on dendritic cells. RENCA‐bearing mice were treated with the tumor vaccinations described above. The two left axillary (vaccination side) DLN were dissected on day 2 after the first tumor vaccination and on day 7 after the third tumor vaccination. All single‐cell suspensions from DLN (n = 3/group) were prepared by mechanical homogenization. The number of cells was determined by counting crushed DLN with a hemocytometer under a light microscope. For phenotype profiles of DC, the cells were washed and blocked with antimouse CD16/32 FcR antibody for 15 min and then analyzed by triple immunostaining using the following monoclonal antibodies: fluorescein isothiocyanate‐conjugated anti‐CD86, phycoerithrin (PE)‐conjugated anti‐CD80, APC‐conjugated anti‐CD11c, and isotype controls (all from eBioscience, San Diego, CA, USA) for 30 min at room temperature. Analysis was carried out using a FACS Calibur with CellQuest software (BD Pharmingen). Data were collected on 40 000 viable cells.
In vitro cytotoxicity assay. Splenocytes were prepared from dead RENCA‐bearing mice 7 days after the third indicated tumor vaccination as described above. The vaccination groups included irRC/AdV/GFP, irRC/AdV/GM, irRC/SeV/GFP, and irRC/SeV/GM cells. To generate RENCA‐specific effector cells, splenocytes (4 × 106 cells/well) depleted of erythrocytes with ammonium chloride were cocultured with mitomycin C (100 µg/mL, 90 min, 37°C)‐treated RENCA cells at a ratio of 10:1 in 1 mL CM in 24‐well plates at 37°C in 5% CO2. Two days later, recombinant human IL‐2 (PeproTech EC, London, UK), at a concentration of 30 U in 500 µL fresh CM, was added to each well. Splenocytes were harvested on day 6 and used as effector cells in a standard 5‐h 51Cr release assay to examine antitumor cytolytic activity.( 25 ) Briefly, both RENCA cells as the tumor target and WEHI‐3B cells as the cold target (1 × 106 cells) were labeled with 3.7 MBq 100 µCi Na2 51CrO4 (PerkinElmer, Boston, MA, ISA) in 200 µL CM for 90 min at 37°C. After three washes with PBS, the labeled target cells (1 × 104 cells/well) were incubated with the effector cells for 5 h at 37°C in 96‐well round‐bottomed microtiter plates at the indicated effector:target ratios. The plates were then centrifuged at 50g for 5 min, and the radioactivity of the supernatants was measured with a γ counter from Auto Well Gamma Systems (Aloka, Tokyo, Japan). The maximum and spontaneous release were determined from samples incubated with 1% Triton X‐100 and medium alone, respectively. Cytolytic activity was calculated using the following formula: specific 51Cr release (%) = (experimental release – spontaneous release) × 100/(maximum release – spontaneous release). Assays were carried out in triplicate. The spontaneous release in all assays was <10% of the maximum release.
Detection of splenic cytotoxic T lymphocyte activity using the CD107a mobilization assay. To monitor the cytolytic activity of putative tumor‐specific CTL (CD3+CD8+ T), the CD107a mobilization assay was carried out to detect CTL degranulation.( 26 , 27 , 28 ) Briefly, splenocytes (1 × 104 cells/well) harvested as described previously were cocultured in CM with RENCA cells at a ratio of 20:1 for 72 h. The cell suspension was then collected and restimulated with or without RENCA cells or WEHI‐3B cells at the indicated ratio for an additional 5–6 h in the presence of phycoerithrin (PE)‐conjugated antimouse CD107a antibody or isotype IgG controls. Cells were washed and blocked with antimouse CD16/32 FcR antibody for 15 min and then surface stained with fluorescence‐conjugated antimouse CD3, CD8, and CD107a markers (all from eBioscience).
Interferon‐γ and interleukin‐4 ELISPOT assay for splenocytes of mice immunized with tumor vaccine cells. On day 6 after the second tumor vaccination in the therapeutic model described above, mice were killed and their splenocytes were tested for mouse IFN‐γ and IL‐4 secretion using an ELISPOT assay kit (Cytokine ELISPOT Set; BD Pharmingen). ImmunoSpot ELISPOT 96‐well plates were coated with 5 µg/mL purified antimouse IFN‐γ or antimouse IL‐4 monoclonal antibody and incubated overnight at 4°C. Wells were washed with PBS containing 0.05% Tween 20 and incubated for 2 h with blocking buffer (RPMI‐1640 with 10% FBS) at room temperature. Red blood cell (RBC)‐depleted splenocytes (1 × 105) were incubated for 20 h at 37°C with 5% CO2 in the presence or absence of irradiated RENCA cells or WEHI‐3B cells at the indicated splenocyte:irradiated tumor cell ratios (100:1 and 50:1) in a total volume of 200 µL. As a positive control, 20 ng/mL phorbol 12‐myristate 13‐acetate (PMA) (Sigma, St Louis, MO, USA), known as a mitogen for T‐cell stimulation, was added to the indicated wells. After the plates were washed, the wells were incubated with 2 µg/mL biotinylated antimouse IFN‐γ or antimouse IL‐4 monoclonal antibody for 2 h at room temperature. The plates were then washed extensively, incubated with streptavidin–horseradish peroxidase solution for 1 h at room temperature, washed twice, incubated with Final Substrate Solution (AEC substrate mixed with AEC Chromogen; BD Pharmingen), and then monitored for spot development for 5 min at room temperature. After drying, spots indicating IFN‐γ‐ or IL‐4‐secreting cells were enumerated manually under a dissecting Axiovert microscope (Zeiss, Jena, Germany) and expressed as the mean number of spots + SD of quadruplicated determinations.
Cytometric bead array and enzyme‐linked immunosorbent assays for the quantification of mouse cytokines produced from splenocytes of mice immunized with tumor vaccine cells. Similar to the ELISPOT analysis, RBC‐depleted mouse splenocytes (5 × 106) harvested on day 6 after the second tumor vaccination were incubated in the presence or absence of irradiated RENCA cells at a 10:1 ratio in a total volume of 1.0 mL at 37°C for 20 h. Cell supernatants were collected, and the concentrations of mouse IL‐2, IL‐4, IL‐5, TNF‐α, and IFN‐γ were measured using the BD Mouse Th1/Th2 Cytokine Cytometric Bead Array Kit (BD Pharmingen) according to the manufacturer's protocol. The concentration of IL‐6 was measured using a mouse IL‐6 immunoassay kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's procedure.
Immunohistochemistry. On day 6 after the second indicated tumor vaccination in the therapeutic RENCA model described above, established RENCA tumors (n = 3/group) were snap frozen by overlaying with OCT compound (Sakura Fine Technical, Tokyo, Japan). All samples were stored at –80°C until analysis. Serial cryostat (8–10‐µm) frozen sections were adhered to Superfrost slide glasses (Matsunami, Osaka, Japan), fixed in acetone at room temperature for 10 min, air‐dried, and rinsed in distilled water to remove the embedding medium. Staining for TIL was conducted following standard procedures. Briefly, sections were incubated sequentially overnight at 4°C with the appropriately diluted primary antibodies mouse CD4 (GK1.5), CD8 (53–6.7), CD11c (N418), and FoxP3 (FJK‐16s) (all from eBioscience) following the manufacturer's instructions, followed by a 1‐h incubation with biotinylated antirat or antihamster secondary antibody (eBioscience). After a 30‐min incubation with streptavidin–peroxidase (Dako Japan, Kyoto, Japan), antigen–antibody reactions were developed using 3,3′‐diaminobenzidine (Nakalai Tesque, Kyoto, Japan) substrate. Slides were washed three times with PBS between each incubation step, counterstained with Mayer's hematoxylin, and dehydrated in a sequentially graded alcohol and xylene series prior to mounting. All incubations were conducted in a humid chamber. Photographs were taken with an Axiovert microscope. The stained cells were visualized in a series of high‐power fields and counted microscopically at ×200 magnification in 30–70 high‐power fields. The percentages of positive cells were calculated, and the results are expressed as the mean ± SD.
Immunofluorescence analysis for tumor‐infiltrating cytolytic effector cells. At the same time as the in vitro cytotoxicity assay, established RENCA tumors (n = 3/group) were dissected. For flow cytometric analysis, we placed tumors in six‐well plates and minced them finely. They were transferred to 15‐mL tubes, incubated for 90 min under continuous rotation in RPMI‐1640‐containing collagenase (Gibco), and passed through a 70‐µm strainer, washed and resuspended in PBS. Viable lymphocytes were enriched and collected using centrifugation over Lympholyte‐M (Cedarlane Laboratories, Barlington, ON, Canada) at 1000× for 20 min and counted using a hemocytometer. Subsequently, to quantify the cytolytic effector cells in TIL, the cell suspensions were stained with fluorescence‐conjugated antimouse CD8, anti‐DX‐5, and antimouse CD107a antibodies for 30 min. The cells were washed twice in staining buffer and analyzed on a FACS Calibur.
Statistical analysis. For statistical analysis, a two‐tailed Student's t‐test was used. The P‐values were obtained from two‐tailed tests of statistical significance. Survival was plotted using Kaplan–Meier curves and statistical relevance was determined by a log‐rank comparison using Statview software. A probability value was considered significant when P < 0.05.
Results
Transduction efficiency of the SeV/dF/GFP vector into various human and mouse tumor‐cell lines. Nine human and five mouse tumor‐cell lines propagated in vitro were collected, transduced by SeV/dF/GFP, and examined for gene transduction efficiency. Flow cytometric analyses showed dose‐dependent GFP expression, and optimal expression was obtained at MOI of 10–100; >90% GFP‐positive tumor‐cell lines were detected at MOI over 10 (Fig. 2a,b).
Figure 2.
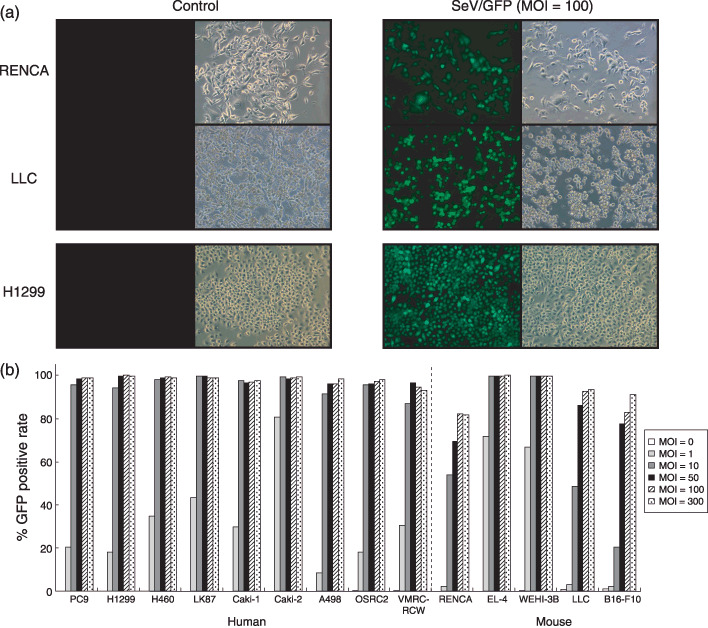
Transduction of various mouse and human cell lines with the SeV/dF/GFP vector. (a) RENCA (row 1), Lewis lung carcinoma (LLC) (row 2), and H1299 cells (row 3) were transduced with SeV/dF/GFP at a multiplicity of infection (MOI) of 100. Fluorescence microscopy of transduced cells was carried out 48 h later (green fluorescent protein [GFP] phases are displayed in column 3). The background fluorescence of transduced cells (control; column 1) was determined in non‐transduced cultures. Phase contrast pictograms are displayed in columns 2 and 4. (b) Nine mouse and five human cell lines were transduced with SeV/dF/GFP at MOI of 0, 1, 10, 50, 100, and 300. The percentage of GFP‐expressing cells was determined by flow cytometric analysis. The bar graph depicts the percentage of GFP‐positive cells at 48 h after transduction with or without SeV/dF/GFP.
Continuous in vitro granulocyte macrophage colony‐stimulating factor expression was obtained with SeV/dF/mGM‐ and SeV/dF/hGM‐transduced tumor‐cell lines. Next, we quantified that levels of GM‐CSF produced from mouse or human GM‐CSF‐transduced tumor cell lines (by SeV/dF/mGM or SeV/dF/hGM, respectively) at the indicated MOI. As shown in Figure 3a, mouse GM‐CSF levels produced from four histologically different mouse tumor cell lines that were SeV/dF/mGM tranduced were maximized to more than 300 ng/106 cells/24 h, at MOI over 50. Likewise, four human cell lines (two non‐small‐cell lung cancer and two RCC cell lines) that were transduced with SeV/dF/hGM produced sufficiently high GM‐CSF levels in a MOI‐dependent manner for at least 7 days after transduction (Fig. 3b). Taken together, these findings demonstrate that SeV/dF/GFP, SeV/dF/mGM, and SeV/dF/hGM vectors have highly successful and continuous gene transduction in various tumor cell lines.
Figure 3.
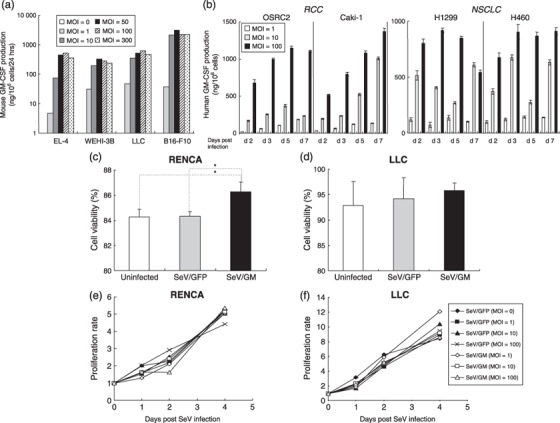
Granulocyte macrophage colony‐stimulating factor (GM‐CSF) production from mouse or human tumor cell lines transduced with SeV/dF/mGM or SeV/dF/hGM, and the viability and proliferation of Sendai virus (SeV)‐transduced cells. (a) One million cells from four mouse tumor cell lines were transduced with SeV/dF/mGM at multiplicities of infection (MOI) of 0, 1,10, 50, 100, and 300 for 90 min in serum‐free RPMI‐1640 and incubated for 10% fetal bovine serum (FBS)/RPMI in 6‐well plates for 24 h. Mouse GM‐CSF levels produced in each supernatant were measured by enzyme‐linked immunosorbent assays. (b) Human GM‐CSF levels produced by four human cell lines (two for non‐small‐cell lung cancer [NSCLC] and two for renal cell carcinoma [RCC]) transduced with SeV/dF/hGM at MOI of 1, 10, and 100 on days 2, 3, 5, and 7 after transduction were measured by enzyme‐linked immunosorbent assays. Cell viability after SeV infection was evaluated by trypan blue exclusion. (c,d) Two million parental RENCA or Lewis lung carcinoma (LLC) cells were transduced with SeV/dF/GFP (MOI = 100) or SeV/dF/mGM (MOI = 100) for 90 min, and cultured for 48 h. The number of trypan blue‐positive and ‐negative cells was counted under a light microscope, and the percentage of cells excluding trypan blue is represented as an index of cell viability. (e,f) RENCA and LLC cells were cultured separately in 96‐well microplates at 1 × 104 cells/well. They were transduced with SeV/dF/GFP (MOI = 1, 10, or 100) or SeV/dF/mGM (MOI = 1, 10, or 100) for 90 min in serum‐free medium, and cultured for 1, 2, and 4 days in RPMI‐1640 with 10% FBS or Dulbecco's modified Eagle's medium with 10% FBS, respectively. At each time point (day 0, 1, 2, or 4 after SeV transduction), the number of viable cells was estimated spectrophotometrically by the incorporation of tetrazolium dye. Representative data from three independent experiments are shown.
SeV/dF vectors did not inhibit the proliferation or viability of transduced tumor cell lines. To exclude the possibility that the SeV‐transduced exogenous genes and constitutive SeV viral components affected the survival and growth of tumor cells, parental RENCA or LLC tumor cells transduced with either SeV/dF/GFP or SeV/dF/mGM at the indicated MOI were cultured in vitro under the same conditions, and cell viability and proliferation were evaluated. SeV transduction (MOI = 100) had little effect on RENCA and LLC cell survival on day 2 when RC/SeV/G cells had rather significantly greater viability than non‐transduced RENCA and RC/SeV/GFP cells (P < 0.05) (Fig. 3c,d). Furthermore, after SeV/dF/GFP transduction at various MOI, both RENCA and LLC cells had the same proliferation rates as non‐transduced cells (control) over 4 consecutive days (P < 0.05) (Fig. 3e,f).
Effects of irradiation on granulocyte macrophage colony‐stimulating factor production from SeV/dF/G‐transduced tumor cells in vitro. To determine the effects of irradiation on the transgene expression of SeV/dF/G‐transduced tumor cells, we measured GM‐CSF production levels from SeV/dF/G‐transduced RENCA (mouse) or A549 (human) cells, with or without irradiation on day 1 (50 Gy and 100 Gy, respectively), at the indicated MOI and times. As shown in Figure 4a,b, irradiated A549 cells produced significantly higher levels of GM‐CSF than non‐irradiated A549 cells, whereas irradiated RENCA cells produced lower (day 2) or similar (days 3 and 5) levels of GM‐CSF than non‐irradiated RENCA cells. The different observations between RENCA and A549 cells seemed to be negligible, and tumor vaccine cells continued to produce abundant GM‐CSF even on day 5 after SeV transduction.
Figure 4.
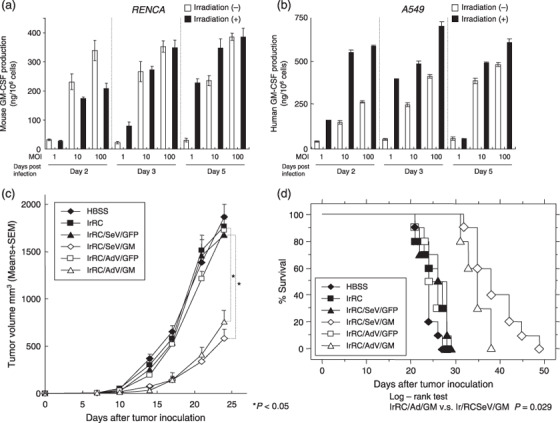
In vitro effects of irradiation on granulocyte macrophage colony‐stimulating factor (GM‐CSF) production from SeV/dF/G‐transduced tumor cells and in vivo effects of irradiated Sendai virus (SeV)‐ or adenovirus‐mediated GM‐CSF‐transduced RENCA vaccine cells against established tumors. (a,b) Levels of GM‐CSF produced from SeV/dF/mGM‐transduced mouse RENCA cells or SeV/dF/hGM‐transduced human H1299 cells with or without irradiation (on day 1) were measured comparatively at multiplicities of infection (MOI) of 1, 10, and 100 on days 2, 3, and 5 after transduction by enzyme‐linked immunosorbent assays. (c) One million of the parental RENCA cells were inoculated subcutaneously into the right flank of BALB/c mice (n = 9), followed by subcutaneous inoculation of 1 × 106 cells of the indicated RENCA vaccine in the left flank weekly for three times (on days 7, 14, 21). The treatment groups included Hank's buffered salt solution (HBSS) only, irRC, irRC/AdV/GFP, irRC/AdV/GM, irRC/SeV/GFP, and irRC/SeV/GM cells. For adenovirus‐ or SeV‐mediated transduction for preparing tumor vaccine cells, RENCA cells were transduced with adenovirus or SeV at a MOI of 300 or 100, respectively. Tumor volume was monitored twice or three times per week. (d) Survival curve of the RENCA‐bearing mice treated with tumor vaccination as described above. Bar graphs depict the means ± SEM. Significant differences are denoted with asterisks (*P < 0.05).
Therapeutic vaccination with irradiated SeV‐ or adenovirus‐mediated granulocyte macrophage colony‐stimulating factor‐transduced RENCA cells retarded established tumor development in vivo. We determined the optimal MOI of adenovirus or SeV vectors for tumor vaccination in preliminary experiments. Irradiated RENCA cells transduced with AdV/mGM or SeV/dF/mGM at a MOI of 300 or 100 produced the highest levels of mouse GM‐CSF in vitro (1250 ± 15.9 ng/106 cells/48 h and 643.98 ± 57.61 ng/106 cells/48 h, respectively), and showed the most effective antitumor efficacies in therapeutic experiments in RENCA‐bearing mice (data not shown). Next, we directly compared the in vivo antitumor therapeutic effects of tumor vaccination between irRC/AdV/GM and irRC/SeV/GM cells. Immunocompetent BALB/c mice were inoculated subcutaneously into the right flank with parental RENCA cells (day 0). On days 7, 14, and 21, 1 × 106 cells of irRC, irRC/AdV/GFP, irRC/SeV/GFP, irRC/AdV/GM, or irRC/SeV/GM were inoculated subcutaneously into the left flank as a tumor vaccination. On day 24, the growth of established RENCA tumors was significantly retarded in mice treated with irRC/AdV/GM or irRC/SeV/GM cells compared with control mice (irRC/AdV/GFP or irRC/SeV/GFP, respectively) (P < 0.05) (Fig. 4c), although tumor development was not eliminated in all treated mice. Mice treated with irRC/SeV/GM cells survived longer than those treated with irRC/AdV/GM cells (P < 0.05) (Fig. 4d), whereas the antitumor effect of irRC/SeV/GM cells was not statistically significant on day 24 compared with that of irRC/AdV/GM cells, suggesting that irRC/SeV/GM vaccination may prevent primary metastases.
Therapeutic vaccination with irradiated SeV‐ or adenovirus‐mediated granulocyte macrophage colony‐stimulating factor‐transduced RENCA cells enhanced the expression of the costimulatory markers CD80 and CD86 on dendritic cells in vivo. We next examined the expression levels of the costimulatory markers CD80 (B7‐1) and CD86 (B7‐2) on DC (CD11c+) in DLN during therapy with tumor vaccination. As shown in Figure 5a,b, the total numbers of CD80+, CD86+, and CD80+CD86+ DC in DLN from mice treated with irRC/AdV/GM or irRC/SeV/GM cells were increased significantly compared with those of mice treated with their respective controls, at the earlier phase on day 2 after first tumor vaccination. The only exception was the CD80+ DC comparison between irRC/SeV/GFP and irRC/SeV/GM therapy (P < 0.05). Furthermore, at the later phase, on day 7 after the third tumor vaccination as a booster, the total numbers of CD80+, CD86+, and CD80+CD86+ DC in DLN from mice treated with irRC/AdV/GM or irRC/SeV/GM cells were increased significantly, amplified almost 10 times compared with those observed at the earlier phase and those treated with their respective controls (P < 0.05). Similar to these results, the percentages of CD80+‐, CD86+‐, and CD80+CD86+‐stained cells on DC from mice treated with irRC/AdV/GM or irRC/SeV/GM cells were also higher than those of mice treated with their respective controls at the same two time points (data not shown). Collectively, these results suggest that the costimulatory markers were markedly upregulated by GM‐CSF‐transduced RENCA vaccines.
Figure 5.
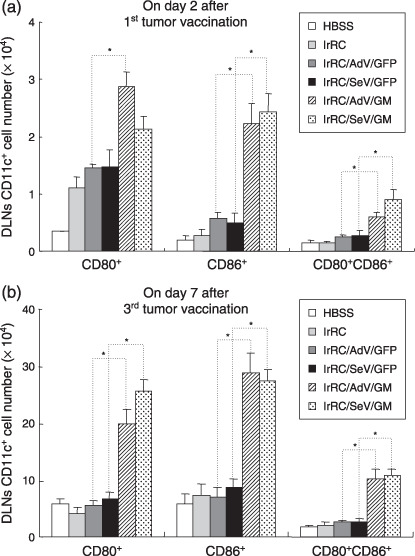
Enhanced recruitment of activated dendritic cells (DC) in draining lymph nodes (DLN) induced with Sendai virus (SeV)‐ or adenovirus (AdV)‐mediated GM‐CSF‐transduced RENCA vaccine cells. The two left axillary DLN were harvested on (a) day 2 after the first tumor vaccination and (b) day 7 after the third tumor vaccination. The total numbers of CD11c+ cells (DC) expressing costimulatory markers (CD80+, CD86+, CD80+ CD86+) in DLN of mice treated with the indicated tumor vaccination are shown. Bar graphs depict the means ± SEM. Significant differences are denoted with asterisks (*P < 0.05). Representative data from two independent experiments are shown.
Splenocytes from mice treated with irRC/AdV/GM or irRC/SeV/GM cells showed tumor‐specific cytotoxicitiy against RENCA cells. To compare the in vitro cytolytic activity against RENCA cells, we next carried out a 51Cr‐release assay using splenocytes from each immunized mouse. Prior to the cytotoxicity assay, we evaluated MHC class I (H‐2Kd) surface expression on RENCA and WEHI‐3B tumor cells by flow cytometry, as MHC class I expression on tumor cells is required for CTL recognition in cancer immunotherapy.( 29 ) MHC class I expression was high on RENCA cells and moderate on WEHI‐3B cells (data not shown). Mice were killed 7 days after the last‐indicated tumor vaccination and splenocytes were harvested. Prepared splenocytes were restimulated in vitro with mitomycin (MMC)‐treated RENCA cells for 6 days, and cytolytic activity was measured. The results showed that cytotoxicity against RENCA cells from mice treated with irRC/AdV/GM or irRC/SeV/GM cells were elevated and superior to those from mice treated with their respective controls. Intriguingly, a relatively low level of cytotoxicity was observed in mice treated with irRC/SeV/GFP cells (Fig. 6a). In contrast, when control syngeneic WEHI‐3B cells were used as a target, they showed negative results (Fig. 6b). These results indicate that GM‐CSF but not GFP substantially contributes to the induction of RENCA‐specific antitumor activity.
Figure 6.
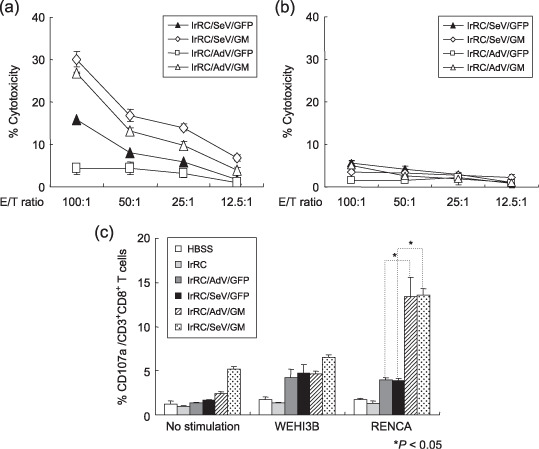
In vitro cytotoxicity assays and the effector cells contributing to the antitumor effects induced by the irradiated Sendai virus (SeV)‐ or adenovirus (AdV)‐mediated granulocyte macrophage colony‐stimulating factor (GM‐CSF)‐transduced RENCA vaccine cells. (a,b) Seven days after the third tumor vaccination with irRC/AdV/GFP, irRC/SeV/GFP, irRC/AdV/GM, or irRC/SeV/GM cells, mice were killed to harvest splenocytes. Splenocytes were restimulated with mitomycin C‐treated RENCA cells for 6 days and used as effector cells in a 51Cr‐release assay. (a) 51Cr‐labeled RENCA cells used as target cells and (b) WEHI‐3B cells used as non‐specific target cells were cocultured with effector cells at the indicated effector to target (E : T) ratios for 5 h. (c) CD107a mobilization of splenic CD8+ T cells from mice treated with the indicated tumor vaccinations. Splenocytes were restimulated in vitro for 72 h with RENCA cells. The cells were then cultured with or without RENCA or WEHI‐3B cells for an additional 5–6 h. The percentages of CD107a‐expressing CD3+CD8+ T cells are indicated. The values represent the means ± SEM of the percentage cytotoxicity. Representative data from three independent experiments are shown.
In order to examine whether the cytolytic effector cells consisted mainly of CD8+ T cells (CTL), a CD107a (lysosomal membrane glycoprotein 1) mobilization assay (a surrogate for lytic degranulation)( 26 , 27 , 28 ) was carried out using restimulated splenocytes with RENCA cells in vitro. CD107a mobilization of CD8+ T cells against RENCA cells was increased significantly in mice treated with irRC/AdV/GM and irRC/SeV/GM cells compared with those treated with their respective controls, whereas the mobilization of CD8+ T cells incubated with WEHI‐3B cells remained at basal levels (P < 0.05) (Fig. 6c). The results suggest that tumor‐specific CD8+ T cells were generated in vivo and possessed the capacity to release an abundant amount of cytolytic granules, including perforin and granzyme B.
In vitro inflammatory cytokine production profile of splenocytes from mice treated with granulocyte macrophage colony‐stimulating factor‐transduced RENCA vaccine cells. For characterization of the immunomodulatory effects of GM‐CSF‐transduced RENCA vaccination, we examined in vitro inflammatory cytokine production of splenocytes cocultured with or without irradiated RENCA cells using immunocompetent mice immunized with RENCA vaccine cells. First, to quantify the number of IL‐4‐ or IFN‐γ‐producing splenocytes, splenocytes harvested from mice either untreated or treated with irRC/AdV/GFP, irRC/SeV/GFP, irRC/AdV/GM, or irRC/SeV/GM cells were subjected to an in vitro ELISPOT assay for IFN‐γ and IL‐4. When cocultured in the presence of irradiated RENCA cells, the number of splenocytes from RENCA‐bearing mice treated with either irRC/AdV/GM or irRC/SeV/GM cells that produced both IFN‐γ and IL‐4 was significantly higher than those from each control group (irRC/AdV/GFP or irRC/SeV/GFP) (P < 0.05). In addition, the number of splenocytes from mice treated with irRC/SeV/GM cells that produced IFN‐γ was significantly higher than those from mice treated with irRC/AdV/GM cells (P < 0.05) (Fig. 7a). These enhanced IFN‐γ‐producing cells were tumor‐antigen specific, as the numbers of IFN‐γ‐ or IL‐4‐producing cells in the presence of an irrelevant antigen (irradiated WEHI‐3B cells) were as low as those in the absence of antigen (Fig. 7b).
Figure 7.
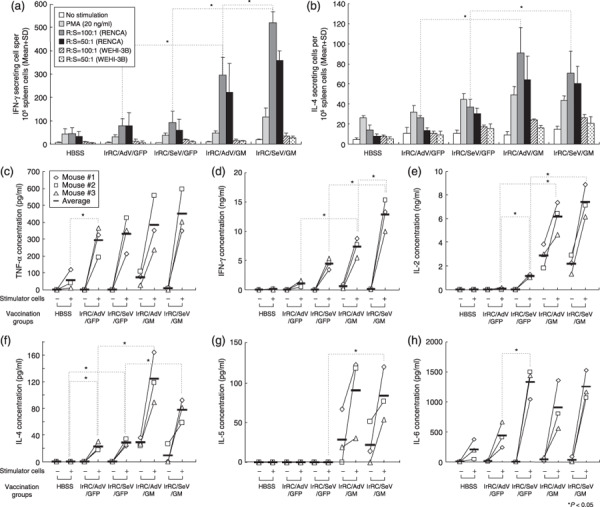
In vitro inflammatory cytokine production profiles of splenocytes from mice treated with granulocyte macrophage colony‐stimulating factor (GM‐CSF)‐transduced RENCA vaccine cells. (a,b) Interferon (IFN)‐γ and interleukin (IL)‐4 production by splenocytes from mice immunized with irRC/AdV/GM or irRC/SeV/GM cells were evaluated using mouse (a) IFN‐γ and (b) IL‐4 ELISPOT assays. Ten thousand splenocytes, as responder cells (R), from RENCA‐bearing mice treated with the indicated tumor vaccines were incubated for 20 h with or without stimulator cells (S) or PMA at the indicated R : S ratios. Bound cytokines were visualized by incubation with biotinylated anti‐IFN‐γ and anti‐IL‐4 monoclonal antibodies, followed by streptavidin–horseradish peroxidase, and the premixed peroxidase substrate 3‐amino‐9‐ethylcarbazole (AEC). Results are expressed as the mean number of spot‐forming cells + SD from quadruplicate determinations per 1 × 105 splenocytes. (c–h) Splenocytes were harvested from mice 5 days after the last inoculation of the indicated tumor vaccines and then cocultured with or without irradiated RENCA stimulator cells. Twenty hours after the mixed lymphocyte and tumor incubation, the concentrations of mouse (c) tumor necrosis factor (TNF)‐α, (d) IFN‐γ, (e) IL‐2, (f) IL‐4, (g) IL‐5, and (h) IL‐6 in the culture supernatants were measured by (c–g) cytometric bead array and (h) enzyme‐linked immunosorbent assays. *P < 0.05 represents significant difference compared with indicated group.
We next quantified various inflammatory cytokines produced by splenocytes from mice either left untreated (HBSS only) or treated with irRC/AdV/GFP, irRC/SeV/GFP, irRC/AdV/GM, or irRC/SeV/GM cells. After a 20‐h coculture with or without irradiated RENCA cells, supernatants were collected, and the following cytokines were measured: IL‐2, IL‐4, IL‐5, IL‐6, IFN‐γ, and TNF‐α. The IFN‐γ, IL‐2 (Th1), and IL‐4 (Th2) levels produced by splenocytes in the presence of stimulator cells, from mice treated with irRC/AdV/G or irRC/SeV/G cells were significantly higher than those from their respective GFP controls (P < 0.05) (Fig. 7d–f). In particular, only the IFN‐γ production of restimulated splenocytes from mice treated with irRC/SeV/GM cells was greater than those treated with irRC/AdV/GM cells, which were similar to the results of the ELISPOT assay (P < 0.05) (Fig. 7d). Intriguingly, both the IL‐2 and IL‐6 production levels of restimulated splenocytes from mice treated with irRC/SeV/GFP cells were significantly higher than those treated with irRC/AdV/GFP cells (P < 0.05) (Fig. 7e,h). Although the TNF‐α and IL‐6 production levels of restimulated splenocytes from all mice treated (except for the HBSS‐treated group) were markedly elevated, there was no significant difference between those seen in each GFP‐treated group and GM‐CSF‐treated group (Fig. 7c,h). The IL‐5 production levels of restimulated splenocytes from mice treated with irRC/AdV/GM or irRC/SeV/GM cells were higher than those treated with their respective GFP controls (irRC/SeV/GM vs irRC/SeV/GFP; P < 0.05) (Fig. 7g).
Characterization of tumor‐infiltrating leukocytes induced by irradiated granulocyte macrophage colony‐stimulating factor‐transduced RENCA vaccine cells. The tumor microenvironment is composed of an elaborate mixture of tumor‐ and host‐derived cells. To identify the key immune cells that induced the antitumor effects by irradiated GM‐CSF‐transduced RENCA vaccine cells, the distribution profiles of TIL in RENCA‐bearing mice either untreated or treated with the indicated vaccination (irRC/AdV/GFP, irRC/SeV/GFP, irRC/AdV/GM, or irRC/SeV/GM) were assessed by immunohistochemistry. The results showed more infiltrating CD8+ T cells in tumors of mice treated with irRC/AdV/GM or irRC/SeV/GM cells than those treated with their respective controls (irRC/AdV/GFP and irRC/SeV/GFP) (P < 0.05). In addition, more infiltrating CD4+ T cells were observed in tumors of mice treated with irRC/SeV/GM cells than in those treated with irRC/SeV/GFP cells (P < 0.05) (Fig. 8a). CD11c (DC) and FoxP3 (regulatory T cells) staining was not significantly different among each tumor vaccination group.
Figure 8.
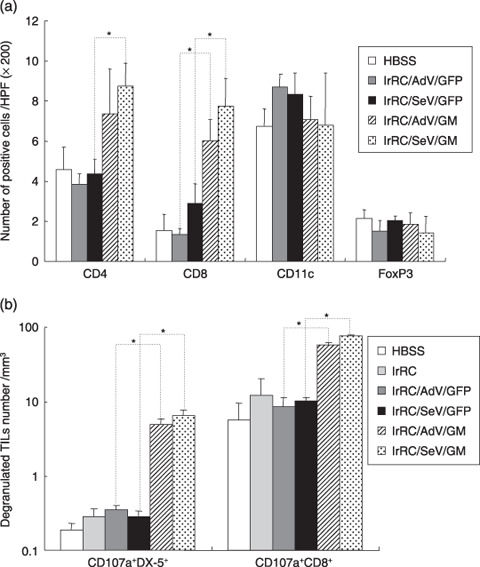
Immunophenotypic analyses of tumor‐infiltrating leukocytes by immunohistochemistry and flow cytometry. (a) RENCA‐bearing mice were either left untreated (HBSS) or treated with indicated tumor vaccine cells (irRC/AdV/GFP, irRC/SeV/GFP, irRC/AdV/G, and irRC/SeV/G). Resected RENCA tumors were then subjected to immunohistochemical evaluation. To evaluate the distribution of CD4+ T, CD8+ T, CD11c+, and FoxP3+ cells in tumors, positively stained cells were enumerated microscopically at ×200 magnification in 30–70 high‐power fields. (b) Enriched viable lymphocytes from mice treated with the tumor vaccination indicated were stained with anti‐CD8, anti‐DX‐5, and antimouse CD107a antibodies and then subjected to flow cytometry. The cell density (divided by the indicated tumor volume [mm3]) of natural killer cells or CD8+ T cells coexpressing degranulated marker of CD107a (CD107a+NK+ or CD107a+CD8+ T cells) in tumor‐infiltrating leukocytes is shown. Bar graphs depict the means ± SEM. Significant differences are denoted with asterisks (*P < 0.05).
To confirm whether the tumor‐infiltrating effector cells, CD8+ T cells (CTL), and NK (DX‐1+) cells were in functionally cytolytic conditions, we next quantified comparatively the cell number density of CD107a‐expressing CD8+ T and NK cells in tumors during therapeutic tumor vaccination. As shown in Figure 8b, tumor vaccination with irRC/AdV/GM or irRC/SeV/GM cells induced significantly enhanced recruitment of both CD107a+CD8+ T cells and CD107a+ NK cells into local tumors compared with those induced by their respective controls (irRC/AdV/GFP and irRC/SeV/GFP) (P < 0.05).
Discussion
In the present study, we demonstrated that non‐transmissible SeV‐mediated GM‐CSF‐transduced RENCA tumor vaccine cells were effective and well tolerated in mouse therapeutic tumor models, indicating that this novel SeV is a promising gene‐delivery vector for clinical GM‐CSF‐transduced tumor vaccines. The in vitro GM‐CSF levels produced from various mouse and human tumor cell lines transduced with SeV/dF/mGM or SeV/dF/hGM were equivalent to those transduced with the corresponding adenoviral vectors, which are known to efficiently deliver GM‐CSF transgenes (in vitro adenovirus transduction; data not shown). Interestingly, although the in vitro GM‐CSF level produced by irRC/SeV/G cells (643.98 ± 57.61 ng/106 cells/48 h) was approximately half the amount of that produced by irRC/AdV/GM cells (AdV/G 1250 ± 15.9 ng/106 cells/48 h), irRC/SeV/GM cells exerted an equivalent antitumor effect compared to that of irRC/AdV/GM cells, and resulted in longer survival than that of irRC/AdV/GM cells in the RENCA‐bearing mouse model. Our finding that SeV transduction itself did not have inhibitory effects on the proliferation or viability of RENCA cells in vitro could further support the in vivo antitumor effect.
The key role of GM‐CSF as an immunomodulator is its ability to recruit and activate functional APC( 4 ) such as DC.( 2 , 3 , 6 ) In the present study, vaccination with irradiated SeV‐ or adenovirus‐mediated GM‐CSF‐transduced RENCA cells enhanced the expression of the costimulatory markers CD80 and CD86 on DC in DLN, which elicited lymphadenopathy with marked expansion of these activated DC numbers, whereas differences in the numbers observed between irRC/AdV/GM and irRC/SeV/GM cells were mild. Besides, the total cell numbers of NK (DX‐5+), CD3+CD4+ T, and CD3+CD8+ T cells in DLN were also increased when treated with these GM‐CSF‐transduced RENCA vaccine cells (data not shown). Hence, the ability of overexpressed endogenous GM‐CSF to recruit massive numbers of mature DC with enhanced tumor antigen presentation and immunostimulatory functions( 1 , 6 , 30 ) as well as other lymphocytes into DLN presumably, in a coordinated manner, activated succeeding effector cells, partially because these relative increases in both CD80 and CD86 expression on DC may lessen the amount of antigen required to trigger T‐cell proliferation.( 31 )
Our results from the in vitro cytotoxicity assay, ELISPOT, and enzyme‐linked immunosorbent assay using splenocytes showed that the tumor‐specific antitumor immunity of the GM‐CSF‐based immunotherapy was induced through cytolytic CTL and systemic greater production of immunostimulatory cytokines, including IL‐2, IFN‐γ (Τh1 cytokines),( 32 , 33 ) IL‐4,( 33 , 34 ) IL‐5,( 32 , 34 ) IL‐6 (Th2 cytokines),( 35 ) and TNF‐α. These findings, taken together with studies assessing the efficacy of GM‐CSF‐based tumor vaccines in cytokine‐deficient mice,( 36 ) suggest principal roles for both Th1 and Th2 immune responses in provoking the antitumor effects of GM‐CSF. Among the cytokines measured, elevated IL‐5 production induced by GM‐CSF‐based tumor vaccines suggests that GM‐CSF systemically activates eosinophils, which are considered to be involved in GM‐CSF‐induced antitumor responses.( 9 , 11 , 34 , 37 )
Our immunohistochemical analysis showed a significant increase in infiltrating CD4+ and CD8+ T cells in tumors treated with irRC/SeV/GM or irRC/AdV/GM cells compared to those treated with their respective GFP controls, consistent with the previous findings that underscored the significance of the number of tumor‐infiltrating CD4+ or CD8+ T lymphocytes in antitumor immunity.( 36 , 38 , 39 ) In particular, Dranoff et al. reported that CD4+ and CD8+ T cells are required for optimal antitumor efficacy elicited by GM‐CSF‐producing tumor vaccines.( 1 , 3 , 40 ) The achievement of immunotherapeutic strategies against cancer depends on the generation of tumor‐specific T cells, which can efficiently enter the tumor tissues and interact with target tumor cells mainly by their releasing perforin and granzyme B.( 26 , 27 , 41 , 42 ) Indeed, significantly increased numbers of CD107a‐expressing CD8+ T and NK cells were observed in tumors of mice vaccinated with irRC/AdV/GM or irRC/SeV/GM cells (Fig. 8b). In conjunction with our results from the cytotoxicity assay, these results indicate that the GM‐CSF‐based tumor vaccines promoted to generate both of the functional CTL (adaptive immunity) and NK cells (innate immunity), and these cells are considered to interact to induce antitumor effects in vivo.( 9 , 37 , 40 , 43 )
Whereas remarkable differences between SeV‐mediated gene transduction and adenovirus‐mediated gene transduction were not observed in our immunological assays, significantly higher IL‐6 production by restimulated splenocytes was observed when they were treated with the vaccination of irRC/SeV/GFP cells compared with irRC/AdV/GFP cells. IL‐6 is a multifunctional cytokine that controls various immune responses, including inflammation.( 35 ) Grohmann et al. reported that IL‐6 plays a critical role in mediating the effects of CD40 ligation in DC and enhancing their immunogenicity.( 44 ) Kurooka et al. reported that HVJ‐E (Sendai virus‐envelope) alone eradicates tumors, and speculated that the mechanisms of the antitumor effect of HVJ‐E may include a rescue from regulatory T cell‐mediated immunosuppression, through dominant IL‐6 secretion from DC stimulated with F glycoprotein of HVJ‐E.( 45 , 46 ) Accordingly, our finding that upregulated IL‐6 production by splenocytes (including DC) when treated with SeV/dF‐based vectors may be one of the advantages of SeV/dF‐based vectors over adenovirus‐based vectors and may provide us with an encouraging rationale to use them for cancer immune gene therapy. Another expected advantage of the use of SeV/dF‐based vectors is that SeV/dF is considered to be safe as it can mediate gene transfer to a cytoplasmic location, evading possible malignant transformation due to nuclear mutations of host cells.( 18 ) Furthermore, actual preclinical achievements in DC‐based tumor immunotherapy( 22 , 25 ) and cancer gene therapy( 47 , 48 ) using novel SeV vectors have recently been reported.
Despite these beneficial characteristics of SeV, the use of SeV vectors as well as adenovirus vectors has been limited by elevated immune responses to their viral components when administered in vivo. However, in the present study, the method of ex vivo SeV/dF transduction into autologous tumor cells followed by the removal of nonabsorbed virus could avoid or minimize the intensive immune responses to SeV/dF in vivo. Before translating the SeV/dF‐mediated autologous GM‐CSF‐transduced vaccine into a clinical setting, we need to confirm the ex vivo transduction efficiencies and the GM‐CSF levels produced by primary specimens resected from several cancer patients. Our study showed that irradiated A549 (human lung cancer) cells produced significantly higher levels of GM‐CSF in vitro than did non‐irradiated A549 cells (Fig. 4b). It could be explained by the report that irradiation enhanced the transcription of various genes, including p53 and nuclear factor‐κB,( 49 ) as well as their transfection and transduction efficiencies and transgene integration.( 50 , 51 , 52 ) The different effects of irradiation between RENCA and A549 cells are inferred to be dependent on the type or species of tumor cells. In some cases, irradiation may be useful to produce GM‐CSF‐transduced tumor vaccines from patients’ tumors.( 53 )
In conclusion, we have demonstrated that non‐transmissible SeV‐mediated GM‐CSF‐transduced tumor vaccines have antitumor effects on RENCA‐bearing mice. Consequently, our results imply, for the first time, that non‐transmissible SeV/dF/G could emerge as an alternative, safe vector for cytoplasmic GM‐CSF‐gene‐transduced tumor immunotherapy, although further preclinical investigations using various tumor‐cell types are needed.
Acknowledgments
We thank Mrs Michiyo Okada for excellent technical assistance. We thank Mrs Moboru Kohno, Eisaku Suzuki, Akihiro Tagawa, Takumi Kanaya, Ms Natsuko Kurosawa, and Dr Takashi Hironaka for the preparation and production of SeV vectors. This work was supported by grants from the Ministry of Health, Labour, and Welfare and the Ministry of Education, Culture, Sports, Science and Technology, and Japan Society for the Promotion of Science, Japan.
References
- 1. Dranoff G, Jaffee E, Lazenby A et al . Vaccination with irradiated tumor cells engineered to secrete murine granulocyte‐macrophage colony‐stimulating factor stimulates potent, specific, and long‐lasting anti‐tumor immunity. Proc Natl Acad Sci USA 1993; 90: 3539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mach N, Dranoff G. Cytokine‐secreting tumor cell vaccines. Curr Opin Immunol 2000; 12: 571–5. [DOI] [PubMed] [Google Scholar]
- 3. Dranoff G. GM‐CSF‐based cancer vaccines. Immunol Rev 2002; 188: 147–54. [DOI] [PubMed] [Google Scholar]
- 4. Tazi A, Bouchonnet F, Grandsaigne M, Boumsell L, Hance AJ, Soler P. Evidence that granulocyte macrophage‐colony‐stimulating factor regulates the distribution and differentiated state of dendritic cells/Langerhans cells in human lung and lung cancers. The J Clin Invest 1993; 91: 566–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inaba K, Inaba M, Romani N et al . Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony‐stimulating factor. J Exp Med 1992; 176: 1693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller G, Pillarisetty VG, Shah AB, Lahrs S, Xing Z, DeMatteo RP. Endogenous granulocyte‐macrophage colony‐stimulating factor overexpression: in vivo results in the long‐term recruitment of a distinct dendritic cell population with enhanced immunostimulatory function. J Immunol 2002; 169: 2875–85. [DOI] [PubMed] [Google Scholar]
- 7. Steinman RM, Witmer‐Pack M, Inaba K. Dendritic cells: antigen presentation, accessory function and clinical relevance. Adv Exp Med Biol 1993; 329: 1–9. [DOI] [PubMed] [Google Scholar]
- 8. Ellem KA, O’Rourke MG, Johnson GR et al . A case report: immune responses and clinical course of the first human use of granulocyte/macrophage‐colony‐stimulating‐factor‐transduced autologous melanoma cells for immunotherapy. Cancer Immunol Immunother 1997; 44: 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soiffer R, Lynch T, Mihm M et al . Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte‐macrophage colony‐stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc Natl Acad Sci USA 1998; 95: 13 141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simons JW, Jaffee EM, Weber CE et al . Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte‐macrophage colony‐stimulating factor gene transfer. Cancer Res 1997; 57: 1537–46. [PMC free article] [PubMed] [Google Scholar]
- 11. Tani K, Azuma M, Nakazaki Y et al . Phase I study of autologous tumor vaccines transduced with the GM‐CSF gene in four patients with stage IV renal cell cancer in Japan: clinical and immunological findings. Mol Ther 2004; 10: 799–816. [DOI] [PubMed] [Google Scholar]
- 12. Simons JW, Mikhak B, Chang JF et al . Induction of immunity to prostate cancer antigens: results of a clinical trial of vaccination with irradiated autologous prostate tumor cells engineered to secrete granulocyte‐macrophage colony‐stimulating factor using ex vivo gene transfer. Cancer Res 1999; 59: 5160–8. [PubMed] [Google Scholar]
- 13. Jaffee EM, Hruban RH, Biedrzycki B et al . Novel allogeneic granulocyte‐macrophage colony‐stimulating factor‐secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol 2001; 19: 145–56. [DOI] [PubMed] [Google Scholar]
- 14. Salgia R, Lynch T, Skarin A et al . Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte‐macrophage colony‐stimulating factor augments antitumor immunity in some patients with metastatic non‐small‐cell lung carcinoma. J Clin Oncol 2003; 21: 624–30. [DOI] [PubMed] [Google Scholar]
- 15. Nagai Y. Paramyxovirus replication and pathogenesis. Reverse genetics transforms understanding. Rev Med Virol 1999; 9: 83–99. [DOI] [PubMed] [Google Scholar]
- 16. Markwell MA, Svennerholm L, Paulson JC. Specific gangliosides function as host cell receptors for Sendai virus. Proc Natl Acad Sci USA 1981; 78: 5406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bitzer M, Armeanu S, Lauer UM, Neubert WJ. Sendai virus vectors as an emerging negative‐strand RNA viral vector system. J Gene Med 2003; 5: 543–53. [DOI] [PubMed] [Google Scholar]
- 18. Yonemitsu Y, Kitson C, Ferrari S et al . Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat Biotechnol 2000; 18: 970–3. [DOI] [PubMed] [Google Scholar]
- 19. Moyer SA, Baker SC, Lessard JL. Tubulin: a factor necessary for the synthesis of both Sendai virus and vesicular stomatitis virus RNAs. Proc Natl Acad Sci USA 1986; 83: 5405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaneda Y, Nakajima T, Nishikawa T et al . Hemagglutinating virus of Japan (HVJ) envelope vector as a versatile gene delivery system. Mol Ther 2002; 6: 219–26. [DOI] [PubMed] [Google Scholar]
- 21. Li HO, Zhu YF, Asakawa M et al . A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J Virol 2000; 74: 6564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoneyama Y, Ueda Y, Akutsu Y et al . Development of immunostimulatory virotherapy using non‐transmissible Sendai virus‐activated dendritic cells. Biochem Biophys Res Commun 2007; 355: 129–35. [DOI] [PubMed] [Google Scholar]
- 23. Inoue M, Tokusumi Y, Ban H et al . Nontransmissible virus‐like particle formation by F‐deficient sendai virus is temperature sensitive and reduced by mutations in M and HN proteins. J Virol 2003; 77: 3238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abe J, Wakimoto H, Yoshida Y, Aoyagi M, Hirakawa K, Hamada H. Antitumor effect induced by granulocyte/macrophage‐colony‐stimulating factor gene‐modified tumor vaccination: comparison of adenovirus‐ and retrovirus‐mediated genetic transduction. J Cancer Res Clin Oncol 1995; 121: 587–92. [DOI] [PubMed] [Google Scholar]
- 25. Shibata S, Okano S, Yonemitsu Y et al . Induction of efficient antitumor immunity using dendritic cells activated by recombinant Sendai virus and its modulation by exogenous IFN‐β gene. J Immunol 2006; 177: 3564–76. [DOI] [PubMed] [Google Scholar]
- 26. Rubio V, Stuge TB, Singh N et al . Ex vivo identification, isolation and analysis of tumor‐cytolytic T cells. Nat Med 2003; 9: 1377–82. [DOI] [PubMed] [Google Scholar]
- 27. Betts MR, Brenchley JM, Price DA et al . Sensitive and viable identification of antigen‐specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Meth 2003; 281: 65–78. [DOI] [PubMed] [Google Scholar]
- 28. Zhou P, L’Italien L, Hodges D, Schebye XM. Pivotal roles of CD4+ effector T cells in mediating agonistic anti‐GITR mAb‐induced‐immune activation and tumor immunity in CT26 tumors. J Immunol 2007; 179: 7365–75. [DOI] [PubMed] [Google Scholar]
- 29. Vertuani S, De Geer A, Levitsky V, Kogner P, Kiessling R, Levitskaya J. Retinoids act as multistep modulators of the major histocompatibility class I presentation pathway and sensitize neuroblastomas to cytotoxic lymphocytes. Cancer Res 2003; 63: 8006–13. [PubMed] [Google Scholar]
- 30. Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow‐derived cells in presenting MHC class I‐restricted tumor antigens. Science 1994; 264: 961–5. [DOI] [PubMed] [Google Scholar]
- 31. Murtaza A, Kuchroo VK, Freeman GJ. Changes in the strength of co‐stimulation through the B7/CD28 pathway alter functional T cell responses to altered peptide ligands. Int Immunol 1999; 11: 407–16. [DOI] [PubMed] [Google Scholar]
- 32. Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, Dranoff G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte‐macrophage colony‐stimulating factor or Flt3‐ligand. Cancer Res 2000; 60: 3239–46. [PubMed] [Google Scholar]
- 33. Nakazaki Y, Hase H, Inoue H et al . Serial analysis of gene expression in progressing and regressing mouse tumors implicates the involvement of RANTES and TARC in antitumor immune responses. Mol Ther 2006; 14: 599–606. [DOI] [PubMed] [Google Scholar]
- 34. Ellyard JI, Simson L, Parish CR. Th2‐mediated anti‐tumour immunity: friend or foe? Tissue Antigens 2007; 70: 1–11. [DOI] [PubMed] [Google Scholar]
- 36. Hung K, Hayashi R, Lafond‐Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J Exp Med 1998; 188: 2357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chu Y, Xia M, Lin Y et al . Th2‐dominated antitumor immunity induced by DNA immunization with the genes coding for a basal core peptide PDTRP and GM‐CSF. Cancer Gene Ther 2006; 13: 510–19. [DOI] [PubMed] [Google Scholar]
- 38. Baskar S, Glimcher L, Nabavi N, Jones RT, Ostrand‐Rosenberg S. Major histocompatibility complex class II+B7‐1+ tumor cells are potent vaccines for stimulating tumor rejection in tumor‐bearing mice. J Exp Med 1995; 181: 619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mumberg D, Monach PA, Wanderling S et al . CD4+ T cells eliminate MHC class II‐negative cancer cells in vivo by indirect effects of IFN‐γ. Proc Natl Acad Sci USA 1999; 96: 8633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grohmann U, Fallarino F, Bianchi R et al . IL‐6 inhibits the tolerogenic function of CD8α+ dendritic cells expressing indoleamine 2,3‐dioxygenase. J Immunol 2001; 167: 708–14. [DOI] [PubMed] [Google Scholar]
- 35. Hirano T. Interleukin 6 and its receptor: Ten years later. Int Rev Immunol 1998; 16: 249–84. [DOI] [PubMed] [Google Scholar]
- 40. Dranoff G. GM‐CSF‐secreting melanoma vaccines. Oncogene 2003; 22: 3188–92. [DOI] [PubMed] [Google Scholar]
- 41. Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev 2003; 3: 35–45. [DOI] [PubMed] [Google Scholar]
- 42. Gilboa E. The promise of cancer vaccines. Nat Rev 2004; 4: 401–11. [DOI] [PubMed] [Google Scholar]
- 43. Hege KM, Jooss K, Pardoll D. GM‐CSF gene‐modified cancer cell immunotherapies: of mice and men. Int Rev Immunol 2006; 25: 321–52. [DOI] [PubMed] [Google Scholar]
- 45. Kurooka M, Kaneda Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res 2007; 67: 227–36. [DOI] [PubMed] [Google Scholar]
- 46. Suzuki H, Kurooka M, Hiroaki Y, Fujiyoshi Y, Kaneda Y. Sendai virus F glycoprotein induces IL‐6 production in dendritic cells in a fusion‐independent manner. FEBS Lett 2008; 582: 1325–9. [DOI] [PubMed] [Google Scholar]
- 47. Iwadate Y, Inoue M, Saegusa T et al . Recombinant Sendai virus vector induces complete remission of established brain tumors through efficient interleukin‐2 gene transfer in vaccinated rats. Clin Cancer Res 2005; 11: 3821–7. [DOI] [PubMed] [Google Scholar]
- 48. Kinoh H, Inoue M, Washizawa K et al . Generation a recombinant Sendai virus that is selectively activated and lyses human tumor cells expressing matrix metalloproteinases Gene Ther 2004; 11: 1137–45. [DOI] [PubMed] [Google Scholar]
- 49. Vereecque R, Saudemont A, Wickham TJ et al . Gamma‐irradiation enhances transgene expression in leukemic cells. Gene Ther 2003; 10: 227–33. [DOI] [PubMed] [Google Scholar]
- 50. Stevens CW, Zeng M, Cerniglia GJ. Ionizing radiation greatly improves gene transfer efficiency in mammalian cells. Human Gene Ther 1996; 7: 1727–34. [DOI] [PubMed] [Google Scholar]
- 51. Zeng M, Cerniglia GJ, Eck SL, Stevens CW. High‐efficiency stable gene transfer of adenovirus into mammalian cells using ionizing radiation. Human Gene Ther 1997; 8: 1025–32. [DOI] [PubMed] [Google Scholar]
- 52. Teh BS, Aguilar‐Cordova E, Vlachaki MT et al . Combining radiotherapy with gene therapy (from the bench to the bedside): a novel treatment strategy for prostate cancer. Oncologist 2002; 7: 458–66. [DOI] [PubMed] [Google Scholar]
- 53. Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High‐dose granulocyte‐macrophage colony‐stimulating factor‐producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res 2004; 64: 6337–43. [DOI] [PubMed] [Google Scholar]