

Abstract
Alterations of DNA methylation, which result in chromosomal instability and silencing of tumor‐related genes, are among the most consistent epigenetic changes observed in human cancers. Analysis of tissue specimens has revealed that DNA methylation alterations participate in multistage carcinogenesis, even from the early and precancerous stages, especially in association with chronic inflammation and/or persistent viral infection, such as chronic hepatitis or liver cirrhosis resulting from infection with hepatitis B or C virus. DNA methylation alterations can account for the histological heterogeneity and clinicopathological diversity of human cancers. Overexpression of DNA methyltransferase 1 is not a secondary result of increased cell proliferative activity, but is significantly correlated with accumulation of DNA hypermethylation in CpG islands of tumor‐related genes. Alteration of DNA methyltransferase 3b splicing may result in chromosomal instability through DNA hypomethylation in pericentromeric satellite regions. Genome‐wide analysis of DNA methylation status has revealed that the DNA methylation profile at the precancerous stage is basically inherited by the corresponding cancers developing in individual patients. DNA methylation status is not simply altered at the precancerous stage; rather, DNA methylation alterations at the precancerous stage may confer vulnerability to further genetic and epigenetic alterations, generate more malignant cancers, and thus determine patient outcome. Therefore, genome‐wide DNA methylation profiling may provide optimal indicators for carcinogenetic risk estimation and prognostication, and thus provide an avenue for cancer prevention and therapy on an individual basis. (Cancer Sci 2009)
Abbreviations
- BAC
bacterial artificial chromosome
- BAMCA
BAC array‐based methylated CpG island amplification
- CIMP
CpG island methylator phenotype
- DNMT
DNA methyltransferase
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- LOH
loss of heterozygosity
- PanIN
pancreatic intraductal neoplasia
- PCNA
proliferating cell nuclear antigen
- RCC
renal cell carcinoma
- UC
urothelial carcinoma
DNA methylation, a covalent chemical modification resulting in addition of a methyl group at the carbon five position of the cytosine ring in CpG dinucleotides, is one of the most consistent epigenetic changes observed in human cancers.( 1 ) DNMTs transfer methyl groups from S‐adenosylmethionine to cytosines.( 2 ) The preference of DNMT1, a major and well‐known DNMT, for hemimethylated over unmethylated substrates in vitro,( 3 ) and its targeting of replication foci by binding to PCNA,( 4 , 5 ) are believed to allow copying of the DNA methylation pattern on the parental strand to the newly synthesized daughter DNA strand. Thus, DNMT1 has been recognized as a “maintenance” DNMT,( 6 ) whereas DNMT3a and DNMT3b show de novo DNA methylation activity.( 7 ) DNA methylation normally promotes a highly condensed heterochromatin structure associated with deacetylation of histones H3 and H4, loss of histone H3, lysine 4 (H3K4) methylation, and gain of H3K9 and H3K27 methylation.( 8 ) When methyl‐CpG‐binding proteins, such as MeCP2( 9 , 10 ) and MBD2,( 11 ) bind to methylated CpG dinucleotide, their transcriptional repression domain recruits a co‐repressor complex containing histone deacetylases. However, histone methyltransferases, such as G9A( 12 ) and SUV39H1,( 13 ) are required to recruit DNMTs. DNA methylation is a stable modification inherited throughout consecutive cell divisions, being essential for the normal development and function of adult organs, particularly for X‐chromosome inactivation, genome imprinting, silencing of transposons and other parasitic elements, and proper expression of genes.( 14 )
Reduction of DNMT1 activity in genetically engineered animals alters the number of tumors or the timing of tumor development, suggesting a causal relationship between DNA methylation alterations and tumorigenesis.( 15 , 16 ) In 1995, when the RB and VHL genes were the only tumor suppressor genes known to be silenced by DNA methylation, we showed that the E‐cadherin tumor suppressor gene is silenced by DNA methylation around the promoter region.( 17 ) The list of tumor‐related genes whose expression levels are altered due to DNA hypo‐ or hypermethylation is increasing.( 18 , 19 , 20 , 21 , 22 ) Transcriptionally repressive chromatin modifications within the promoters of tumor‐related genes silenced by DNA methylation are known to resemble the chromatin modifications of these genes in normal embryonic stem cells, for example, polycomb complex binding and H3K27 methylation.( 23 ) These genes also have an active marker, H3K4 methylation, in normal stem cells, and this bivalent state is converted to a primary active or repressive chromatin conformation after differentiation cues have been received.( 23 ) During carcinogenesis, such modifications may render the genes vulnerable to errors, resulting in aberrant DNA methylation.( 24 ) DNA hypomethylation induces chromosomal instability through decondensation of heterochromatin and enhancement of chromosomal recombination during carcinogenesis.( 25 ) Translational epigenetics have come of age,( 26 , 27 ) and empirical analysis of DNA methylation status in clinical tissue samples in connection with the clinicopathological diversity of human cancers is assuming increasing importance for the diagnosis, prevention, and therapy of cancers.( 28 , 29 )
Alterations of DNA methylation during multistage carcinogenesis
Alterations of DNA methylation at the precancerous stage. DNA methylation alterations play a key role in the early steps of human carcinogenesis. In the 1990s, although LOH on chromosome 16 was frequently detected by classical Southern blotting in HCCs that were poorly differentiated, large in size, and associated with metastasis,( 30 ) only a few of the molecular events occurring in the earlier stage of hepatocarcinogenesis were known. Since DNA methylation alterations may be correlated with chromosomal instability, we examined the DNA methylation status on chromosome 16 using Southern blotting with a DNA methylation‐sensitive restriction enzyme. DNA methylation alterations at multiple loci on chromosome 16, compared to normal liver tissue samples, were frequently revealed even in samples of non‐cancerous liver tissue showing chronic hepatitis or liver cirrhosis,( 31 , 32 ) which are widely considered to be precancerous conditions,( 33 ) indicating that DNA methylation alterations are a very early event during multistage hepatocarcinogenesis. This was one of the earliest reports of DNA methylation alterations at the precancerous stage.( 31 )
DNA hypermethylation around the promoter region of the E‐cadherin tumor suppressor gene (16q22.1), which encodes a Ca2+‐dependent cell‐cell adhesion molecule,( 34 ) has been detected even in samples of non‐cancerous liver tissue showing chronic hepatitis or cirrhosis.( 35 ) Heterogeneous E‐cadherin expression in such non‐cancerous liver tissue, which is associated with small focal areas of hepatocytes showing only slight E‐cadherin immunoreactivity, might be due, at least partly, to DNA hypermethylation.( 35 ) Reduction of E‐cadherin expression due to DNA methylation around the promoter region may participate even in the very early stage of hepatocarcinogenesis through loss of intercellular adhesiveness and destruction of tissue morphology.
Studies of LOH by PCR using microsatellite markers have been reported, using specimens microdissected from precancerous lesions in several organ types. Whether aberrant DNA methylation precedes chromosomal instability during hepatocarcinogenesis was re‐examined using microdissected specimens obtained from lobules, pseudo lobules or regenerative nodules in non‐cancerous liver tissue from patients with HCCs by bisulfite modification. Although no degree of DNA methylation of any of the examined C‐type CpG islands, which are generally methylated in a cancer‐specific but not age‐dependent manner, was ever detected in normal liver tissue from patients without HCCs, DNA hypermethylation of such islands was frequently found, even in microdissected specimens of non‐cancerous liver tissue showing no remarkable histological changes obtained from patients with HCCs in which LOH was never detected.( 36 ) Thus it was confirmed that aberrant DNA methylation is an earlier event preceding chromosomal instability during hepatocarcinogenesis.
As another example of inflammation‐associated carcinogenesis, ductal carcinomas of the pancreas frequently develop after chronic damage due to pancreatitis. At least a proportion of peripheral pancreatic ductal epithelia with an inflammatory background may be at the precancerous stage. When the DNA methylation status of the p14, p15, p16, p73, APC, hMLH1, MGMT, BRCA1, GSTP1, TIMP‐3, E‐cadherin, and DAPK‐1 genes was examined, the average number of methylated tumor‐related genes and the incidence of DNA methylation of at least one gene were increased in peripheral pancreatic ductal epithelia with an inflammatory background and in another precancerous lesion, PanIN, in comparison with normal peripheral pancreatic duct epithelia.( 37 )
UCs of the urinary bladder, renal pelvis, and ureter are clinically remarkable because of their multicentricity and tendency to recur (Fig. 1a).( 38 ) A possible mechanism for such multiplicity is the “field effect.” Even non‐cancerous urothelia showing no remarkable histological changes obtained from patients with UCs can be considered precancerous, because they may have been exposed to carcinogens in the urine. When the DNA methylation status of multiple C‐type CpG islands was examined, the average number of methylated C‐type CpG islands was increased in non‐cancerous urothelia showing no remarkable histological changes obtained from patients with UCs, in comparison with normal urothelia obtained from patients without UCs.( 39 )
Figure 1.
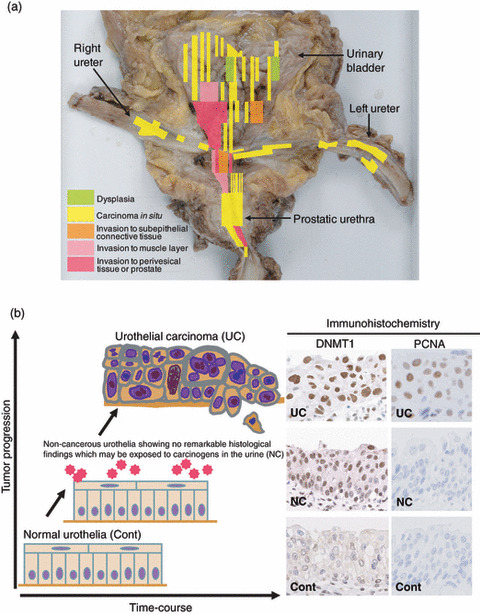
Overexpression of DNA methyltransferase (DNMT) 1 protein during multistage urothelial carcinogenesis. (a) Specimen obtained by radical cystectomy for multiple urothelial carcinomas (UCs) of the urinary bladder, bilateral ureters, and prostatic urethra. UCs are clinically remarkable because of their multicentricity and tendency to recur: synchronously or metachronously multifocal UCs often develop in individual patients.( 38 ) A possible mechanism for such multiplicity is the “field effect.” Even non‐cancerous urothelia showing no remarkable histological changes obtained from patients with UCs can be considered precancerous, because they may be exposed to carcinogens in the urine. (b) Immunohistochemical examination for DNMT1 and proliferating cell nuclear antigen (PCNA) in tissue specimens. The incidence of nuclear DNMT1 immunoreactivity had already increased in non‐cancerous urothelia showing no remarkable histological changes obtained from patients with UCs (NC), where the PCNA labeling index had not yet increased, compared to that in normal urothelia obtained from patients without UCs (Cont), indicating that DNMT1 overexpression preceded any increase of cell proliferative activity.( 56 ) The intensity of nuclear DNMT1 immunoreactivity was further increased in UCs.( 56 )
Cigarette smoking is another background factor associated with alterations of DNA methylation during multistage carcinogenesis. DNA hypermethylation at the D17S5 locus, where the HIC (hypermethylated‐in‐cancer)‐1 tumor suppressor gene was identified, is observed even in non‐cancerous lung tissue, which may contain progenitor cells for cancers, obtained from patients with non‐small‐cell lung cancers. The incidence of DNA hypermethylation in non‐cancerous lung tissue obtained from patients with non‐small‐cell lung cancers is significantly correlated with both smoking history and the extent of pulmonary anthracosis, as an index of the cumulative effects of smoking.( 40 ) Thus, DNA methylation alterations are frequently found even at the precancerous stage in various organs, especially in association with chronic inflammation( 41 , 42 ) and/or persistent infection with viruses( 43 , 44 , 45 ) or other pathogenic microorganisms, and with cigarette smoking.
DNA methyltransferase 1 overexpression and regional DNA hypermethylation. With respect to the molecular backgrounds of DNA methylation alterations,( 46 ) it has been reported that levels of DNMT1 mRNA expression are significantly higher in samples of non‐cancerous liver tissue showing chronic hepatitis or cirrhosis than in normal liver tissue, and are even higher in HCCs.( 47 , 48 ) The incidence of DNMT1 overexpression in HCCs is significantly correlated with poorer tumor differentiation and portal vein involvement.( 49 ) Moreover, the recurrence‐free and overall survival rates of patients with HCCs showing DNMT1 overexpression are significantly lower than those of patients with HCCs that do not.( 49 )
As mentioned above, at least a proportion of peripheral pancreatic ductal epithelia with an inflammatory background may be at the precancerous stage. The incidence of DNMT1 protein expression increases with progression from peripheral pancreatic ductal epithelia with an inflammatory background, to PanIN, to well‐differentiated ductal carcinoma, and finally to poorly differentiated ductal carcinoma of the pancreas, in comparison with normal peripheral pancreatic duct epithelia.( 50 ) DNMT1 overexpression in ductal carcinomas of the pancreas is significantly correlated with the extent of invasion to the surrounding tissue, an advanced stage, and poorer patient outcome.( 50 ) The average number of methylated tumor‐related genes in microdissected specimens of peripheral pancreatic ductal epithelia with an inflammatory background, PanIN, and ductal carcinoma was significantly correlated with the level of DNMT1 protein expression examined immunohistochemically in precisely microdissected areas.( 37 )
Expression levels of DNMT1 mRNA and protein are significantly correlated with poorer differentiation and the CIMP, a cancer phenotype characterized by accumulation of DNA methylation of C‐type CpG islands,( 51 , 52 ) in stomach cancers,( 53 ) but no such association has been observed for the expression of DNMT2, DNMT3a, or DNMT3b.( 54 ) Epstein–Barr virus infection in stomach cancers is significantly associated with marked accumulation of DNA methylation of C‐type CpG islands and overexpression of DNMT1 protein.( 53 ) Helicobacter pylori infection, another etiologic factor for stomach carcinogenesis, has also been reported to strongly promote regional DNA hypermethylation( 55 ) but is not correlated with DNMT1 expression levels.( 53 )
It is debatable whether increased DNMT1 expression is due to an increase in the proportion of dividing cells or to an acute increase of DNMT1 expression per individual cancer cell. Immunohistochemical examinations have clearly revealed that the incidence of nuclear DNMT1 immunoreactivity is already higher in non‐cancerous urothelia showing no remarkable histological changes obtained from patients with UCs, which may already be exposed to carcinogens in the urine but in which the PCNA labeling index had not yet increased, compared to that in normal urothelia from patients without UCs, indicating that DNMT1 overexpression preceded increased cell proliferative activity (Fig. 1b).( 56 ) The incidence of nuclear DNMT1 immunoreactivity showed a further and progressive increase in dysplastic urothelia, and during transition to UCs (Fig. 1b).( 56 ) Among all examined microdissected specimens of non‐cancerous urothelia showing no remarkable histological changes from patients with UCs, or dysplastic urothelia and UCs, accumulation of DNA methylation of C‐type CpG islands was significantly correlated with the level of DNMT1 protein expression.( 39 )
Thus DNMT1 overexpression participates not only in the precancerous stage but also in the malignant progression of various cancers, and has a prognostic impact on patients. DNMT1 overexpression is frequently associated with CIMP of cancers. Although the maintenance activities of DNMT1 are related to its in vitro preference for hemimethylated substrates, excessive amounts of DNMT1 in comparison to PCNA may participate in de novo methylation of CpG islands. The molecular mechanisms that target DNMT1 to unmethylated substrates in cancers need to be clarified.
Splicing alteration of DNMT3b and DNA hypomethylation in pericentromeric satellite regions. DNA hypomethylation in pericentromeric satellite regions is known to result in centromeric decondensation and enhanced chromosome recombination. In HCCs( 57 ) and UCs,( 58 ) DNA hypomethylation of these regions is correlated with copy number alterations on chromosomes 1 and 9, respectively, where satellite regions are rich. DNMT3b is required for DNA methylation of pericentromeric satellite regions in early mouse embryos, and germline mutations of the DNMT3b gene have been reported in patients with immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome, a rare recessive autosomal disorder characterized by DNA hypomethylation of pericentromeric satellite regions.( 59 ) The major splice variant of DNMT3b in normal liver tissue samples is DNMT3b3, which possesses the conserved catalytic domains.( 60 ) DNMT activity of human DNMT3b3 has been confirmed in vitro.( 61 ) In contrast, DNMT3b4 lacks the conserved catalytic domains, although it retains the N‐terminal domain required for targeting to heterochromatin sites. Samples of normal liver tissue show only a trace level of DNMT3b4 expression.( 60 ) The levels of DNMT3b4 mRNA expression and the ratio of DNMT3b4 mRNA to DNMT3b3 in samples of non‐cancerous liver tissue obtained from patients with HCCs, and in HCCs themselves, are significantly correlated with the degree of DNA hypomethylation in pericentromeric satellite regions.( 60 ) DNA demethylation on satellite 2 has been observed in DNMT3b4‐transfected human epithelial 293 cells.( 60 ) As DNMT3b4 lacking DNMT activity competes with DNMT3b3 for targeting to pericentromeric satellite regions, DNMT3b4 overexpression may lead to chromosomal instability through induction of DNA hypomethylation in such regions.
Furthermore, the growth rate of DNMT3b4 transfectants is approximately double that of mock‐transfectants soon after the introduction of DNMT3b4, when chromosomal instability may not yet have accumulated.( 62 ) Genes implicated in interferon signaling including signal transducer and activator of transcription (STAT) 1, which acts as an effector of interferon signaling, are upregulated in DNMT3b4 transfectants,( 62 ) suggesting that DNMT3b may act to maintain the DNA methylation status of not only pericentromeric satellite regions but also specific genes, probably in cooperation with DNMT1, in cancer cells.
Genome‐wide DNA methylation profiling
DNA methylation profiles in precancerous conditions are inherited by cancers. The above findings that DNA methylation alterations are associated with multistage carcinogenesis have prompted us to carry out genome‐wide DNA methylation analysis of tissue specimens. Recently, analysis on a genomic‐wide scale has become possible using DNA methylation‐sensitive restriction enzyme‐based or anti‐methyl‐cytosine antibody affinity techniques that enrich methylated and unmethylated fractions of genomic DNA.( 63 , 64 ) These fractions can then be hybridized to DNA microarrays or sequenced. Ultra‐high‐throughput DNA sequencing technologies are being introduced for the direct sequencing of enriched, methylated fragments or for bisulfite‐converted genomic sequencing.( 65 )
We have used BAMCA.( 66 , 67 , 68 , 69 ) Many researchers in this field use the promoter arrays to identify genes that are methylated in cancer cells. However, the promoter regions of specific genes are not the only target of DNA methylation alterations in human cancers. DNA methylation status in genomic regions not directly participating in gene silencing, such as the edges of CpG islands, may be altered at the precancerous stage before the alterations of the promoter regions themselves occur.( 70 ) Genomic regions in which DNA hypomethylation affects chromosomal instability may not be contained in promoter arrays. Moreover, aberrant DNA methylation of large chromosome regions, which are regulated in a coordinated manner in human cancers due to a process of long‐range epigenetic silencing, has recently attracted attention.( 71 ) Therefore, we used a BAC array that may be suitable, not for focusing on specific promoter regions, but for overviewing the DNA methylation status of individual large regions among all chromosomes.
When BAMCA methods were applied to samples of non‐cancerous renal tissue obtained from patients with clear cell RCCs, many BAC clones showed DNA hypo‐ or hypermethylation in comparison to normal renal tissue samples from patients without any primary renal tumors.( 72 ) RCCs are usually well demarcated and covered by a fibrous capsule, and hardly ever contain fibrous stroma between cancer cells (Fig. 2a). We were therefore able to obtain cancer cells of high purity from surgical specimens, avoiding contamination with either non‐cancerous epithelial cells or stromal cells (Fig. 2a). Therefore, the DNA methylation alterations observed in samples of non‐cancerous renal tissue from patients with RCCs cannot be attributable to contamination during sampling. Moreover, DNA methylation alterations in non‐cancerous renal tissue did not depend on the distance from the RCC itself to the site from which the non‐cancerous renal tissue samples were taken. Because of the lack of any remarkable histological changes or any association with chronic inflammation and persistent infection with viruses or other pathogenic microorganisms, precancerous conditions in the kidney have rarely been described. However, from the viewpoint of DNA methylation, we can consider that non‐cancerous renal tissue from patients with RCCs is already at the precancerous stage, showing genome‐wide DNA methylation alterations.
Figure 2.
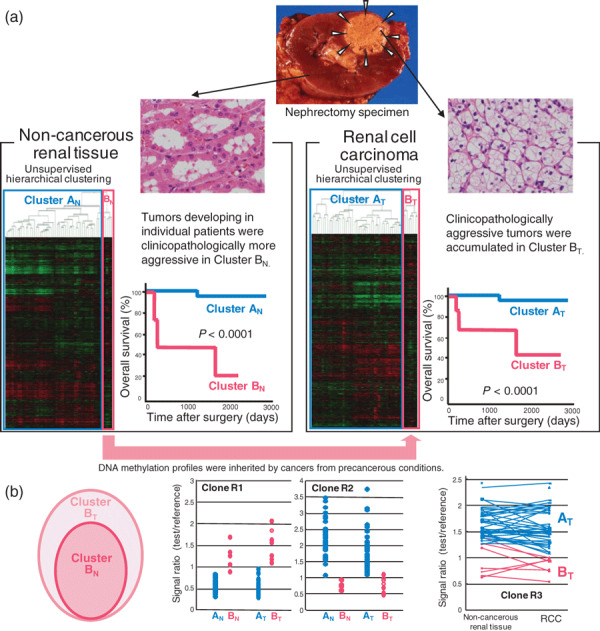
DNA methylation profiles in precancerous conditions and renal cell carcinomas (RCCs). (a) Bacterial artificial chromosome array‐based methylated CpG island amplification (BAMCA) data for tissue samples obtained from patients with RCCs (arrowheads). Using unsupervised hierarchical clustering analysis based on BAMCA data for samples of their non‐cancerous renal tissue, patients with RCCs were clustered into two subclasses, Clusters AN and BN. ( 72 ) Clinicopathologically aggressive RCCs were accumulated in Cluster BN, and the overall survival rate of patients in Cluster BN was significantly lower than that of patients in Cluster AN.( 72 ) Using unsupervised hierarchical clustering analysis based on BAMCA data for their RCCs, patients were clustered into two subclasses, Clusters AT and BT.( 72 ) Clinicopathologically aggressive clear cell RCCs were accumulated in Cluster BT, and the overall survival rate of patients in Cluster BT was significantly lower than that of patients in Cluster AT.( 72 ) (b) Correlation between DNA methylation profiles of precancerous conditions and those of RCCs. Cluster BN was completely included in Cluster BT (left panel). The majority of the bacterial artificial chromosome (BAC) clones, 724 in all, significantly discriminating Cluster BN from Cluster AN, also discriminated Cluster BT from Cluster AT.( 72 ) In 311 of the 724 BAC clones, where the average signal ratio of Cluster BN was higher than that of Cluster AN, such as Clone R1 in the middle panel, the average signal ratio of Cluster BT was also higher than that of Cluster AT without exception.( 72 ) In 413 of the 724 BAC clones, where the average signal ratio of Cluster BN was lower than that of Cluster AN, such as Clone R2 in the middle panel, the average signal ratio of Cluster BT was also lower than that of Cluster AT without exception.( 72 ) As shown in the scattergram of the signal ratios in non‐cancerous renal tissue samples and RCCs for all examined patients for a representative BAC clone, Clone R3, the DNA methylation status of the non‐cancerous renal tissue was basically inherited by the corresponding RCC in individual patients (right panel).( 72 )
We then carried out two‐dimensional unsupervised hierarchical clustering analysis based on BAMCA data for non‐cancerous renal tissue samples. The patients with RCCs were clustered into two subclasses, clusters AN and BN (Fig. 2a). The corresponding RCCs of patients in Cluster BN showed more frequent macroscopically evident renal vein tumor thrombi, microscopically evident vascular involvement, and higher pathological TNM stages than those in Cluster AN.( 72 ) The overall survival rate of patients in Cluster BN was significantly lower than that of patients in Cluster AN (Fig. 2a).( 72 ) Tumor aggressiveness and even patient outcome might thus be determined by DNA methylation profiles at the precancerous stage.
In RCCs themselves, more BAC clones showed DNA hypo‐ or hypermethylation, and its degree was increased in comparison with samples of non‐cancerous renal tissue obtained from patients with RCCs. Two‐dimensional unsupervised hierarchical clustering analysis based on BAMCA data for RCCs was able to group patients into two subclasses, Clusters AT and BT (Fig. 2a). RCCs in Cluster BT more frequently showed renal vein tumor thrombi, vascular involvement, and higher pathological TNM stages than those in Cluster AT.( 72 ) The overall survival rate of patients in Cluster BT was significantly lower than that of patients in Cluster AT (Fig. 2a).( 72 )
Patients who were grouped in Cluster BN on the basis of BAMCA data for non‐cancerous renal tissue were also grouped in Cluster BT on the basis of BAMCA data for RCC themselves. That is, Cluster BN was completely included in Cluster BT (Fig. 2b).( 72 ) The majority of the BAC clones significantly discriminating Cluster BN from Cluster AN also discriminated Cluster BT from Cluster AT.( 72 ) Among BAC clones characterizing both clusters BN and BT, where the average signal ratio of Cluster BN was higher than that of Cluster AN, the average signal ratio of Cluster BT was also higher than that of Cluster AT without exception (Fig. 2b). Among BAC clones characterizing both clusters BN and BT, where the average signal ratio of Cluster BN was lower than that of Cluster AN, the average signal ratio of Cluster BT was also lower than that of Cluster AT without exception (Fig. 2b). Comparison between the signal ratios of each BAC clone characterizing both clusters BN and BT in non‐cancerous renal tissue and those in the corresponding RCCs for all patients revealed that the DNA methylation status of the non‐cancerous renal tissue was basically inherited by the corresponding RCC in each individual patient (Fig. 2b).( 72 )
In non‐cancerous renal tissue showing no remarkable histological changes and consisting mainly of renal tubules with specialized functions, no progenitor cell is able to gain a growth advantage, and clonal expansion is unable to occur. Therefore, the distinct DNA methylation profile of Cluster BN, which is clinicopathologically valid, cannot be established through the selection of one of a number of random DNA methylation profiles in non‐cancerous renal tissue in patients with clear cell RCCs, and instead may be established through distinct target mechanisms. As the DNA methylation profiles in Cluster BT are shared by phenotypically similar patients, who all suffer from clinicopathologically aggressive tumors and show a poor outcome, DNA methylation alterations in at least a proportion of the BAC regions characterizing Cluster BT cannot be passenger changes. It is clear that cancer itself can induce alterations in DNA methylation. However, DNA methylation alterations of BAC regions characterizing Cluster BT may significantly participate in carcinogenesis, as the DNA methylation profile in Cluster BN was established at a very early and precancerous stage of carcinogenesis and inherited during progression of the cancers themselves as Cluster BT. At least a proportion of DNA methylation alterations at the precancerous stage may be “epigenetic gatekeepers”( 21 ) and which allow time for further epigenetic and genetic alterations including genetic gatekeeper mutations (Fig. 3).
Figure 3.
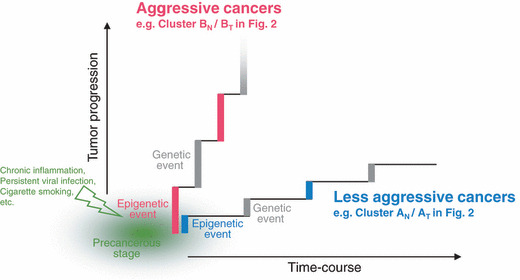
Significance of DNA methylation alterations at the precancerous stage. Chronic inflammation, persistent infection with viruses or other pathogenic microorganisms, cigarette smoking, exposure to chemical carcinogens, and other unknown factors may participate in the establishment of particular DNA methylation profiles, such as Cluster BN in Fig. 2. Such DNA methylation alterations in precancerous conditions may not occur randomly, but may be prone to further accumulation of epigenetic and genetic alterations (regional DNA hypermethylation of C‐type CpG islands and copy number alterations were accumulated in Cluster BT in Fig. 2),( 72 ) thus generating more malignant cancers, such as the renal cell carcinomas in patients belonging to Cluster BT.
In fact, when the DNA methylation status of C‐type CpG islands was examined,( 73 ) the average number of methylated CpG islands was significantly higher in Cluster BT based on BAMCA than in Cluster AT. The frequency of CIMP in Cluster BT was significantly higher than that in Cluster AT. Genome‐wide DNA methylation alterations consisting of both hypo‐ and hypermethylation revealed by BAMCA in Cluster BT were associated with regional DNA hypermethylation of C‐type CpG islands. For comparison with their DNA methylation status, we also examined copy number alterations by array‐based comparative genomic hybridization. By unsupervised hierarchical clustering analysis based on copy number alterations, RCCs were clustered into the two subclasses, clusters ATG and BTG. Loss of chromosome 3p and gain of chromosomes 5q and 7 were frequent in both clusters ATG and BTG. Loss of chromosomes 1p, 4, 9, 13q, and 14q was frequent only in Cluster BTG, and not in Cluster ATG.( 74 ) RCCs showing higher histological grades, renal vein tumor thrombi, vascular involvement and higher pathological TNM stages were accumulated in Cluster BTG. The recurrence‐free and overall survival rates of patients in Cluster BTG were significantly lower than those of patients in Cluster ATG.( 74 ) A subclass of Cluster BT based on BAMCA data was completely included in Cluster BTG showing accumulation of copy number alterations. Genetic and epigenetic alterations are not mutually exclusive during renal carcinogenesis, and particular DNA methylation profiles may be closely related to chromosomal instability. DNA methylation alterations at the precancerous stage, which may not occur randomly but may foster further epigenetic and genetic alterations, can generate more malignant cancers and even determine patient outcome (Fig. 3).
Carcinogenetic risk estimation and prognostication based on DNA methylation status. In samples of non‐cancerous liver tissue obtained from patients with HCCs, many BAC clones show DNA hypo‐ or hypermethylation in comparison with normal liver tissue from patients without HCCs (Fig. 4a).( 75 ) The effectiveness of surgical resection for HCC is limited, unless the disease is diagnosed early at the asymptomatic stage. Therefore, surveillance at the precancerous stage is a priority for patients with HBV or HCV infection. To reveal the baseline liver histology, microscopic examination of liver biopsy specimens is carried out in patients with HBV or HCV infection prior to interferon therapy.( 76 , 77 ) Carcinogenetic risk estimation using such liver biopsy specimens is advantageous for close follow‐up of patients who are at high risk of HCC development. To establish an indicator for carcinogenetic risk estimation, we first omitted potentially insignificant BAC clones associated only with inflammation and/or fibrosis and focused on BAC clones for which DNA methylation status was altered at the precancerous stage in comparison to normal liver tissue and was inherited by HCCs themselves from the precancerous stage (Fig. 4b). Among the BAC clones studied, a bioinformatics approach further identified the top 25 for which DNA methylation status was able to discriminate non‐cancerous liver tissue from patients with HCCs in the learning cohort from normal liver tissue with sufficient sensitivity and specificity.( 75 ) By two‐dimensional hierarchical clustering analysis using these 25 BAC clones, samples of normal liver tissue and samples of non‐cancerous liver tissue obtained from patients with HCCs in the learning cohort were successfully subclassified into different subclasses without any error (Fig. 4c). The criteria established using a combination of the DNA methylation status of the 25 BAC clones (Fig. 4d) diagnosed non‐cancerous liver tissue from patients with HCCs in the learning cohort as being at high risk of carcinogenesis with a sensitivity and specificity of 100%.( 75 ) The sensitivity and specificity in the validation cohort were both 96%, and thus our criteria were successfully validated.( 75 )
Figure 4.
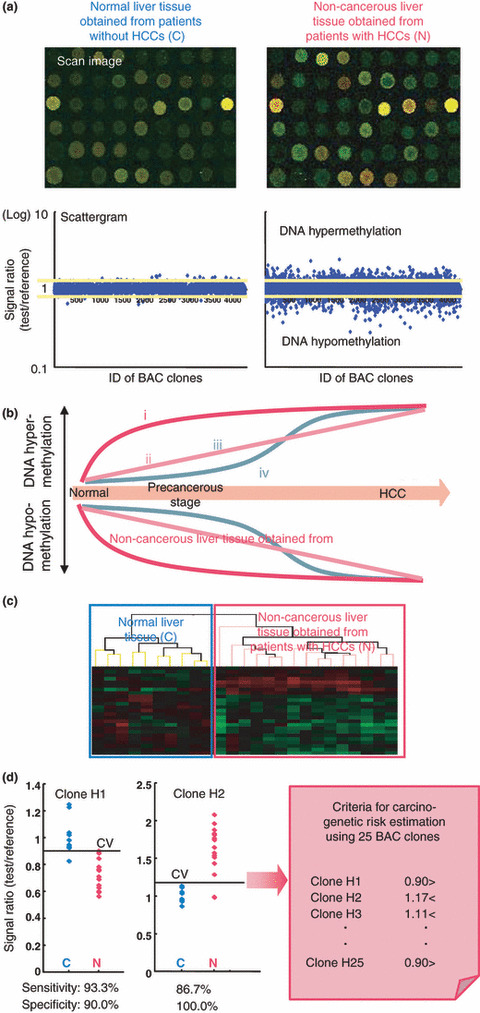
Risk estimation of hepatocellular carcinoma (HCC) development based on DNA methylation status. (a) Examples of scan images and scattergrams of signal ratios in normal liver tissue obtained from patients without HCCs (C) and non‐cancerous liver tissue obtained from patients with HCCs (N). In N samples, many bacterial artificial chromosome (BAC) clones showed DNA hypo‐ or hypermethylation compared to C samples.( 75 ) (b) Four patterns of DNA methylation alterations seen in BAC clones during multistage hepatocarcinogenesis: (i) DNA methylation alterations occurred at the chronic hepatitis and liver cirrhosis stage, and DNA methylation status did not alter in HCCs from the chronic hepatitis and liver cirrhosis stage; (ii) DNA methylation alterations occurred at the chronic hepatitis and liver cirrhosis stage and further altered in HCCs; (iii) although DNA methylation alterations occurred at the chronic hepatitis and liver cirrhosis stage, the DNA methylation status returned to normal in HCCs; and (iv) DNA methylation alterations occurred only in HCCs. In order to establish criteria for carcinogenetic risk estimation, we focused on BAC clones whose DNA methylation status was inherited by HCCs from the precancerous stage (groups i and ii), whereas group iii may only reflect inflammation and/or fibrosis, and group iv may participate only in the malignant progression stage. (c) Two‐dimensional hierarchical clustering analysis using BAC clones that were selected as the top 25 for which DNA methylation status was able to discriminate N from C with sufficient sensitivity and specificity by Wicoxon test and the support vector machine algorithm.( 75 ) C and N samples in the learning cohort were successfully subclassified into different subclasses without any error.( 75 ) (d) Scattergrams of the signal ratios in C and N samples in the learning cohort for representative BAC clones, Clone H1 and Clone H2. Using the cut‐off values (CV) in each panel, N samples in the learning cohort were discriminated from C samples with sufficient sensitivity and specificity.( 75 ) Based on a combination of DNA methylation status for the 25 BAC clones, the criteria for carcinogenetic risk estimation were established. Using these criteria, the sensitivity and specificity for diagnosis of N samples in the learning cohort as being at high risk of carcinogenesis were both 100%.( 75 ) The sensitivity and specificity in the validation cohort were both 96%, and thus the criteria were successfully validated.( 75 )
It was confirmed that there were no significant differences in the number of BAC clones satisfying our criteria between samples of non‐cancerous liver tissue showing chronic hepatitis and samples of non‐cancerous liver tissue showing cirrhosis, indicating that our criteria were not associated with the degree of inflammation or fibrosis.( 75 ) In addition, the average numbers of BAC clones satisfying our criteria were significantly lower in liver tissue samples from patients with HBV or HCV infection but without HCCs than in samples of non‐cancerous liver tissue obtained from patients with HCCs.( 75 ) Therefore, our criteria may be applicable for classifying liver tissue samples obtained from patients who are being followed up because of HBV or HCV infection, chronic hepatitis, or cirrhosis into those that may generate HCCs and those that will not. We intend to validate the reliability of such risk estimation prospectively using liver biopsy specimens obtained prior to interferon therapy from a large cohort of patients with HBV or HCV infection.
To establish criteria for prognostication of patients with HCCs, in the learning cohort, patients who had survived more than 4 years after hepatectomy and patients who had suffered recurrence within 6 months and died within a year after hepatectomy were defined as a favorable‐outcome group and a poor‐outcome group, respectively. Wilcoxon test revealed that the signal ratios of 41 BAC clones differed significantly between the two groups.( 75 ) Two‐dimensional hierarchical clustering analysis using the 41 BAC clones successfully subclassified HCCs in the favorable‐outcome group and the poor‐outcome group into different subclasses without any error (Fig. 5a). We also established cut‐off values for the 41 BAC clones that allowed discrimination of samples between the poor‐outcome and favorable‐outcome groups with sufficient sensitivity and specificity (Fig. 5b). Multivariate analysis revealed that satisfying our criteria for 32 or more BAC clones was a predictor of overall patient outcome and was independent of parameters that are already known to have prognostic significance,( 75 ) such as histological differentiation, and presence of portal vein tumor thrombi, intrahepatic metastasis, and multicentricity.( 33 ) The cancer‐free and overall survival rates of patients with HCCs satisfying the criteria for 32 or more BAC clones in the validation cohort were significantly lower than those of patients with HCCs satisfying the criteria for less than 32 BAC clones (Fig. 5c).( 75 ) Such prognostication using liver biopsy specimens obtained before transarterial embolization, transarterial chemoembolization, and radiofrequency ablation may be advantageous even for patients who undergo such therapies.
Figure 5.
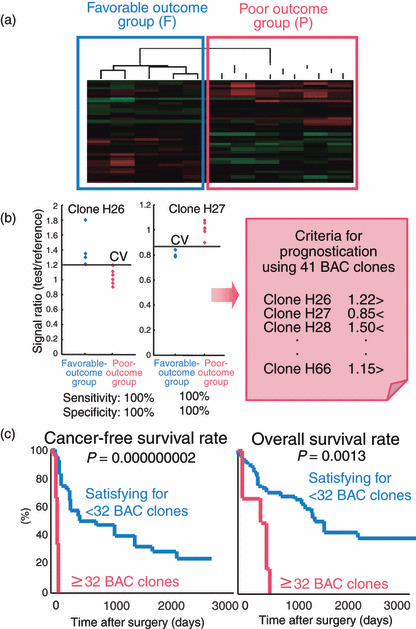
Prognostication of patients with HCC development based on DNA methylation status. (a) Two‐dimensional hierarchical clustering analysis using 41 bacterial artificial chromosome (BAC) clones selected as those for which DNA methylation status was able to discriminate a poor‐outcome group (P), who suffered recurrence within 6 months and died within a year after hepatectomy, from a favorable‐outcome group (F), who survived for more than 4 years after hepatectomy, with sufficient sensitivity and specificity by Wilcoxon test.( 75 ) F and P patients in the learning cohort were successfully subclassified into different subclasses without any error.( 75 ) (b) Scattergrams of the signal ratios in F and P patients in the learning cohort for representative BAC clones, Clone H26 and Clone H27. Using the cut‐off values (CV) in each panel, P patients in the learning cohort were discriminated from F patients with 100% sensitivity and specificity.( 75 ) Based on a combination of the DNA methylation status of the 41 BAC clones, criteria for prognostication were established. (c) The cancer‐free and overall survival rates of patients with HCCs in the validation cohort. Patients with HCCs satisfying the criteria for 32 or more BAC clones showed significantly poorer outcome than patients with HCCs satisfying the criteria for less than 32 BAC clones.( 75 )
As mentioned above, even non‐cancerous urothelia showing no remarkable histological changes obtained from patients with UCs may be exposed to carcinogens in urine. In fact, genome‐wide DNA methylation profiles of non‐cancerous urothelia obtained from patients with nodular invasive UCs showing an aggressive clinical course were inherited by the nodular invasive UCs themselves, suggesting that DNA methylation alterations that were correlated with the development of more malignant invasive cancers had already accumulated in non‐cancerous urothelia.( 78 ) These findings prompted us to estimate the degree of carcinogenetic risk based on DNA methylation profiles in non‐cancerous urothelia. We were able to identify BAC clones for which DNA methylation status was able to completely discriminate non‐cancerous urothelia from patients with UCs from normal urothelia and diagnose them as having a high risk of urothelial carcinogenesis.( 78 ) If it were possible to identify individuals who are at high risk of urothelial carcinogenesis, then strategies for the prevention or early detection of UCs, such as smoking cessation or repeated urine cytology examinations, might be applicable.
In order to start adjuvant systemic chemotherapy immediately in patients who have undergone total cystectomy and are still at high risk of recurrence and metastasis of UCs, prognostic indicators have been explored. Subclassification based on unsupervised two‐dimensional hierarchical clustering analysis using BAMCA data for UCs was significantly correlated with recurrence after surgery due to metastasis to pelvic lymph nodes or distant organs.( 78 ) These data prompted us to establish criteria for predicting recurrence of UCs based on DNA methylation status, and we successfully identified BAC clones for which DNA methylation status completely discriminated patients who suffered recurrence from patients who did not, whereas high histological grade, invasive growth, and vascular or lymphatic involvement were unable to achieve such complete discrimination.( 78 )
It is well known that patients with UCs of the renal pelvis and ureter frequently develop metachronous UC in the urinary bladder after nephroureterectomy. Therefore, such patients need to undergo repeated urethrocystoscopic examinations for detection of intravesical metachronous UCs. To decrease the need for such invasive urethrocystoscopic examinations, indicators for intravesical metachronous UCs are needed. DNA methylation profiles of non‐cancerous urothelia obtained by nephroureterectomy from patients with UCs of the renal pelvis or ureter, which may be exposed to the same carcinogens in the urine as non‐cancerous urothelia from which metachronous UCs originate, were correlated with the risk of intravesical metachronous UC development.( 78 ) In non‐cancerous urothelia from nephroureterectomy specimens, we are able to identify BAC clones for which DNA methylation status was able to completely discriminate patients with UCs of the renal pelvis or ureter who developed intravesical metachronous UCs from patients who did not.( 78 ) After prospective validation, combination of such BAC clones may be an optimal indicator for the development of intravesical metachronous UC.
Perspective
On the basis of DNA methylation profiling, translational epigenetics has clearly come of age. The incidence of DNA methylation alterations is generally high during multistage carcinogenesis in various organs. DNA methylation alterations are stably preserved on DNA double strands by covalent bonds, and these can be detected using highly sensitive methodology. Therefore, they may be better diagnostic indicators than mRNA and protein expression profiles, which can be easily affected by the microenvironment of cancer cells or precursor cells. Genome‐wide DNA methylation profiling can provide optimal indicators for carcinogenetic risk estimation and prognostication using samples of urine, sputum, and other body fluids, and also biopsy and surgically resected specimens.
However, most of the recently developed detection technologies such as promoter arrays, CpG‐island arrays and high‐throughput sequencing are sequence‐based methods and cannot comprehensively measure the DNA methylation status of repetitive sequences and gene bodies. The dynamics of DNA methylation at such non‐unique sequences still remain to be determined.( 79 ) Our BAC array‐based methods do not focus only on specific promoter regions and CpG islands, and have successfully identified the chromosomal regions in which coordinated DNA methylation alterations have clinicopathological impact. Evaluation of the correlation between the methylation status of each CpG site in selected BAC clones and the clinicopathological diversity of cancers may provide new insights into the roles of DNA methylation during multistage carcinogenesis. Subclassification of cancers based on DNA methylation profiling may provide clues for clarification of distinct target mechanisms and molecules for prevention and therapy in patients belonging to specific clusters.
Acknowledgments
This study was supported by a Grant‐in‐Aid for the Third Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare of Japan, a Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labor and Welfare of Japan, a Grant from the New Energy and Industrial Technology Development Organization (NEDO), and the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NiBio).
References
- 1. Delcuve GP, Rastegar M, Davie JR. Epigenetic control. J Cell Physiol 2009; 219: 243–50. [DOI] [PubMed] [Google Scholar]
- 2. Hermann A, Gowher H, Jeltsch A. Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol Life Sci 2004; 61: 2571–87. [DOI] [PubMed] [Google Scholar]
- 3. Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl‐terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol 1988; 203: 971–83. [DOI] [PubMed] [Google Scholar]
- 4. Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. Human DNA‐(cytosine‐5) methyltransferase‐PCNA complex as a target for p21WAF1. Science 1997; 277: 1996–2000. [DOI] [PubMed] [Google Scholar]
- 5. Baylin SB. Tying it all together: epigenetics, genetics, cell cycle, and cancer. Science 1997; 277: 1948–9. [DOI] [PubMed] [Google Scholar]
- 6. Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet 2000; 9: 2395–402. [DOI] [PubMed] [Google Scholar]
- 7. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99: 247–57. [DOI] [PubMed] [Google Scholar]
- 8. Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10: 295–304. [DOI] [PubMed] [Google Scholar]
- 9. Jones PL, Veenstra GJ, Wade PA et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 1998; 19: 187–91. [DOI] [PubMed] [Google Scholar]
- 10. Nan X, Ng HH, Johnson CA et al. Transcriptional repression by the methyl‐CpG‐binding protein MeCP2 involves a histone deacetylase complex. Nature 1998; 393: 386–9. [DOI] [PubMed] [Google Scholar]
- 11. Kanai Y, Ushijima S, Nakanishi Y, Hirohashi S. Reduced mRNA expression of the DNA demethylase, MBD2, in human colorectal and stomach cancers. Biochem Biophys Res Commun 1999; 264: 962–6. [DOI] [PubMed] [Google Scholar]
- 12. Esteve PO, Chin HG, Smallwood A et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 2006; 20: 3089–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res 2003; 31: 2305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9: 465–76. [DOI] [PubMed] [Google Scholar]
- 15. Laird PW, Jackson‐Grusby L, Fazeli A et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995; 81: 197–205. [DOI] [PubMed] [Google Scholar]
- 16. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300: 455. [DOI] [PubMed] [Google Scholar]
- 17. Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E‐cadherin invasion‐suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci USA 1995; 92: 7416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 19. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction? Nat Rev Cancer 2006; 6: 107–16. [DOI] [PubMed] [Google Scholar]
- 20. Gronbaek K, Hother C, Jones PA. Epigenetic changes in cancer. Apmis 2007; 115: 1039–59. [DOI] [PubMed] [Google Scholar]
- 21. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128: 683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358: 1148–59. [DOI] [PubMed] [Google Scholar]
- 23. Shibata D. Inferring human stem cell behaviour from epigenetic drift. J Pathol 2009; 217: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ohm JE, McGarvey KM, Yu X et al. A stem cell‐like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39: 237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pogribny IP, Beland FA. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci 2009; 66: 2249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003; 3: 253–66. [DOI] [PubMed] [Google Scholar]
- 27. Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res 2009; 15: 3938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanai Y, Hirohashi S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis 2007; 28: 2434–42. [DOI] [PubMed] [Google Scholar]
- 29. Kanai Y. Alterations of DNA methylation and clinicopathological diversity of human cancers. Pathol Int 2008; 58: 544–58. [DOI] [PubMed] [Google Scholar]
- 30. Tsuda H, Zhang WD, Shimosato Y et al. Allele loss on chromosome 16 associated with progression of human hepatocellular carcinoma. Proc Natl Acad Sci USA 1990; 87: 6791–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Sugimura T, Hirohashi S. Aberrant DNA methylation on chromosome 16 is an early event in hepatocarcinogenesis. Jpn J Cancer Res 1996; 87: 1210–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Hirohashi S. Aberrant DNA methylation precedes loss of heterozygosity on chromosome 16 in chronic hepatitis and liver cirrhosis. Cancer Lett 2000; 148: 73–80. [DOI] [PubMed] [Google Scholar]
- 33. Hirohashi S, Ishak KG, Kojiro M et al. Hepatocellular carcinoma. In: Hamilton SR, Altonen LA, eds. World Health Organization classification of tumours. Pathology and genetics. Tumours of the digestive system. Lyon: IARC Press, 2000; 159–72. [Google Scholar]
- 34. Hirohashi S, Kanai Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci 2003; 94: 575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanai Y, Ushijima S, Hui AM et al. The E‐cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer 1997; 71: 355–9. [DOI] [PubMed] [Google Scholar]
- 36. Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis – a comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology 2000; 32: 970–9. [DOI] [PubMed] [Google Scholar]
- 37. Peng DF, Kanai Y, Sawada M et al. DNA methylation of multiple tumor‐related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis 2006; 27: 1160–8. [DOI] [PubMed] [Google Scholar]
- 38. Kakizoe T. Development and progression of urothelial carcinoma. Cancer Sci 2006; 97: 821–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakagawa T, Kanai Y, Ushijima S, Kitamura T, Kakizoe T, Hirohashi S. DNA hypermethylation on multiple CpG islands associated with increased DNA methyltransferase DNMT1 protein expression during multistage urothelial carcinogenesis. J Urol 2005; 173: 1767–71. [DOI] [PubMed] [Google Scholar]
- 40. Eguchi K, Kanai Y, Kobayashi K, Hirohashi S. DNA hypermethylation at the D17S5 locus in non‐small cell lung cancers: its association with smoking history. Cancer Res 1997; 57: 4913–5. [PubMed] [Google Scholar]
- 41. Kanai Y, Ushijima S, Ochiai A, Eguchi K, Hui A, Hirohashi S. DNA hypermethylation at the D17S5 locus is associated with gastric carcinogenesis. Cancer Lett 1998; 122: 135–41. [DOI] [PubMed] [Google Scholar]
- 42. Hodge DR, Peng B, Cherry JC et al. Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res 2005; 65: 4673–82. [DOI] [PubMed] [Google Scholar]
- 43. Kanai Y, Hui AM, Sun L et al. DNA hypermethylation at the D17S5 locus and reduced HIC‐1 mRNA expression are associated with hepatocarcinogenesis. Hepatology 1999; 29: 703–9. [DOI] [PubMed] [Google Scholar]
- 44. Sawada M, Kanai Y, Arai E, Ushijima S, Ojima H, Hirohashi S. Increased expression of DNA methyltransferase 1 (DNMT1) protein in uterine cervix squamous cell carcinoma and its precursor lesion. Cancer Lett 2007; 251: 211–9. [DOI] [PubMed] [Google Scholar]
- 45. Burgers WA, Blanchon L, Pradhan S, De Launoit Y, Kouzarides T, Fuks F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007; 26: 1650–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kanai Y, Ushijima S, Nakanishi Y, Sakamoto M, Hirohashi S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett 2003; 192: 75–82. [DOI] [PubMed] [Google Scholar]
- 47. Sun L, Hui AM, Kanai Y, Sakamoto M, Hirohashi S. Increased DNA methyltransferase expression is associated with an early stage of human hepatocarcinogenesis. Jpn J Cancer Res 1997; 88: 1165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Expression of mRNA for DNA methyltransferases and methyl‐CpG‐binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology 2001; 33: 561–8. [DOI] [PubMed] [Google Scholar]
- 49. Saito Y, Kanai Y, Nakagawa T et al. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int J Cancer 2003; 105: 527–32. [DOI] [PubMed] [Google Scholar]
- 50. Peng DF, Kanai Y, Sawada M et al. Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci 2005; 96: 403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toyota M, Ahuja N, Ohe‐Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer 2004; 4: 988–93. [DOI] [PubMed] [Google Scholar]
- 53. Etoh T, Kanai Y, Ushijima S et al. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am J Pathol 2004; 164: 689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanai Y, Ushijima S, Kondo Y, Nakanishi Y, Hirohashi S. DNA methyltransferase expression and DNA methylation of CPG islands and peri‐centromeric satellite regions in human colorectal and stomach cancers. Int J Cancer 2001; 91: 205–12. [DOI] [PubMed] [Google Scholar]
- 55. Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol 2007; 40: 142–50. [DOI] [PubMed] [Google Scholar]
- 56. Nakagawa T, Kanai Y, Saito Y, Kitamura T, Kakizoe T, Hirohashi S. Increased DNA methyltransferase 1 protein expression in human transitional cell carcinoma of the bladder. J Urol 2003; 170: 2463–6. [DOI] [PubMed] [Google Scholar]
- 57. Wong N, Lam WC, Lai PB, Pang E, Lau WY, Johnson PJ. Hypomethylation of chromosome 1 heterochromatin DNA correlates with q‐arm copy gain in human hepatocellular carcinoma. Am J Pathol 2001; 159: 465–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nakagawa T, Kanai Y, Ushijima S, Kitamura T, Kakizoe T, Hirohashi S. DNA hypomethylation on pericentromeric satellite regions significantly correlates with loss of heterozygosity on chromosome 9 in urothelial carcinomas. J Urol 2005; 173: 243–6. [DOI] [PubMed] [Google Scholar]
- 59. Hansen RS, Wijmenga C, Luo P et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA 1999; 96: 14412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Overexpression of a splice variant of DNA methyltransferase 3b, DNMT3b4, associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc Natl Acad Sci USA 2002; 99: 10060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Soejima K, Fang W, Rollins B. DNA methyltransferase 3b contributes to oncogenic transformation induced by SV40T antigen and activated Ras. Oncogene 2003; 22: 4723–33. [DOI] [PubMed] [Google Scholar]
- 62. Kanai Y, Saito Y, Ushijima S, Hirohashi S. Alterations in gene expression associated with the overexpression of a splice variant of DNA methyltransferase 3b, DNMT3b4, during human hepatocarcinogenesis. J Cancer Res Clin Oncol 2004; 130: 636–44. [DOI] [PubMed] [Google Scholar]
- 63. Estecio MR, Yan PS, Ibrahim AE et al. High‐throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res 2007; 17: 1529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Beck S, Rakyan VK. The methylome: approaches for global DNA methylation profiling. Trends Genet 2008; 24: 231–7. [DOI] [PubMed] [Google Scholar]
- 65. Meissner A, Mikkelsen TS, Gu H et al. Genome‐scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454: 766–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Inazawa J, Inoue J, Imoto I. Comparative genomic hybridization (CGH)‐arrays pave the way for identification of novel cancer‐related genes. Cancer Sci 2004; 95: 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Misawa A, Inoue J, Sugino Y et al. Methylation‐associated silencing of the nuclear receptor 1I2 gene in advanced‐type neuroblastomas, identified by bacterial artificial chromosome array‐based methylated CpG island amplification. Cancer Res 2005; 65: 10233–42. [DOI] [PubMed] [Google Scholar]
- 68. Tanaka K, Imoto I, Inoue J et al. Frequent methylation‐associated silencing of a candidate tumor‐suppressor, CRABP1, in esophageal squamous‐cell carcinoma. Oncogene 2007; 26: 6456–68. [DOI] [PubMed] [Google Scholar]
- 69. Sugino Y, Misawa A, Inoue J et al. Epigenetic silencing of prostaglandin E receptor 2 (PTGER2) is associated with progression of neuroblastomas. Oncogene 2007; 26: 7401–13. [DOI] [PubMed] [Google Scholar]
- 70. Maekita T, Nakazawa K, Mihara M et al. High levels of aberrant DNA methylation in Helicobacter pylori‐infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 2006; 12: 989–95. [DOI] [PubMed] [Google Scholar]
- 71. Clark SJ. Action at a distance: epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet 2007; 16 (Spec No 1): R88–95. [DOI] [PubMed] [Google Scholar]
- 72. Arai E, Ushijima S, Fujimoto H et al. Genome‐wide DNA methylation profiles in both precancerous conditions and clear cell renal cell carcinomas are correlated with malignant potential and patient outcome. Carcinogenesis 2009; 30: 214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Arai E, Kanai Y, Ushijima S, Fujimoto H, Mukai K, Hirohashi S. Regional DNA hypermethylation and DNA methyltransferase (DNMT) 1 protein overexpression in both renal tumors and corresponding nontumorous renal tissues. Int J Cancer 2006; 119: 288–96. [DOI] [PubMed] [Google Scholar]
- 74. Arai E, Ushijima S, Tsuda H et al. Genetic clustering of clear cell renal cell carcinoma based on array‐comparative genomic hybridization: its association with DNA methylation alteration and patient outcome. Clin Cancer Res 2008; 14: 5531–9. [DOI] [PubMed] [Google Scholar]
- 75. Arai E, Ushijima S, Gotoh M et al. Genome‐wide DNA methylation profiles in liver tissue at the precancerous stage and in hepatocellular carcinoma. Int J Cancer 2009; 125: 2854–62. [DOI] [PubMed] [Google Scholar]
- 76. Arase Y, Ikeda K, Suzuki F et al. Comparison of interferon and lamivudine treatment in Japanese patients with HBeAg positive chronic hepatitis B. J Med Virol 2007; 79: 1286–92. [DOI] [PubMed] [Google Scholar]
- 77. Yoshida H, Tateishi R, Arakawa Y et al. Benefit of interferon therapy in hepatocellular carcinoma prevention for individual patients with chronic hepatitis C. Gut 2004; 53: 425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nishiyama N, Arai E, Chihara Y et al. Genome‐wide DNA methylation profiles in urothelial carcinomas and urothelia at the precancerous stage. Cancer Sci 2009; doi: DOI: 10.1111/j.1349-7006.2009.01330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mohn F, Schubeler D. Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends Genet 2009; 25: 129–36. [DOI] [PubMed] [Google Scholar]