

Abstract
Mouse glioma 261 (Gl261) cells are used frequently in experimental glioblastoma therapy; however, no detailed description of the Gl261 tumor model is available. Here we present that Gl261 cells carry point mutations in the K‐ras and p53 genes. Basal major histocompatibility complex (MHC)I, but not MHCII, expression was detected in Gl261 cells. The introduction of interferon‐γ‐encoding genes increased expression of both MHCI and MHCII. A low amount of B7‐1 and B7‐2 RNA was detected in wild‐type cells, but cytokine production did not change expression levels. Gl261 cells were transduced efficiently by adenoviral vectors; the infectivity of retroviral vectors was limited. Low numbers of transplanted Gl261 cells formed both subcutaneous and intracranial tumors in C57BL/6 mice. The cells were moderately immunogenic: prevaccination of mice with irradiated tumor cells 7 days before intracranial tumor challenge prevented tumor formation in approximately 90% of mice. When vaccination was carried out on the day or 3 days after tumor challenge, no surviving animals could be found. In vitro‐growing cells were radiosensitive: less than 2 Gy was required to achieve 50% cell mortality. Local tumor irradiation with 4 Gy X‐rays in brain tumor‐bearing mice slowed down tumor progression, but none of the mice were cured off the tumor. In conclusion, the Gl261 brain tumor model might be efficiently used to study the antitumor effects of various therapeutic modalities, but the moderate immunogenicity of the cells should be considered. (Cancer Sci 2006; 97: 546 – 553)
Abbreviations:
- CAR
coxsackie‐adenovirus receptor
- CTL
cytotoxic T lymphocyte
- DMEM
Dulbecco's Modified Eagle's Medium
- FCS
fetal calf serum
- GFP
green fluorescent protein
- Gl261
glioma 261
- GM‐CSF
Granulocyte‐Macrophage Colony Stimulating Factor
- IFN
interferon
- IL
interleukin
- MHC
major histocompatibility complex
- MOI
multiplicity of infection
- MoMLV
Moloney murine leukemia virus
- PCR
polymerase chain reaction
- RT
reverse transcription.
Malignant brain tumors have a poor outcome, with a median survival of 9 months and only 5–10% of patients surviving 2 years. The conventional therapies for glioblastoma multiforme are surgical removal of the bulk tumor mass, followed by radiotherapy. As complete surgical removal is almost impossible, radiotherapy continues to play a major role in the treatment of malignant gliomas, but these tumors are often radioresistant. The combination of chemotherapy with other modalities provides only modest improvements in survival rates.( 1 , 2 , 3 )
The application of suitable animal models in glioma research is necessary for the development of new therapeutic approaches. An ideal animal model of human glioma should reproduce the major characteristics of this neoplasm: predictable and reproducible in vitro and in vivo growth patterns; infiltrative, but non‐metastatic, tumor growth; and poor immunogenicity. A number of animal brain tumor models are being used; however, no model currently available simulates human high‐grade gliomas exactly. Tumor models vary in their immunogenicity, growth patterns and invasiveness. For this reason it is important to choose an appropriate animal model depending on the endpoint examined. Most models are derived from rat and several xenograft models, based on the intracerebral transplantation of human brain tumors into immune‐deficient nude mice, are also available. Murine models of malignant brain tumors are used much less frequently, mainly because the number of available models is limited.
One of the most frequently used murine brain tumor models is Gl261. The Gl261 tumor was induced originally by intracranial injection of 3‐methylcholantrene into C57BL/6 mice and maintained by serial intracranial and subcutaneous transplantations of small tumor pieces on the syngeneic mouse strain.( 4 ) Perhaps, because of these serial passages, Gl261 lacks the most important glial differentiation markers, such as glial fibrillary acidic protein.( 5 ) During the mid‐1990s, in vitro‐growing cell cultures were established from the Gl261 tumor, almost in parallel in a few laboratories (including ours) and the Gl261 tumor model was used in many immune therapeutic and gene therapeutic investigations.( 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ) Despite its frequent usage, there is no detailed description available of the growth characteristics of the Gl261 cell line, nor the immunogenicity of the in vivo tumor model.
The Gl261 murine glioma model has been used in our laboratory for more than two decades. Recently, we used this model to investigate the therapeutic effect of autologous, cytokine‐secreting tumor vaccines,( 9 ) and the combined effect of radiation and gene‐directed enzyme pro‐drug therapy on brain tumors.( 7 ) In the present paper we give a detailed description of the Gl261 tumor model, including: carcinogenic alterations present in the Gl261 cells; sensitivity of the cells toward retroviral and adenoviral vectors; immunogenic characteristics of the in vivo‐growing tumor cells; and potential alterations in the immunogenicity of the cells after introduction of various cytokine‐expressing vectors into the cells. We believe this information might be useful for those who are going to use this model in the future.
Materials and Methods
Cell lines
The B16 and NIH3T3 cell lines were bought from ECACC (Salisbury, UK) and cultured in DMEM supplemented with 10% FCS.
Permanent Gl261 cell line
Tumor pieces of Gl261 were received from NCI (National Cancer Institute, Frederick, MD, USA) during the early 1980s and maintained either frozen or with serial passages on syngeneic C57Bl/6 mice. To establish a permanent cell line, a Gl261 tumor growing subcutaneously was removed under aseptic conditions and a cell suspension was made using standard techniques. Cells were plated in 10% FCS containing DMEM and split when they reached confluence. No significant differences in the growth characteristics of Gl261 cells were noticed when cells were cultured in DMEM containing 20% FCS, or in Ham's F12 or in DMEM‐Ham's F12 with 10% FCS.
Radiation sensitivity of Gl261 cells
Gl261 cells were plated on Petri dishes (500 cells/dish) and 6 h later they were irradiated with various doses of 60Co‐γ radiation (Gammatron‐3, dose rate: 0.4781 Gy/min; Siemens, Erlangen, Germany). Cells were cultured for 2 weeks, after which time colonies were stained with Coomassie Brillant Blue and counted. The number of colonies on the irradiated plates was compared to the corresponding number on unirradiated plates.
Adenoviral vectors
AdexCALacZ, AdexCAmIL‐2, AdexCAmIL‐4, AdexCAmIL‐12p35, AdexCAmIL‐12p40, AdexCAmGM‐CSF and AdexCAmIFNγ, encoding the corresponding cytokines, have been described previously.( 9 ) As well as AdexCALacZ, we also used another LacZ‐encoding adenoviral vector (AxCAZ3‐F/RGD) that carried an Arg‐Gly‐Asp (RGD) insertion in the adenovirus fiber protein. This allowed the virus to utilize the integrin receptor during the cell entry process instead of the CAR.( 14 ) Cytokine production was measured in the culture supernatants of transduced cells using conventional enzyme‐linked immunosorbent assay kits (Endogen, Woburn, MA, USA and BioSource Europe, Nivelles, Belgium) at various time points after transduction.
Retrovirus vectors
An NIH3T3 cell line producing a replication‐competent, ecotropic MoMLV retroviral vector (BL29) encoding the GFP gene was kindly provided by Dr D. Klatzmann, Paris, France. Retroviral vectors were produced as described.( 15 ) Logarithmically growing NIH3T3 or Gl261 cells were infected with serial dilutions of BL29 supernatant for 1 h under serum‐free conditions with or without polybrene. Transduction efficiency was analyzed 3 days later by measuring the ratio of GFP‐expressing cells by flow cytometry (FACS Calibur; Becton‐Dickinson, Mountain View, CA USA). Transduction efficiency was followed at 3‐day intervals up to 30 days.
Analysis of carcinogenic alterations
Total cellular RNA and DNA were isolated from the Gl261 cells and from the ‘original’ intracranial tumor. Expression of the H‐ras, K‐ras, N‐ras, myc and p53 genes was investigated by Northern blot. Point mutations in exons 1–2 of the H‐ras, K‐ras and N‐ras oncogenes and in exons 5–8 of the p53 gene were investigated as described.( 16 )
B7‐1 and B7‐2 expression in Gl261 cells
Total cellular RNA was isolated from control and cytokine‐producing Gl261 cells 48 h after adenovirus infection. RNA was reverse transcribed and the cDNAs amplified in one step (Access RT‐PCR System; Promega, Madison, WI, USA), according to the manufacturer's instructions. Briefly, 220 ng RNA was reverse transcribed and amplified in the presence of 0.1 mM dNTPs, 0.1 µM of each primer, 0.75 mM MgSO4 and reaction buffer in 25‐µL final volumes. The enzymes added were: 0.5 U AMV reverse transcriptase (5 U/µL) and 0.5 U Tfl DNA polymerase (5 U/µL). RT was carried out at 48°C for 45 min, followed by an initial denaturation step at 94°C for 2 min, and an amplification of the cDNA product for 30 cycles using the following parameters: denaturation at 94°C for 0.5 min, annealing at 55°C for 0.5 min and primer extension at 68°C for 1 min, followed by a final incubation at 72°C for 7 min. The same cycling parameters and number of cycles were used for both B7‐1 and B7‐2 analysis. As a control, β‐actin expression was measured by RT‐PCR in a separate reaction. Reaction conditions were similar to the B7‐1 and B7‐2 amplification, except that the MgSO4 concentration was 0.5 mM and the annealing temperature was 50°C. The following primers were used: β‐actin: 5′‐GGCTGTATTCCCCTCCATCGT‐3′, 5′‐CGTACATGGCTGGGGTGTTGA‐3′; B7‐1: 5′‐TGCTGTCTGTCATTGCTGGGA‐3′, 5′‐CCCAGGTGAAGTCCTCTGACA‐3′; and B7‐2: 5′‐GTAGACGTGTTCCAGAACTT‐3′, 5′‐TCTCACTGCCTTCACTCTGCAT‐3′.
MHCI and MHCII expression in Gl261 cells
The same RNA was used as above. RT‐PCR was carried out in two steps: 1 µg of total cytoplasmic RNA was mixed with 0.5 µg of oligo(dT)15 primer in a final volume of 5 µL. The reaction mixture was incubated at 70°C for 5 min and chilled immediately on ice. The RT reaction mixture consisted of 3 mM MgCl2, 0.5 mM dNTP mix, ImProm‐II reverse transcriptase (Promega) and reaction buffer supplied by the manufacturer, in a final volume of 15 µL. Five µL of RNA and primer mix was added to the RT reaction mix, and tubes were incubated in a thermal cycler at 25°C for 5 min, followed by a 1‐h incubation at 42°C. The RT enzyme was inactivated by incubation at 70°C for 15 min. For the PCR, 3 µL of the RT reaction was used. PCR conditions were as follows: 3 mM MgCl2, 1 mM dNTPs, 75 pmol of each primer and 2 U of DynaZyme II DNA polymerase (Finnzymes, Espoo, Finland) in a final volume of 100 µL. Cycling conditions were identical to those for β‐actin amplification described above. β‐actin expression was again used as a control. The following primers were used: MHCI: 5′‐AGGCA‐CATGTGACCCATCACC‐3′, 5′‐CCATCTCAGGGTGAGG‐GGCTC‐3′; and MHCII: 5′‐CCTGTGATCAACATCACATGG‐CTC‐3′, 5′‐CACACACCACAGTCTCT GTCAGCT‐3′. RT‐PCR products were analyzed on 5% polyacrylamide gels. Semi‐quantitative measurement of mRNA expression was carried out with the GDS8000 Complete Gel Documentation and Analysis System (UVP International, Cambridge, UK). When the effect of irradiation on MHCI, MHCII, B7‐1 or B7‐2 expression was studied, wild‐type or cytokine‐transduced Gl261 cells were irradiated with 20 Gy Co60‐γ‐radiation 24 h before RNA isolation and RT‐PCR analysis.
Tumor models and tumor treatment
Intracranial and subcutaneous tumor transplantation, treatment of tumor‐bearing mice with cytokine‐secreting tumor cell vaccines, and local tumor irradiation were carried out as described previously.( 7 , 9 ) To investigate the in vivo immunogenicity of Gl261 cells, intracranial tumors were induced by transplantation of 2 × 104 cells. Subcutaneous vaccination was carried out with 1 × 104, 1 × 105 and 1 × 106 Gl261 cells in 100‐µL final volumes, either 7 days or 3 days before, on the day of, or 3 days after tumor transplantation. In vitro‐growing Gl261 cells used in vaccination studies were irradiated with 20 Gy Co60‐γ‐radiation immediately before use to stop cell division. All animal studies were carried out according to Hungarian national regulations under the permission and guidance of the corresponding institutional and national ethical bodies.
Histopathology
Brain tumors were removed, fixed in 4% formaldehyde and embedded in paraffin. Thin sections of 8 µm were cut and stained with hematoxylin‐eosin.
Results
In vitro growth and radiation sensitivity of Gl261 cells
Gl261 cells grew rapidly under in vitro conditions with a population doubling time of 20 h in log phase. Cells did not show contact inhibition. Cell densities of 2.5 × 105/cm2 were reached without substantially affecting cell viability. In various gene therapy protocols, radiation is used to stop cell division of genetically modified cells. Therefore, radiation sensitivity of Gl261 cells was investigated after irradiation with increasing doses of Co60‐γ‐radiation (data not shown). At 2 Gy, survival was approximately 40%, at 10 Gy it was approximately 0.0009% and at 20 Gy no surviving cells were seen, suggesting that irradiation of Gl261 cells with 20 Gy is sufficient to completely and irreversibly stop cell division.
Transduction efficiency with adenoviral and retroviral vectors
First, in vitro‐growing cells were infected with a LacZ‐encoding adenoviral vector (AdexCALacZ) at different MOI. At 10 MOI, approximately 70% of the cells carried the vector, whereas all cells carried the vector at 100 MOI. We also investigated how expression of the therapeutic genes was dependent on the MOI used. When Gl261 cells were transduced with either an IL‐2 or a GM‐CSF‐encoding adenoviral vector, IL‐2 and GM‐CSF production increased linearly up to 500 MOI (data not shown). The fact that cytokine production continued to increase even above 100 MOI, when all of the cells were already infected, suggested that Gl261 cells could pick up more than one copy of the adenoviral vector. It also suggests that increasing the MOI increases the expression levels of the therapeutic genes.
Adenoviruses enter the cells through the CAR. Many cells are deficient in CAR, which results in low transduction efficiency with adenoviral vectors. There are reports that modification of the adenovirus fiber protein, allowing the virus to bind to integrin receptors, can increase transduction efficiency in many cell lines.( 17 ) To test this possibility we used another LacZ‐encoding vector (AxCAZ3‐F/RGD) that enters the cells through integrin receptors. No increase in transduction efficiency was seen in Gl261 cells with this vector (data not shown).
Using adenoviral vectors, the expression of the introduced genes is transient. It is important to know how long the therapeutic genes are expressed in the cells and how irradiation with high doses influences gene expression. In Gl261 cells transduced with an IL‐2‐encoding vector (AdexCA‐IL‐2), cytokine secretion peaked 48 h after infection and remained nearly constant for an additional 4 days (Fig. 1). Irradiating Gl261 cells with a lethal dose (20 Gy) had no substantial effect on cytokine secretion during this interval (Fig. 1). Irradiated cells started to die after 5 days.
Figure 1.

Interleukin (IL)‐2 production in irradiated glioma 261 (Gl261) cells. Cells were transduced with IL‐2 encoding adenoviral vector at 200 multiplicity of infection. Twenty‐four hours after infection cells were irradiated with 10 or 20 Gy Co60‐γ‐radiation. IL‐2 levels were determined in the culture medium by enzyme–linked immunosorbent assay at different time points after irradiation. Values represent the mean of three independent experiments.
We also tested transduction efficiency after infection with retroviral vectors. Gl261 cells or NIH3T3 cells were infected with serial dilutions of an ecotropic MoMLV, replication‐competent, GFP‐encoding retroviral vector (BL29). Because of the replicative ability of the vector, GFP transduction rate was followed up to 30 days after infection. When Gl261 cells were infected with undiluted BL29 vector preparation, approximately 1 or 3% of the cells expressed GFP 3 days after transduction, reaching 5 or 7% at day 30, in the absence or presence of polybrene, respectively. For the NIH3T3‐positive control cells, 90% of transduction was observed at day 3 with 100‐fold dilution of the same vector preparation, and when starting from 1% of transduction at day 3, GFP was propagated in 100% of the cells within 1 week. These results demonstrate the low sensitivity of Gl261 cells toward retroviral infection and propagation.
Carcinogenic alterations in Gl261 cells
We have studied the potential oncogenic alterations in genes that are involved frequently in human carcinogenesis. First, expression of the myc, H‐ras, K‐ras and N‐ras oncogenes and the p53 tumor suppressor gene were investigated at the RNA level both in in vitro‐growing Gl261 cells and in the ‘original’ brain tumor. Compared to normal brain, elevated expression of the myc oncogene and p53 tumor suppressor gene was detected, whereas H‐ras expression decreased. No changes in expression of the K‐ras or N‐ras oncogenes was seen (data not shown).
Next, the presence of point mutations was investigated in exons 1 and 2 of the ras oncogenes and in exons 5–8 of the p53 tumor suppressor gene. No point mutations were detected in the H‐ras and N‐ras genes. In the K‐ras gene one mutation was found at codon 12 of exon 1 (GGT to GTT, replacing glycine with valine). The p53 gene also possessed a homozygous point mutation at codon 153 of exon 5, where the wild‐type CGT sequence mutated to CCT (replacing arginine with proline).
Immunological characteristics of Gl261 cells
Expression of the major histocompatibility antigens MHCI and MHCII was investigated by RT‐PCR in Gl261 wild‐type cells and in cells genetically modified by appropriate adenoviral vectors to secrete the following cytokines: IL‐2, IL‐4, IL‐12, GM‐CSF or IFN‐γ. A well detectable basal MHCI level could be seen in wild‐type cells, which was definitely higher than the MHCI level in healthy brain (used as control). The only cytokine that modified MHCI expression was IFN‐γ. MHCI levels increased in parallel with the amount of IFN‐γ produced by transduced Gl261 cells (Table 1). No detectable MHCII expression was observed in wild‐type Gl261 cells. Infection of Gl261 cells with the IFN‐γ‐encoding adenovirus vector produced pronounced induction of MHCII expression (Table 1). Irradiation with 20 Gy had no effect on MHCI and MHCII expression levels, neither in wild‐type nor in cytokine‐producing cells (data not shown).
Table 1.
Major histocompatibility (MHC) expression in interferon (IFN)‐γ‐producing glioma 261 (Gl261) cells
| IFN‐γ (MOI) | MHCI | MHCII |
|---|---|---|
| 0 | 1 | 0 |
| 10 | 2.17 ± 0.74 | 0.097 ± 0.018 |
| 50 | 3.45 ± 1.25 | 0.231 ± 0.049 |
| 100 | 6.96 ± 1.33 | 0.194 ± 0.05 |
Gl261 cells were transduced at different multiplicity of infection (MOI) with AdexCAmIFNγ. MHCI and MHCII expression was measured at the RNA level by reverse transcription–polymerase chain reaction. All data represent the mean of six independent experiments. MHCI expression values are shown relative to MHC expression in non‐transduced cells. Because there was no detectable MHCII expression in IFN‐γ‐non‐producing cells, the expression values are presented relative to actin expression in uninfected cells.
Expression of B7‐1 and B7‐2 costimulatory molecules was also investigated. A low amount of B7‐1 and B7‐2 RNA could be detected in wild‐type cells, but cytokine production did not change this expression. Irradiation of wild‐type or cytokine‐secreting cells also did not modify B7‐1 or B7‐2 expression levels (data not shown).
Growth characteristics and radiation sensitivity of in vivo‐growing Gl261 cells
Transplanted Gl261 cells can form both subcutaneous and intracranial tumors in the syngeneic host, C57BL/6 mice. Neither the intracranial nor the subcutaneous tumors will give metastases. After subcutaneous implantation large tumor volumes were reached. Animals died either due to cachexia or secondary infections. Intracranial tumors had a rapid growth rate with slightly invasive growth pattern (Fig. 2). Lymphocyte infiltration was hardly detectable in intracranial tumors, but after treatment with a GM‐CSF‐secreting tumor vaccine, strong CD4+ and CD8+ infiltration was detected.( 9 )
Figure 2.

Histopathology of glioma 261 (Gl261) tumors. Sections were cut from formaldehyde‐fixed tumors and stained with hematoxylin–eosin. N, normal brain tissue; T, brain tumor.
After intracranial implantation of 1 × 105, 1 × 104, 1 × 103 and 1 × 102 Gl261 cells, the mean survival time of the animals was 25, 27, 36 and 55 days, respectively (Fig. 3). The animals died due to local tumor growth and increased intracranial pressure. Treatment with 4 Gy local tumor irradiation substantially slowed down tumor progression, but no tumor‐bearing animals were cured by radiation alone (Fig. 3).
Figure 3.

Survival of glioma‐bearing mice. Brain tumors were induced by intracranial injection of various numbers of glioma 261 (Gl261) cells. Tumor‐bearing mice were left untreated or the tumors were irradiated with 4 Gy X‐rays and survival followed. Survival curves represent the pooled data of two independent experiments (10 mice per treatment group). irr, tumors were treated by local irradiation.
Immunogenicity of the in vivo‐growing Gl261 cells
Glioma‐bearing mice were vaccinated (or prevaccinated) with a single subcutaneous injection of irradiated (20 Gy) Gl261 cells at different time points relative to the intracranial tumor transplantation. When subcutaneous vaccination was carried out 7 or 3 days before intracranial tumor transplantation, approximately 80–90% or 30–40% of the animals remained tumor free, respectively. However, if subcutaneous vaccination was done on the day of the intracranial tumor transplantation, or 3 days thereafter, no significant effect was observed compared to non‐vaccinated controls (Fig. 4). These data suggest that Gl261 cells are moderately immunogenic. When surviving animals were rechallenged 9 months later with intracranial transplantation of Gl261 cells, no tumors were formed, showing that the immunity raised by Gl261 vaccination was long lasting.
Figure 4.
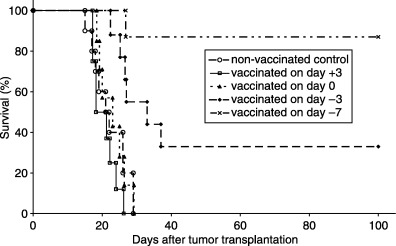
Survival of glioma‐bearing mice vaccinated with autologous tumor cells. Brain tumors were induced by intracranial injection of 2 × 104 glioma 261 (Gl261) cells in 10 µL final volume on day 0. Subcutaneous vaccinations were carried out with 1 × 106 Gl261 cells 7 days before (day −7), 3 days before (day −3), on the day of (day 0), or 3 days after (day +3) tumor transplantation and survival followed. Survival curves represent the pooled data of two independent experiments (10 mice per treatment groups).
The number of cells used for prevaccination was also important, as only animals prevaccinated with at least 1 × 106 Gl261 cells 7 days before the intracranial tumor challenge remained tumor free. When fewer cells (1 × 105) were used for vaccination no effect on mouse survival was found. We also investigated the specificity of this immunity. C57BL/6 mice were prevaccinated with 1 × 106 Gl261 cells 7 days before being challenged by intracranial transplantation of B16 melanoma cells. Gl261 prevaccination failed to result in an increase in median survival of B16‐challenged mice, showing that immune activation was raised specifically against Gl261 cells (data not shown).
Discussion
Gl261 is one of the few available murine glioma models. Originally this tumor was propagated in vivo by transplanting small tumor pieces on its syngeneic host, the C57BL/6 mouse. In the mid‐1990s, a few laboratory groups established a permanent cell line from the tumor.( 6 , 8 , 9 , 10 , 11 , 12 ) In the present paper we report the detailed characterization of the Gl261 cell line and the tumor model.
Gl261 cells are easily adapted to in vitro growth. Cells have a fast growth rate with no contact inhibition. Gl261 tumors grow aggressively in their syngeneic host, the C57BL/6 mouse. The highly aggressive growth characteristic of the in vivo tumor is also supported by the finding that the tumor might be transplanted efficiently onto F1 hybrids of C57BL/6 and DBA2 mice (E. J. Hídvégi, unpublished data, 1986). We showed that as few as 100 tumor cells transplanted intracranially killed all of the animals within 70 days, but implantation of 1 × 105 cells had 100% mortality rate within 25 days. Similar results were obtained by Plautz et al., Natsume et al. and Glick et al. who showed that intracranial injection of 1–4 × 105 Gl261 cells killed all of the mice within 21 days.( 10 , 12 , 18 ) In this context the Gl261 glioma model can be included among the aggressively growing tumors. Rat glioma models, such as 9L, F98 and CNS‐1, also have a high rate of tumor take and a short median survival time. In contrast, the C6 rat glioma( 19 , 20 ) and the 4C8 mouse glioma( 5 ) models are much less aggressive. Some human gliomas transplanted onto nude animals (T98G, SF‐767, SF‐126) have quite low tumor take rates and grow very slowly, whereas others grow well (U‐251 MG, U‐87 MG).( 21 , 22 ) We have never encountered spontaneous regression of the tumors for the Gl261 model, as has been described for the C6 model.( 23 )
The in vitro‐growing Gl261 cells are rather radiosensitive, as less than 2 Gy is required to achieve 50% cell mortality. Other frequently used glioma models are either more radiation resistant (most human glioma cell lines, such as U‐373MG, U‐87MG, SF126 and T98G),( 24 , 25 ) or their radio‐sensitivity is in a similar range to that of Gl261 cells (C6 and RG2).( 26 , 27 ) However, the radiosensitivity of the in vivo‐growing Gl261 tumor does not correlate well with its in vitro radiosensitivity. We showed that if animals are transplanted intracranially with relatively low number of tumor cells and irradiated with 4 Gy at an early time point after tumor induction (3 days), irradiation has no curative effect. Only a mild prolongation in the life time of the irradiated animals could be seen. Similar findings were reported by Taghian and his coworkers, showing that the intrinsic radiosensitivity of glioma cell lines may not correlate with the radiosensitivity of the in vivo‐growing tumor, and thus may not be the major determinant of the poor clinical outcome of glioma patients.( 28 , 29 )
Gl261 cells could be very efficiently transduced with adenoviral vectors (70% transduction efficiency at 10 MOI and 100% at 100 MOI). We even demonstrated that Gl261 cells could take more than one copy of the adenoviral vector. Adenoviral transduction efficiency of other glioma cell lines is lower than that of the Gl261 cells: at 100 MOI the transduction efficiency of the human glioma U373 cells is 75%, for rat C6 cells it is 25% and for 9L cells it is 10% (K. Lumniczky, unpublished data, 2005). Cytokine production after infection with the corresponding cytokine‐encoding adenovirus vectors peaked 48 h after infection and remained barely unchanged for almost 1 week. Irradiation with high doses (20 Gy) hardly changed cytokine secretion during this interval. It is in slight contrast with former reports that in human and murine melanoma cell lines lethal‐dose irradiation increased cytokine expression (IL‐2 and IFN‐γ).( 30 )
In contrast to the high transduction efficiency of adenoviral vectors, MoMLV‐derived ecotropic retroviral vectors show a very limited capability to infect Gl261 cells productively. Much higher retroviral transfection efficiency (20–30%) was otherwise reported for a spontaneously arisen murine astrocytoma cell line (SMA‐560).( 31 )
Both the Gl261 cells and the ‘original’ brain tumor contain several molecular biological alterations characteristic of human gliomas. They harbor a p53 mutation that is accompanied by elevated p53 expression. The p53 gene mutation is a very common alteration in human brain tumors and it is usually accompanied by a bad prognosis.( 32 , 33 ) It is interesting, however, that Blaszczyk‐Thurin and Ertl also reported elevated p53 expression in Gl261 cells, but they did not detect any point mutation in the sequenced cDNAs.( 34 ) We cannot exclude the possibility that the p53 mutation present in our Gl261 tumor was acquired in our laboratory during the early passages, but it should be clarified by additional experiments. Importantly, this p53 mutation is present at a non‐conserved region of the gene (codon 153),( 35 ) suggesting a low functional importance for it. The Gl261 cells also carry a mutated K‐ras gene and activated c‐myc. The K‐ras mutation is located at codon 12 of exon 1, which is one of the most frequently mutated sites of the K‐ras gene. This mutation probably reflects the chemical carcinogen‐induced (3‐methylcholantrene) origin of the tumor. K‐ras mutations are reported to be present in human brain tumors, although with lower frequency than in other human tumor types.( 36 ) Alterations of the c‐myc gene are relatively rare in human gliomas, but elevated c‐myc RNA levels, as well as amplification and rearrangement of the gene,( 37 ) and prolonged half‐life and stabilization of the protein, were described.( 38 )
Several publications report that MHCI and MHCII antigens are downregulated in many gliomas. Recently, Zagzag et al. detected the downregulation of MHCI and MHCII expression in different migrating and invading human and murine glioblastoma cell lines in vitro and in vivo.( 39 ) Rat C6 cells have also been shown to downregulate MHCI and MHCII levels,( 40 ) and 9L cells also expressed low levels of MHCI.( 41 ) Anderson et al. demonstrated that in vitro culturing of established human glioma cell lines leads to an increase in the cell surface expression of MHCI and MHCII antigens and the B7‐2 receptor when comparing ex vivo tumor cells to those after a single or varying passages in vitro.( 42 ) We found that Gl261 cells had slightly elevated MHCI RNA levels compared to RNA isolated from normal brain. MHCII levels were barely detectable, similar to the levels in healthy brain tissue. In concordance with other reports in the literature we found that the only cytokine that was able to modify MHC expression was IFN‐γ.( 31 , 41 , 43 ) In particular, MHCII levels were increased dramatically, as Akbasak et al. have also shown.( 6 ) Irradiation of cytokine‐producing Gl261 cells did not change MHC expression levels. Wen et al. reported the same for 9L cells.( 44 ) Abdel‐Wahab et al., however, detected increased MHC expression in human and murine melanoma cell lines after lethal‐dose irradiation.( 30 ) We found that a very low, but detectable, level of B7‐1 and B7‐2 RNAs were expressed constitutively on Gl261 cells, whereas they were absent in healthy brain tissue. Irradiation or cytokine secretion had no effect on B7‐1 or B7‐2 expression either. Constitutive expression of the B7 costimulatory molecules was not detected in early passage human glioma cultures or in established human glioma cell lines.( 45 )
Gl261 is a moderately immunogenic tumor model. We demonstrated that prevaccination of mice with a minimum of one million Gl261 cells 7 or 3 days prior to intracranial transplantation of the tumor protected the animals from tumor outgrowth. We think that immunogenicity of Gl261 cells might be due to elevated MHCI expression and to the presence of B7 costimulatory molecules on the surface of Gl261 cells, which may render them more susceptible to class I MHC‐dependent cytotoxic T cell recognition and to a certain degree of antigen presentation. Several of the currently used animal glioma models are also immunogenic. In 1974, Blume et al. already reported that 9L rat glioma cells are highly immunogenic, with 89% of the preimmunized animals resistant to subsequent intracranial or subcutaneous tumor challenge.( 46 ) The C6 model is also immunogenic. However, in this case the reason might be the lack of a syngeneic host, as the tumor arose in an outbred Wistar rat.( 47 ) Animal gliomas induced by oncoviruses are also highly immunogenic.( 48 ) Other glioma cell lines, such as F98,( 49 ) CNS‐1( 50 ) and RG2( 51 ) rat cells as well as human high‐grade brain tumors are regarded as being non‐ or weakly immunogenic.
We demonstrated the existence of an immunological memory in mice that have been prevaccinated with irradiated Gl261 cells. When these animals were intracranially rechallenged with wild‐type Gl261 cells, no tumors formed. Smilowitz et al. describes the same in rats who had been vaccinated with 9L cells.( 52 ) In a study where they treated Gl261 bearing mice with a combination of sublethal whole‐body irradiation and intravenous transfer of tumor‐draining lymph node T cells, Plautz et al. showed that cured mice rechallenged 60 days after initial tumor challenge with Gl261 cells rejected the tumor.( 10 ) Long‐term immune memory was also demonstrated in Gl261 tumor‐bearing mice after intratumoral administration of liposomes containing the murine IFN‐β gene.( 13 ) Besides, we also proved the specificity of the immune response, as prevaccinated mice were protected only against Gl261 glioma, and were not protected against intracranially implanted B16 melanoma. This is an interesting point, because it has been shown that malignant melanomas and gliomas share common antigens. Prins et al. reported in a recent publication that Gl261 cells express the melanoma antigens gp100 and tyrosine‐related protein 2. They also found that a gp100‐specific CTL cell line would specifically lyse both Gl261 cells and B16 melanoma cells.( 53 ) However, a highly specific immune response was also detected by Smilowitz et al. who found that rats prevaccinated with 9L cells were not protected against the D74 glioma.( 52 )
In conclusion, the Gl261 murine glioma model harbors the main characteristics of most glioma models. It has an invasive but non‐metastatic growth pattern, a high tumor take rate (where survival time is dependent on the tumor cell number transplanted initially), it carries both p53 and K‐ras mutations, and has elevated c‐myc and p53 expression. Contrary to other animal glioma models, which often downregulate MHCI expression, Gl261 has an elevated basal MHCI expression and also expresses low levels of B7‐1 and B7‐2 RNA, which might be responsible for the immunogenicity of the cells. The Gl261 brain tumor model can be used efficiently to investigate the antitumor effects of several therapeutic modalities, such as immunotherapy, gene therapy, chemotherapy or radiotherapy, and antiangiogenic therapy, but the moderate immunogenicity of the cells should be considered when evaluating the experimental data.
Acknowledgments
This work was supported by the following Hungarian Grants: ETT 494/2003 and 158/2003; OTKA T‐047034, F‐034775, F‐46330, T‐032627; NKFP 1/048/2001 and the EU grant: QLK3‐CT‐1999‐00364. The authors appreciate the expert technical assistance of Erzsébet Fekete, Mária Frigyesi and Rita Lökös.
TS and LK contributed equally to this study.
References
- 1. DeAngelis LM. Brain tumours. N Engl J Med 2001; 344: 114–23. [DOI] [PubMed] [Google Scholar]
- 2. Glioma Meta‐analysis Trialists Group. Chemotherapy in adult high‐grade glioma: a systemic review and meta‐analysis of individual patient data from 12 randomised trials. Lancet 2002; 359: 1011–18. [DOI] [PubMed] [Google Scholar]
- 3. Legler JM, Gloeckler Ries LA, Smith MA et al. Brain and other central nervous system cancers: recent trends in incidence and mortality. J Natl Cancer Inst 1999; 91: 1382–90. [DOI] [PubMed] [Google Scholar]
- 4. Ausman JI, Shapiro WR, Rall DP. Studies on the chemotherapy of experimental brain tumors: development of an experimental model. Cancer Res 1970; 30: 2394–400. [PubMed] [Google Scholar]
- 5. Weiner NE, Pyles RB, Chalk CL et al. A syngeneic mouse glioma model for study of glioblastoma therapy. J Neuropathol Exp Neurol 1999; 58: 54–60. [DOI] [PubMed] [Google Scholar]
- 6. Akbasak A, Oldfield EH, Saris SC. Expression and modulation of major histocompatibility antigens on murine primary brain tumor in vitro . J Neurosurg 1991; 75: 922–9. [DOI] [PubMed] [Google Scholar]
- 7. Désaknai S, Lumniczky K, Ésik O, Hamada H, Sáfrány G. Local tumour irradiation enhances the anti‐tumour effect of a double‐suicide gene therapy system in a murine glioma model. J Gene Med 2003; 5: 377–85. [DOI] [PubMed] [Google Scholar]
- 8. Herrlinger U, Kramm CM, Johnston KM et al. Vaccination for experimental gliomas using GM‐CSF‐transduced glioma cells. Cancer Gene Ther 1997; 4: 345–52. [PubMed] [Google Scholar]
- 9. Lumniczky K, Désaknai S, Mangel L et al. Local tumor irradiation augments the anti‐tumor effect of cytokine producing autologous cancer cell vaccines in a murine glioma model. Cancer Gene Ther 2002; 9: 44–52. [DOI] [PubMed] [Google Scholar]
- 10. Plautz GE, Touhalisky JE, Shu S. Treatment of murine gliomas by adoptive transfer of ex vivo activated tumor‐draining lymph node cells. Cellular Immunol 1997; 178: 101–7. [DOI] [PubMed] [Google Scholar]
- 11. Yu JS, Burwick JA, Dranoff G, Breakefield XO. Gene therapy for metastatic brain tumors by vaccination with granulocyte‐macrophage colony‐stimulating factor‐transduced tumor cells. Hum Gene Ther 1997; 10: 1065–72. [DOI] [PubMed] [Google Scholar]
- 12. Natsume A, Mizuno M, Ryuke Y, Yoshida J. Antitumor effect and cellular immunity activation by murine interferon‐β gene transfer against intracerebral glioma in mouse. Gene Ther 1999; 6: 1626–33. [DOI] [PubMed] [Google Scholar]
- 13. Natsume A, Tsujimura K, Mizuno M, Takahashi T, Yoshida J. IFN‐β gene therapy induces systemic antitumor immunity against malignant glioma. J Neurooncol 2000; 47: 117–24. [DOI] [PubMed] [Google Scholar]
- 14. Nakamura T, Sato K, Hamada H. Effective gene transfer to human melanomas via integrin‐targeted adenoviral vectors. Hum Gene Ther 2002; 13: 613–26. [DOI] [PubMed] [Google Scholar]
- 15. Solly SK, Trajcevski S, Frisen C et al. Replicative retroviral vectors for cancer gene therapy. Cancer Gene Ther 2003; 10: 30–9. [DOI] [PubMed] [Google Scholar]
- 16. Lumniczky K, Antal S, Unger E, Wunderlich L, Hídvégi EJ, Sáfrány G. Carcinogenic alterations in murine liver, lung and uterus tumors induced by in utero exposure to ionizing radiation. Mol Carcinogen 1998; 21: 100–10. [PubMed] [Google Scholar]
- 17. Koizumi N, Mizuguchi H, Hosono T et al. Efficient gene transfer by fiber‐mutant adenoviral vectors containing RGD peptide. Biochim Biophys Acta 2001; 1568: 13–20. [DOI] [PubMed] [Google Scholar]
- 18. Glick RP, Lichtor T, Kim TS, Ilangovan S, Cohen EP. Fibroblasts genetically engineered to secrete cytokines suppress tumor growth and induce antitumor immunity to a murine glioma in vivo . Neurosurgery 1995; 36: 548–55. [DOI] [PubMed] [Google Scholar]
- 19. San‐Galli F, Vrignaud P, Robert J, Coindre JM, Cohadon F. Assessment of the experimental model of transplanted C6 glioblastoma in Wistar rats. J Neurooncol 1989; 7: 299–304. [DOI] [PubMed] [Google Scholar]
- 20. Li Y, Owusu A, Lehnert S. Treatment of intracranial rat glioma model with implant of radiosensitizer and biomodulator drug combined with external beam radiotherapy. Int J Radiat Oncol Biol Phys 2004; 58: 519–27. [DOI] [PubMed] [Google Scholar]
- 21. Rubenstein M, Shaw M, Mirochnik Y et al. In vivo establishment of T98G human glioblastoma. Meth Find Exp Clin Pharmacol 1999; 21: 391–3. [DOI] [PubMed] [Google Scholar]
- 22. Ozawa T, Wang J, Hu LJ, Bollen AW, Lamborn KR, Deen DF. Growth of human glioblastomas as xenografts in the brains of athymic rats. In Vivo 2002; 16: 55–60. [PubMed] [Google Scholar]
- 23. Vince GH, Bendszus M, Schweitzer T et al. Spontaneous regression of experimental gliomas: an immunohistochemical and MRI study of the C6 glioma spheroid implantation model. Exp Neurol 2004; 190: 478–85. [DOI] [PubMed] [Google Scholar]
- 24. Acevedo‐Duncan M, Peariman J, Zachariah B. Sensitivity of human glioma U‐373MG cells to radiation and the protein kinase C inhibitor, calphostin. C Cell Prolif 2001; 34: 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yount GL, Levine KS, Kuriyama H, Haas‐Kogan DA, Israel MA. Fas (APO‐1/CD95) signaling pathway is intact in radioresistant human glioma cells. Cancer Res 1999; 59: 1362–5. [PubMed] [Google Scholar]
- 26. Weizsacker M, Nagamune A, Winkelstroter R, Vieten H, Wechsler W. Radiation and drug response of the rat glioma RG2. Eur J Cancer Clin Oncol 1982; 18: 891–5. [DOI] [PubMed] [Google Scholar]
- 27. Zhang W, Yamada H, Sakai N, Nozawa Y. Sensitization of C6 glioma cells to radiation by stauroporine, a potent protein kinase C inhibitor. J Neurooncol 1993; 15: 1–7. [DOI] [PubMed] [Google Scholar]
- 28. Taghian A, Ramsay J, Allalunis‐Turner J et al. Intrinsic radiation sensitivity may not be the major determinant of the poor clinical outcome of glioblastoma multiforme. Int J Radiat Oncol Biol Phys 1993; 25: 243–9. [DOI] [PubMed] [Google Scholar]
- 29. Taghian A, Dubois W, Budach W, Baumann M, Freeman J, Suit H. In vivo radiation sensitivity of glioblastoma multiforme. Int J Radiat Oncol Biol Phys 1995; 32: 99–104. [DOI] [PubMed] [Google Scholar]
- 30. Abdel‐Wahab Z, Dar MM, Hester D et al. Effect of irradiation on cytokine production, MHC antigen expression, and vaccine potential of interleukin‐2 and interferon‐gamma gene‐modified melanoma cells. Cell Immunol 1996; 171: 246–54. [DOI] [PubMed] [Google Scholar]
- 31. Sampson JH, Ashley DM, Archer GE et al. Characterization of a spontaneous murine astrocytoma and abrogation of its tumorigenicity by cytokine secretion. Neurosurgery 1997; 41: 1365–73. [DOI] [PubMed] [Google Scholar]
- 32. Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 1992; 355: 846–7. [DOI] [PubMed] [Google Scholar]
- 33. Ishii N, Tada M, Hamou MF et al. Cells with TP53 mutations in low grade astrocytic tumors evolve clonally to malignancy and are an unfavorable prognostic factor. Oncogene 1999; 18: 5870–80. [DOI] [PubMed] [Google Scholar]
- 34. Blaszczyk‐Thurin MOI, Ertl HCJ. An experimental vaccine expressing wild‐type p53 induces protective immunity against glioblastoma cells with high levels of endogenous p53. Scand J Immunol 2002; 56: 361–75. [DOI] [PubMed] [Google Scholar]
- 35. Friend S. p53: a glimpse at the puppet behind the shadow play. Nature 1994; 265: 334–5. [DOI] [PubMed] [Google Scholar]
- 36. Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682–9. [PubMed] [Google Scholar]
- 37. Trent J, Meltzer P, Rosenblum M et al. Evidence for rearrangement, amplification and expression of c‐myc in a human glioblastoma. Proc Natl Acad Sci USA 1986; 83: 470–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shindo H, Tani E, Matsumuto T, Hashimoto T, Furuyama J. Stabilization of c‐myc protein in human glioma cells. Acta Neuropathol (Berlin) 1993; 86: 345–52. [DOI] [PubMed] [Google Scholar]
- 39. Zagzag D, Salnikow K, Chiriboga L et al. Downregulation of major histocompatibility antigens in invading glioma cells: stealth invasion of the brain. Lab Invest 2005; 85: 328–41. [DOI] [PubMed] [Google Scholar]
- 40. Oshiro S, Liu Y, Fukushima T, Asotra K, Black KL. Modified immunoregulation associated with interferon‐gamma treatment of rat glioma. Neurol Res 2001; 23: 359–66. [DOI] [PubMed] [Google Scholar]
- 41. Wen PY, Lampson MA, Lampson LA. Effects of γ‐interferon on major histocompatibility complex antigen expression and lymphocytic infiltration in the 9L gliosarcoma brain tumor model: implications for strategies of immunotherapy. J Neuroimmunol 1992; 36: 57–68. [DOI] [PubMed] [Google Scholar]
- 42. Anderson RC, Elder JB, Brown MD et al. Changes in the immunologic phenotype of human malignant glioma cells after passaging in vitro . Clin Immunol 2002; 102: 84–95. [DOI] [PubMed] [Google Scholar]
- 43. Oshiro S, Fukushima T, Tomonaga M, Black KL. Response of MHC class‐1 antigen on rat glioma cells to cytokines. Anticancer Res 2000; 20: 605–10. [PubMed] [Google Scholar]
- 44. Wen P, Loeffler JS, Morris JH, Lampson LA. The effects of irradiation on major histocompatibility complex expression and lymphocytic infiltration in the normal rat brain and the 9L gliosarcoma brain tumor model. J Neuroimmunol 1990; 27: 239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parney IF, Farr‐Jones MA, Chang LJ, Petruk KC. Human glioma immunobiology in vitro: implications for immunogene therapy. Neurosurgery 2000; 46: 1169–77. [DOI] [PubMed] [Google Scholar]
- 46. Blume MR, Wilson CB, Vasquez DA. 1974; Immune response to a transplantable intracerebral glioma in rats. In: Sane K, Ishi S, LeVray D, eds. Recent Progress in Neurologic Surgery. Amsterdam: Excerpta Medica, 1974; 129–34. [Google Scholar]
- 47. Trojan J, Johnson TR, Rudin SD, Ilan J, Tykocinski ML, Ilan J. Treatment and prevention of rat glioblastoma by immunogenic C6 cells expressing antisense insulin‐like growth factor I RNA. Science 1993; 259: 94–7. [DOI] [PubMed] [Google Scholar]
- 48. Watts RG, Merchant RE. Cerebrovascular effects and tumor kinetics after a single intratumoral injection of human recombinant interleukin‐2 alone or in combination with intravenous chemotherapy in a rat model of glioma. Neurosurgery 1992; 31: 89–99. [DOI] [PubMed] [Google Scholar]
- 49. Tzeng JJ, Barth RF, Orosz GC, James SM. Phenotype and functional activity of tumor‐infiltrating lymphocytes isolated from immunogenic and non‐immunogenic rat brain tumors. Cancer Res 1991; 51: 2373–8. [PubMed] [Google Scholar]
- 50. Kruse CA, Molleston MC, Parks EP, Schiltz PM, Kleinschmidt‐DeMasters BK, Hickey WF. A rat glioma model, CNS‐1, with invasive characteristics similar to those of human gliomas: a comparison to 9L gliosarcoma. J Neurooncol 1994; 22: 191–200. [DOI] [PubMed] [Google Scholar]
- 51. Tzeng JJ, Barth RF, Clendenon NR, Gordon WA. Adoptive immunotherapy of a rat glioma using lymphokine‐activated killer cells and interleukin‐2. Cancer Res 1990; 50: 4338–43. [PubMed] [Google Scholar]
- 52. Smilowitz HM, Joel DD, Slatkin DN et al. Long‐term immunological memory in the resistance of rats to transplanted intracerebral 9L gliosarcoma (9LGS) following subcutaneous immunization with 9LGS. J Neurooncol 2000; 46: 193–203. [DOI] [PubMed] [Google Scholar]
- 53. Prins RM, Odesa SK, Liau LM. Immunotherapeutic targeting of shared melanoma‐associated antigens in a murine glioma model. Cancer Res 2003; 63: 8487–91. [PubMed] [Google Scholar]