

Abstract
Germline mutations of the VHL gene are responsible for VHL. Approximately 70% of VHL families display small intragenic mutations detectable by sequencing, whereas partial‐ or whole‐gene deletions have been described in the majority of the remaining families. For such large deletions, complex genetic techniques other than sequencing might have to be used. In this study, we describe an RQ‐PCR assay with TaqMan fluorescent probes to detect germline VHL deletions. The RQ‐PCR primer/probe sets were designed for the three VHL coding exons as well as for the 5′ promoter and 3′ untranslated regions. The RQ‐PCR assay for 30 normal and 10 known VHL deletion control samples demonstrated high sensitivity and specificity. We then screened 29 individuals from 19 classical VHL families (16 type 1, 2 type 2A, and one type 2B) and one PHEO family, as well as four solitary suspected cases, none displaying any sequence changes, for VHL deletions by the RQ‐PCR assay. We detected germline deletions in 17 (89%) classical families including 15 type 1, one type 2A, and one type 2B. We also identified two mutation carriers and two non‐carriers in our family cohort. The one PHEO family and four solitary cases did not display any deletion patterns. These findings indicated that the TaqMan‐based RQ‐PCR assay is a simple and potent technique for the rapid, sensitive, and specific investigation of VHL genetic diagnoses that could be used profitably before more complex large‐deletion detection techniques. (Cancer Sci 2006; 97: 400 –405)
Abbreviations:
- HB
hemangioblastoma
- MIM
Mendelian inheritance in man
- PHEO
pheochromocytoma
- PCR
polymerase chain reaction
- pVHL
VHL protein
- Q‐Southern
quantative Southern blot analysis
- RCC
renal cell carcinoma
- RQ‐PCR
real‐time quantitative polymerase chain reaction
- SSCP
single‐strand conformational polymorphism
- VHL
von Hippel–Lindau disease;
VHL (MIM 193300) is an autosomal dominantly inherited disorder characterized by a predisposition to multiple neoplastic lesions, including retinal angiomas, central nervous system hemangioblastomas, pancreatic tumors, clear‐cell renal carcinomas, PHEO, endolymphatic sac tumors, epididymal cystadenomas, and cystic lesions in various organs.( 1 , 2 ) The gene responsible for VHL is located on chromosome 3p25 and was cloned as a VHL tumor suppressor.( 3 ) Subsequently, the detection of germline VHL alterations as well as genotype–phenotype correlation analyses have been carried out extensively in various countries, and genetic testing based on the detection of germline alterations is now widely applied as a standard diagnostic procedure.( 4 , 5 , 6 , 7 ) Previous studies demonstrated that approximately 70% of VHL families have relatively small intragenic mutations affecting nucleotides of up to 30 bp; such mutations are detectable by routine PCR‐based sequencing of the three VHL coding exons.( 4 ) However, standard sequencing analysis failed to detect germline alteration in the rest of the families. As the majority of these cases have been demonstrated to possess relatively large (Kb‐Mb order) deletions involving the partial or whole VHL gene, various molecular genetic techniques other than sequencing, including pulsed‐field gel electrophoresis, standard or quantitative Southern blot analysis, long‐range or quantitative PCR, and fluorescence in situ hybridization, have been applied to identify such large deletions.( 8 , 9 , 10 , 11 , 12 ) However, these analytic procedures sometimes encounter various problems, such as high technical complexity, the need for specific apparatus and relatively large amounts of DNA samples, and high costs in terms of time or money. The obtained data are occasionally obscure, depending on the mutational types, this is, deletion size and location. As accurate genetic testing for VHL is becoming more important for the clinical management of VHL kindred, there is clearly a growing need for simple, easy, yet reliable detection procedures for such cases.
RQ‐PCR is a powerful technique designed primarily to study mRNA expression. This method is also used to detect changes in the quantity of a targeted genome (i.e. changes associated with gene amplification or deletion) using a small amount of genomic DNA.( 13 , 14 , 15 ) In this study, we have developed an RQ‐PCR assay with TaqMan fluorescent probes as an alternative means of detecting germline deletions in the VHL gene. The RQ‐PCR primer/probes were specifically designed to cover the whole VHL gene, including the three coding exons, and the 5′ promoter and 3′ untranslated regions. We found that the RQ‐PCR assay is a simple, fast, and useful germline deletion assay when applied to Japanese VHL families as well as to patients suspected of having VHL.
Materials and Methods
Patients and DNA preparation
A total of 27 individuals from 19 unrelated Japanese VHL families (y1 to y19) were enroled in the present study (Table 1). One patient sample and two or more kindred samples were obtained from each of 14 and each of five families, respectively. All individuals were clinically diagnosed with the classical VHL criteria.( 1 ) Sixteen families were considered as VHL type 1 (without PHEO), two as type 2A (with PHEO but no RCC), and one as type 2B (PHEO + RCC). In addition, one family (y20) included two patients affected by PHEO only but without any other neoplastic lesions. Also enrolled in the study were four unrelated, solitary patients (y21–24), including a patient with retinal angioma and multiple renal cysts, a 27‐year‐old patient with a cerebellar HB, and two cases with HB and multiple renal cysts but without any other VHL manifestations (Table 1).
Table 1.
Japanese VHL patients and VHL gene dosage values by RQ‐PCR assay
| Family identifier | VHL type | Individual identifier | Clinical status | VHL gene dosage values by RQ‐PCR§ | Q‐Southern¶ | Genetic diagnosis | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vprm5 | Vex1 | Vex2 | Vex3 | Vutr3 | ||||||
| y1 | 1 | y1–1 | AF | 1.06 | 0.92 | 0.51 | 0.91 | 0.87 | x | |
| y2 | 1 | y2–1 | AF | 0.57 | 0.50 | 0.49 | 0.45 | 0.42 | TD | |
| y3 | 1 | y3–1 | AF | 1.06 | 0.96 | 0.50 | 0.91 | 1.00 | PD | |
| y3–2 | AF | 0.99 | 1.05 | 0.51 | 0.96 | 1.06 | PD | |||
| y4 | 1 | y4–1 | AF | 0.49 | 0.51 | 0.41 | 0.55 | 0.43 | Ra | |
| y5 | 1 | y5–1 | AF | 0.83 | 0.52 | 0.58 | 0.52 | 0.49 | TD | |
| y6 | 1 | y6–1 | AF | 0.83 | 0.53 | 0.52 | 0.58 | 0.54 | x | |
| y7 | 1 | y7–1 | AF | 0.99 | 0.58 | 0.54 | 0.54 | 0.53 | TD | |
| y8 | 1 | y8–1 | AF | 1.04 | 0.96 | 1.09 | 1.00 | 0.98 | Norm | |
| y9 | 1 | y9–1 | AF | 1.00 | 0.44 | 0.52 | 0.93 | 0.81 | Ra | |
| y10 | 1 | y10–1 | AF | 0.98 | 0.48 | 0.96 | 0.95 | 0.84 | x | |
| y10–2 | nAF | 1.03 | 0.93 | 1.04 | 1.03 | 0.99 | x | non‐carrier | ||
| y10–3 | AF | 0.82 | 0.49 | 0.89 | 1.01 | 0.79 | x | |||
| y10–4 | AF | 0.98 | 0.46 | 0.78 | 0.93 | 0.80 | x | |||
| y11 | 1 | y11–1 | AF | 1.04 | 0.91 | 0.51 | 0.91 | 0.87 | PD | |
| y12 | 1 | y12–1 | AF | 0.58 | 0.46 | 0.45 | 0.48 | 0.51 | x | |
| y12–2 | AF | 0.59 | 0.51 | 0.48 | 0.44 | 0.59 | TD | |||
| y13 | 2B | y13–1 | AF | 1.04 | 0.90 | 0.86 | 0.50 | 0.46 | PD | |
| y13–2 | nAF | 1.06 | 0.80 | 0.83 | 0.42 | 0.47 | PD | carrier | ||
| y13–3 | nAF | 0.98 | 0.85 | 0.93 | 0.81 | 1.07 | x | non‐carrier | ||
| y14 | 1 | y14–1 | AF | 0.95 | 0.89 | 0.80 | 0.54 | 0.55 | PD | |
| y15 | 2A | y15–1 | AF | 1.14 | 0.79 | 0.79 | 0.92 | 1.05 | Norm | |
| y16 | 2A | y16–1 | AF | ND | 0.50 | 0.91 | 0.80 | ND | x | |
| y17 | 1 | y17–1 | AF | 1.11 | 0.86 | 0.43 | 0.56 | 0.52 | x | |
| y18 | 1 | y18–1 | AF | 1.00 | 0.52 | 0.49 | 0.52 | 0.56 | x | |
| y18–2 | nAF | 1.11 | 0.52 | 0.52 | 0.53 | 0.57 | x | carrier | ||
| y19 | 1 | y19–1 | AF | 1.11 | 0.49 | 0.50 | 0.47 | 0.88 | x | |
| y20 | PHEO | y20–1 | AF | 1.11 | 0.85 | 0.86 | 0.80 | 1.07 | Norm | |
| y20–2 | AF | 1.08 | 0.88 | 0.82 | 0.90 | 0.97 | x | |||
| y21 | RA/RC | y21–1 | AF | 1.04 | 1.02 | 0.91 | 1.04 | 0.82 | x | |
| y22 | HB | y22–1 | AF | 0.91 | 0.92 | 0.87 | 0.94 | 0.81 | x | |
| y23 | HB/RC | y23–1 | AF | 1.00 | 0.90 | 0.91 | 1.03 | 1.03 | Norm | |
| y24 | HB/RC | y24–1 | AF | 1.10 | 0.80 | 0.87 | 0.87 | 0.86 | x | |
AF, affected; HB, central nervous system hemangioblastoma only; HB/RC, hemangioblastoma and multiple renal cysts; nAF; not affected; PHEO, pheochromocytoma only; RA/RC, retinal angioma and multiple renal cysts.
Genomic DNA was prepared from EDTA‐containing peripheral blood samples using standard procedures. All individuals were examined using standard mutation detection techniques, that is, DNA‐SSCP followed by sequencing or direct sequencing of the three VHL coding exons; no germline sequence alteration was identified.( 16 , 17 ) Standard Southern blotting with EcoRI or HindIII digestion was performed in 12 families (y1 to y11), three of which (y3, y4, and y9) were suggested to have germline VHL rearrangements.( 16 , 17 ) Southern analysis was not done in the remaining cases. In addition, DNA samples from 30 unrelated, healthy volunteers were used as VHL deletion‐negative controls. For VHL deletion‐positive controls, we used DNA samples of 10 patients in whom germline deletion of the entire VHL gene had been confirmed previously by quantitative Southern analysis.( 10 ) Written informed consent for genetic diagnosis was obtained from all individuals, and the study protocol was approved by the institutional ethics committee of the Yokohama City University School of Medicine (Yokohama, Japan).
RQ‐PCR for VHL deletion detection
We designed RQ‐PCR primers and TaqMan probes (Nippon EGT Co., Ltd., Tokyo, Japan) for the three VHL coding exons and/or surrounding intronic regions. In addition, we assigned two primer/probe sets, one within the 5′ promoter region and the other in the 3′ untranslated region, to screen functionally important regions (Fig. 1).( 3 , 18 ) The human serum albumin gene (ALB) (MIM +103600) located on chromosome 4q13.3, G protein‐coupled receptor 15 (GPR15) (MIM 601166), and zinc finger protein 80 (ZNF80) (MIM 194553) genes, on chromosome 3q12.1 and 3q13.31, respectively, were chosen as internal reference controls.( 15 , 19 ) All primer and probe sequences were designed using Primer Express software (Applied Biosystems, Foster City, CA, USA) and carefully checked to exclude other homologous genomic sequences by using the National Center for Biotechnology Information BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/) (Table 2). We chose FAM/TAMRA (5′‐fluorescence/3′‐quencher) for each TaqMan probe.
Figure 1.
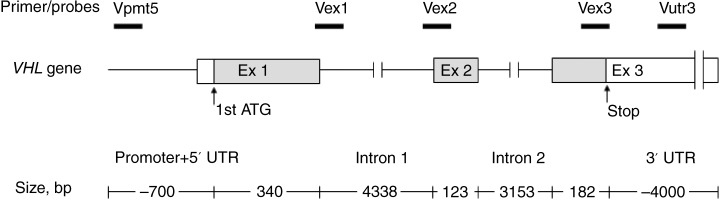
VHL gene structure and the locations of the five primer/probe sets for RQ‐PCR assay.
Table 2.
Primers and probes for RQ‐PCR assay of the VHL gene
| Name | Forward (location) | Reverse (location) | Probe (5′‐FAM, 3′‐TAMRA) (location) | Amplicon size (bp) |
|---|---|---|---|---|
| Vpmt5 | TGATTGGGTGTTCCCGTGT (−675 to −656) | TCATGTCAGACGCGCAATGT (−619 to −599) | TGCGCCACCCTCGAACCTTGTT (−651 to −630) | 76 |
| Vex1 | GCATCCACAGCTACCGAGGT (323 to IVS1 +2) | CCGTGCTATCGTCCCTGCT (IVS1 +29 to IVS1 +47) | CCCGGCGCTTAGGCCCGA (IVS1 +8 to IVS1 +25) | 65 |
| Vex2 | CACCGGTGTGGCTCTTTAACA (IVS1–41 to IVS1–21) | GTTAACCAGAAGCCCATCGTGT (373 to 394) | CCCGATAGGTCACCTTTGGCTCTTCAGA (IVS1–8 to 363) | 94 |
| Vex3 | GGTCGCTCTACGAAGATCTGGA (545 to 568) | TCTTCAATCTCCCATCCGTTG (625 to *3) | ACCCAAATGTGCAGAAAGACCTGGAGC (572 to 599) | 101 |
| Vutr3 | TCTTTGAGACCCCAGTGCCT (*468 to *487) | AGTGTCCCCTGGTTTGTTCCT (*489 to *508) | CACATCATGAGCCTTCAGTCAGGGTTTGT (*512 to *532) | 74 |
| ALB | ACCAAATGCTGCACAGAATCC | CGTATGTTTCATCGACTTCCAGAG | TGGTGAACAGGCGACCATGCTTTTC | 73 |
| Ex 12 | ||||
| GPR15 | TGCTACGAGCCCAAACTCTGA | AGACTGGAAGGAAGACAGAGGTGT | ATCAGGGAGACCCACTCCCATGTTCC | 74 |
| ZNF80 | GCAGGTCTCCACAGGAGACAAT | CCCCTCACGAACCAAAGTGT | CCATGAATGTGACTCCCAGGGACCAAGTA | 76 |
Primer and probe locations were considered 1st ATG codon of the VHL gene as nucleotide position 1 and stop codon as nucleotide position 645.
The RQ‐PCR assay was optimized basically by following the manufacturer's instructions (Applied Biosystems). In brief, the optimal RQ‐PCR conditions were arrived at by trying PCR reactions using 1.25, 2.5, 5, 10, and 20 ng of DNA along with 50, 100, 150, 200, and 300 nM of the reverse primers; each 25’l reaction used 300 nM forward primer and a 200 nM probe concentration. The optimal reverse primer concentration for all primers was found to be 300 nM, and DNA concentrations between 20 ng and 2.5 ng were within the linear dynamic range of the system. DNA aliquots of 10 ng/’l were prepared for RQ‐PCR. Each 25’l reaction contained 1 × qPCR Mastermix Plus (Eurogentec, Seraing, Belgium), 5 mM MgCl2, 300 nM each of the forward and reverse primers, 200 nM TaqMan probe, 1’l of DNA, and a final concentration of 3% dimethyl sulfoxide (Sigma‐Aldrich, St Louis, MO, USA). PCR amplification was performed using an iCycler iQ Real‐Time PCR Detection System (Bio‐Rad Laboratories, Hercules, CA, USA). The optimized PCR conditions for real‐time analysis consisted of 1 cycle at 50°C for 2 min and denaturation at 95°C for 10 min, followed by 40 cycles at 95°C for 20 s and 60°C for 60 s. The amount of product was measured by interpolation from a standard DNA sample. DNAs for a standard curve were serially diluted to obtain five standard solutions for use in the PCR reaction to generate the reference curve, which was obtained by using iCycler data analysis software (Bio‐Rad Laboratories). At least three independent PCR reactions were performed to obtain the mean signal value for each primer/probe set. After normalization against the mean signal value of internal reference controls, this method allowed determination of VHL gene dosage. Samples retaining the homozygous VHL allele were expected to have gene dosage values close to 1.0, and samples with a hemizygous VHL allele were expected to have gene dosage values close to 0.5.
Southern blot analysis for the VHL gene
Q‐Southern with EcoRI and AseI double digestions followed by hybridization with VHL g7cDNA for detecting germline deletion or rearrangement was carried out according to the methods described previously.( 10 )
Statistical analysis
The Kruskal–Wallis H‐test was used to examine differences between groups. All statistical analysis was performed with spss software version 10.0J (SPSS, Chicago, IL). All statistical tests were two‐sided and were considered to be statistically significant at P < 0.05.
Results
Our assay is theoretically based on the signal dosage changes between the target VHL gene and the reference gene detected by RQ‐PCR, therefore the reference gene dosage value is critically important. We chose three genes, ALB, GPR15, and ZNF80, as candidate internal references in our VHL/RQ‐PCR assay; each has a unique DNA sequence and single locus on the human genome, and none of these gene loci are considered to be involved in the VHL disease.( 15 , 19 ) We initially checked the signal dosages of the three reference genes in DNA samples from 30 normal healthy controls and 10 VHL deletion controls by RQ‐PCR. As expected, the RQ‐PCR signal ratios for ALB/GPR15, GPR15/ZNF80, and ZNF80/ALB were approximately 1.0 both in the normal control and deletion control groups, and there were no significant differences between the two groups in any of the three ratios (Table 3). These data showed that any three reference genes can be used as an internal control in our VHL/RQ‐PCR assay. We therefore used ALB as an internal reference control for the following RQ‐PCR assay.
Table 3.
Signal ratios for ALB/GPR15, GPR15/ZNF80, and ZNF80/ALB in normal and VHL deletion‐positive control samples detected by RQ‐PCR
| Samples (n) | Reference genes | Signal ratio, mean ± SD | P‐value † |
|---|---|---|---|
| Normal controls (30) | ALB/GPR15 | 1.01 ± 0.14 | 0.437 |
| GPR15/ZNF80 | 1.01 ± 0.16 | ||
| ZNF80/ALB | 1.05 ± 0.17 | ||
| VHL deletion controls (10) | ALB/GPR15 | 0.98 ± 0.05 | 0.802 |
| GPR15/ZNF80 | 1.03 ± 0.15 | ||
| ZNF80/ALB | 1.01 ± 0.13 |
Kruskal–Wallis H‐test; SD, standard deviation.
To assess the effectiveness of the RQ‐PCR assay, we initially examined 30 mutation‐negative control DNA samples from healthy volunteers as well as 10 germline VHL deletion‐positive controls. The VHL gene dosage values for the 5′ promoter region, exons 1, 2, and 3, and the 3′ untranslated region detected by the RQ‐PCR assay in 30 healthy controls ranged from 0.82 to 1.19, 0.81–1.07, 0.79–1.14, 0.78–1.16, and 0.85–1.20, respectively (Fig. 2). However, the 10 known deletion controls displayed gene dosage values for the five primer/probe sets ranging from 0.51 to 0.60, 0.45–0.58, 0.47–0.56, 0.42–0.59, and 0.49–0.59, respectively (Fig. 2). The gene dosage values were consistent with a normal distribution, and all VHL deletion controls displayed a dosage value of at least 3.5 standard deviations for each primer/probe set from the mean of the 30 healthy controls (Fig. 2). The sensitivity and the specificity for the RQ‐PCR assay in the preliminary normal and VHL deletion‐positive control samples were therefore considered to be 100% each.
Figure 2.
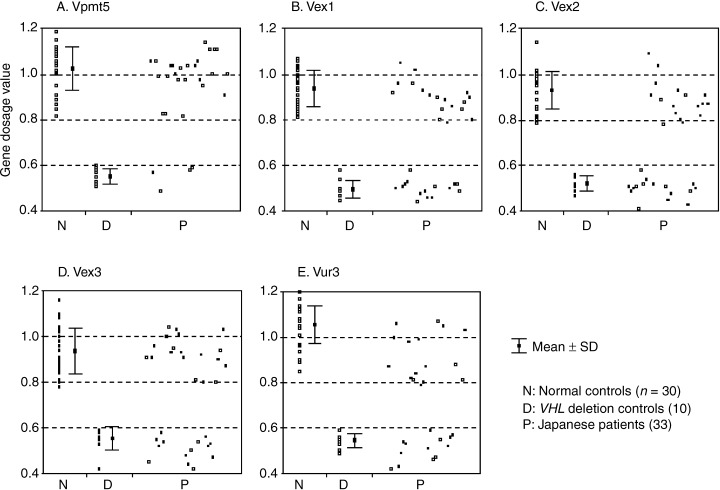
VHL gene dosage values detected by RQ‐PCR assay for the five primer/probe sets in 30 normal controls, 10 VHL deletion controls, and 33 Japanese patients. DNA samples of 30 healthy controls and patients, each of whom retained two copies of the VHL gene, displayed gene dosage values of approximately 1.0 (∼0.8 to ∼1.2), whereas the samples from 10 deletion controls and patients who each had a single copy of VHL displayed values of approximately 0.5 (∼0.4 to ∼0.6).
After confirmation of the quality of the RQ‐PCR assay, we then applied the procedures to the Japanese kindred from 19 unrelated VHL families and one PHEO family, and to the four solitary patient samples. As expected, the detected gene dosage values for each of the primer/probe sets in the Japanese patients converged into two separate ranges coinciding with those found in the normal and known‐positive control samples (Table 1 and Fig. 2). We therefore decided that VHL deletions were present in 23 individuals from 17 families (89% of the VHL families), including 15 type 1, one type 2A, and one type 2B. The two families presenting Southern rearrangements in the previous study also showed some deletion patterns. However, individuals from two families, including one type 1 and one type 2A (y8 and y15), did not show any deletion patterns.
Of the 17 VHL families displaying deletion patterns, nine (53%) showed partial deletions affecting one or two coding exons, whereas the remaining eight were suggested to have larger deletions encompassing all three coding exons. Furthermore, three of the eight families showed deletions for all five primer/probe sets, suggesting loss of the entire VHL locus. It should be noted that all of the deleted cases were impaired in at least one of the three coding exons and we found no cases that showed deletion in either the 5′ promoter or 3′ untranslated region only.
Germline deletion is rarely found in VHL type 2 families.( 4 , 10 ) We found partial deletions in two type 2 families in our Japanese patient cohort. Interestingly, one type 2A (HB + PHEO only) family (y16) seemed to have a partial deletion that include the exon 1 region, whereas the type 2B family (HB + PHEO + RCC) (y13) showed a partial loss of exon 3 and the downstream region.
Of the 17 families with deletion, we analyzed two or more kindred samples in five families (y3, y10, y12, y13, and y18) (Table 1). We detected identical deletion patterns in the individuals from within each of these five families. Although no apparent VHL manifestations were detected by clinical surveillance in four individuals, we considered two (y13–2 and y18–2) as deletion mutation carriers and two (y10–2 and y13–3) as non‐carriers as a result of the RQ‐PCR assay.
The two affected patients in the PHEO family (y20) and the four solitary cases (y21 to y24) did not display any aberrant gene dose values for any of the primer/probe sets examined.
Finally, we confirmed the germline VHL deletion status in Japanese patients by Q‐Southern. Because the DNA samples were of insufficient quantity or quality in 23 cases, we were only able to successfully analyze a total of 10 individuals by Q‐Southern (Table 1). The Q‐Southern demonstrated that all six cases showing deletion patterns by RQ‐PCR also demonstrated either VHL total‐ or partial‐deletion patterns, whereas four cases without any RQ‐PCR change did not have any Q‐Southern aberration (Table 1). Therefore, the sensitivity and the specificity for germline VHL deletion detection in the RQ‐PCR were equal to those in the Q‐Southern in our Japanese patient samples, although the sample numbers were relatively small.
Discussion
In the present study, we have developed an RQ‐PCR assay employing TaqMan probes to assess large germline deletions of the VHL gene. The RQ‐PCR assay in 30 deletion‐negative and 10 known deletion‐positive control samples was proven to be sufficient both in sensitivity and specificity. We then applied the RQ‐PCR assay to 27 individuals from 19 Japanese VHL families, who clinically met the diagnostic criteria without any sequencing aberrations in the VHL coding region. As a result, we successfully identified partial or whole VHL germline deletions in 89% of the families. In addition, we genetically diagnosed two mutation carriers and two non‐carriers among family members without clinical manifestations. The sensitivity and the specificity for our RQ‐PCR assay were comparable with those in the Q‐Southern, which is considered one of the standard detection methods for VHL large deletions( 10 , 19 ) Moreover, our RQ‐PCR assay can easily estimate which exon(s) or region of the gene is actually deleted. The data suggested that RQ‐PCR is a simple, rapid, and useful germline deletion assay for VHL.
The majority of the families in this study were VHL type 1 (HB + RCC without PHEO) (16/19; 84%) Within this group, we found partial or whole VHL loss in 15 families (94%). In these type 1 families with large deletions, crucial disruption or total loss of the pVHL function is likely to occur. Subsequently, this disruption will be responsible for the development of the type 1 disease phenotype, including HB and RCC, as previously reported.( 4 , 10 )
We also detected partial deletions in a type 2A family and in a type 2B family. Almost all the mutations previously characterized in type 2 families (with PHEO) were missense mutations at specific codons in the pVHL, and large deletions were very rare. A previous international surveillance study demonstrated that only two of 78 VHL type 2 families (2.6%) showed large genomic deletions,( 4 ) whereas Stolle et al. reported that five of 26 type 2 families (19%) have quantitative Southern rearrangements.( 10 ) In our survey for Japanese VHL, we screened a total of 17 type 2 families. Subsequently, we found that 14 families have intragenic mutations (M. Yoshida et al., unpublished data, 2000). In addition, we newly identified two families (11.8%) with large deletions using the RQ‐PCR assay. From the functional analyses for these missense mutated pVHL in the type 2 families, not only ‘loss of function’ but also ‘gain of function’ of pVHL with the occurrence of PHEO have been proposed, although the mechanisms underlying these functional changes have yet to be elucidated in detail.( 20 , 21 ) It is worth noting that our data suggested that one type 2A family had lost the exon 1 region, whereas the 3′ region involving exon 3 and the 3′ untranslated region had disappeared in the type 2B family. It is noteworthy that Stolle et al. also reported five type 2 families displaying VHL rearrangements, as determined by Q‐Southern; these probably represented partial deletions, as were found in our families.( 10 ) Further studies of actual deletion regions and deduced pVHL conformational and functional changes in these families will be of interest with respect to the development of VHL‐associated tumors, including PHEO.
We did not identify any deletions in two classical VHL families, one being type 1 and the other type 2A. One possibility is that, although VHL is a relatively small gene that encompasses approximately 13 kb of genomic DNA, the number and amplicon size of the RQ‐PCR primers in the analysis may not have been sufficient to screen the entire VHL locus (Fig. 1). We may add more primer/probe sets on the appropriate locations to improve the mutation detection rate. Secondly, other alteration mechanisms may be involved that are not detectable by either PCR‐based sequencing or RQ‐PCR. For example, VHL mosaicism has been reported in such cases. Although VHL mosaicism is relatively rare, it is a considerable mechanism in de novo cases.( 22 , 23 ) Notably, our two cases are also de novo occurrences. These patients, together with the other members of their families, should be further analyzed to target the mosaicism.
The one PHEO family also did not display germline VHL mutation or deletion. Several genes other than VHL, including RET (MIM 164761), SDHB (185470), and SDHD (602690), have been implicated in familial PHEO.( 24 , 25 ) The possible involvement of these susceptible genes should be examined as a next step.
We did not detect any germline change either in sequencing or the RQ‐PCR assay in the four solitary suspected cases. Based on the genetic study and the clinical manifestations, these cases are likely to be sporadic, non‐VHL occurrences. However, we have to consider the limitation of the RQ‐PCR assay mentioned above. Careful follow‐up is still needed for these cases.
While the present study was under way, Hoebeeck et al. reported a procedure for germline VHL deletion using RQ‐PCR with SYBR Green I dye.( 19 ) Even though their analysis was performed independently of ours, without any sharing of information, and despite several differences between the studies in the RQ‐PCR conditions and the family cohort, the basic strategies and the results were very similar. This agreement strongly suggests the validation of RQ‐PCR as a reliable means of detecting VHL gene dosage changes. However, because the assay is still somewhat very delicate, that is, the accurate detection of two‐fold quantitative change, we believe that the RQ‐PCR with TaqMan probe should be theoretically more specific and ideal compared to those with the SYBR Green dye combined with dissociation curves. Our assay presented here can be used profitably before employing more expensive and complex large‐deletion detection techniques such as pulsed‐field gel electrophoresis, Q‐Southern, and fluorescence in situ hybridization analysis.
Acknowledgments
We would like to thank the Japanese VHL patients and their families, without whose cooperation this work would have been impossible. We are also indebted to our colleagues at the different hospitals who availed us of blood samples and clinical information about their patients. We also thank Yoko Nakamura, Takako Yamaki, and Rie Shimizu for their excellent technical assistance. This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (#17659508).
References
- 1. Lonser RR, Glenn GM, Walther M et al. Von Hippel–Lindau disease. Lancet 2003; 361: 2059–67. [DOI] [PubMed] [Google Scholar]
- 2. Maher ER. Von Hippel–Lindau disease. Curr Mol Med 2004; 4: 833–42. [DOI] [PubMed] [Google Scholar]
- 3. Latif F, Tory K, Gnarra J et al. Identification of the von Hippel‐Lindau disease tumor suppressor gene. Science 1993; 260: 1317–20. [DOI] [PubMed] [Google Scholar]
- 4. Zbar B, Kishida T, Chen F et al. Germline mutations in the Von Hippel–Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat 1996; 8: 348–57. [DOI] [PubMed] [Google Scholar]
- 5. Zbar B, Kaelin W, Maher E, Richard S. Third International Meeting on von Hippel–Lindau disease. Cancer Res 1999; 59: 2251–3. [PubMed] [Google Scholar]
- 6. Decker HJ, Neuhaus C, Jauch A et al. Detection of a germline mutation and somatic homozygous loss of the von Hippel–Lindau tumor‐suppressor gene in a family with a de novo mutation. A combined genetic study, including cytogenetics, PCR/SSCP, FISH, and CGH. Hum Genet 1996; 97: 770–6. [DOI] [PubMed] [Google Scholar]
- 7. Hes F, Zewald R, Peeters T et al. Genotype–phenotype correlations in families with deletions in the von Hippel–Lindau (VHL) gene. Hum Genet 2000; 106: 425–31. [DOI] [PubMed] [Google Scholar]
- 8. Yao M, Latif F, Kuzmin I et al. Von Hippel–Lindau disease: identification of deletion mutations by pulsed‐field gel electrophoresis. Hum Genet 1993; 92: 605–14. [DOI] [PubMed] [Google Scholar]
- 9. Richards FM, Phipps ME, Latif F et al. Mapping the Von Hippel–Lindau disease tumour suppressor gene: identification of germline deletions by pulsed field gel electrophoresis. Hum Mol Genet 1993; 2: 879–82. [DOI] [PubMed] [Google Scholar]
- 10. Stolle C, Glenn G, Zbar B et al. Improved detection of germline mutations in the von Hippel–Lindau disease tumor suppressor gene. Hum Mutat 1998; 12: 417–23. [DOI] [PubMed] [Google Scholar]
- 11. Cybulski C, Krzystolik K, Maher ER et al. Long polymerase chain reaction in detection of germline deletions in the von Hippel–Lindau tumour suppressor gene. Hum Genet 1999; 105: 333–6. [DOI] [PubMed] [Google Scholar]
- 12. Pack SD, Zbar B, Pak E et al. Constitutional von Hippel–Lindau (VHL) gene deletions detected in VHL families by fluorescence in situ hybridization. Cancer Res 1999; 59: 5560–4. [PubMed] [Google Scholar]
- 13. Fink L, Seeger W, Ermert L et al. Real‐time quantitative RT‐PCR after laser‐assisted cell picking. Nat Med 1998; 4: 1329–33. [DOI] [PubMed] [Google Scholar]
- 14. Bieche I, Olivi M, Champeme MH, Vidaud D, Lidereau R, Vidaud M. Novel approach to quantitative polymerase chain reaction using real‐time detection: application to the detection of gene amplification in breast cancer. Int J Cancer 1999; 78: 661–6. [DOI] [PubMed] [Google Scholar]
- 15. Sieber OM, Lamlum H, Crabtree MD et al. Whole‐gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or ‘multiple’ colorectal adenomas. Proc Natl Acad Sci USA 2002; 99: 2954–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kondo K et al. Clinical Research Group for VHL in Japan: germline mutations in the von Hippel–Lindau disease (VHL) gene in Japanese VHL. Hum Mol Genet 1995; 4: 2233–7. [DOI] [PubMed] [Google Scholar]
- 17. Yoshida M, Ashida S, Kondo K et al. Germ‐line mutation analysis in patients with von Hippel–Lindau disease in Japan: an extended study of 77 families. Jpn J Cancer Res 2000; 91: 204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zatyka M, Morrissey C, Kuzmin I et al. Genetic and functional analysis of the von Hippel–Lindau (VHL) tumour suppressor gene promoter. J Med Genet 2002; 39: 463–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoebeeck J, Van Der Luijt R, Poppe B et al. Rapid detection of VHL exon deletions using real‐time quantitative PCR. Laboratory Invest 2005; 85: 24–33. [DOI] [PubMed] [Google Scholar]
- 20. Hes FJ, Hoppener JW, Lips CJM. Pheochromocytoma in von Hippel–Lindau disease. J Clin Endocrinol Metab 2003; 88: 969–74. [DOI] [PubMed] [Google Scholar]
- 21. Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol 2004; 22: 4991–5004. [DOI] [PubMed] [Google Scholar]
- 22. Murgia A, Martella M, Vinanzi C, Polli R, Perilongo G, Opocher G. Somatic mosaicism in von Hippel–Lindau Disease. Hum Mutat 2000; 15: 114. [DOI] [PubMed] [Google Scholar]
- 23. Sgambati MT, Stolle C, Choyke PL et al. Mosaicism in von Hippel–Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet 2000; 66: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neumann HP, Bausch B, McWhinney SR et al. Germ‐line mutations in nonsyndromic pheochromocytoma. New Eng J Med 2002; 346: 1459–66. [DOI] [PubMed] [Google Scholar]
- 25. Maher ER, Eng C. The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Genet 2002; 11: 2347–54. [DOI] [PubMed] [Google Scholar]