

Abstract
It is desirable to find more appropriate therapeutic opportunities in non‐small‐cell lung cancer (NSCLC) due to the current poor prognosis of affected patients. Recently, several histone deacetylase (HDAC) inhibitors, including suberoylanilide hydroxamic acid (SAHA), have been reported to exhibit antitumor activities against NSCLC. S‐1, a novel oral fluorouracil anticancer drug, has been developed for clinical use in the treatment of NSCLC in Japan. Using an MTT assay, we analyzed the growth‐inhibitory effect of 5‐fluorouracil (5‐FU), S‐1, and SAHA against three NSCLC cell lines, as well as the breast cancer cell line MCF7 which is known to be highly sensitive to 5‐FU. Combined treatment with low‐dose SAHA enhanced 5‐FU‐ and S‐1‐mediated cytotoxicity and resulted in synergistic effects, especially in 5‐FU‐resistant cells. Both the mRNA and protein expression levels of thymidylate synthase (TS), dihydropyrimidine dehydrogenase (DPD), and orotate phosphoribosyltransferase (OPRT), which are associated with 5‐FU sensitivity/response, were analyzed in the cells undergoing treatment. 5‐Fluorouracil‐resistant lung cancer cells displayed high expression of TS mRNA and protein. Suberoylanilide hydroxamic acid down‐regulated TS mRNA and protein expression, as well as repressed the rapid induction of this factor during 5‐FU treatment, in all examined cell types. We also examined the status of the Rb‐E2F1 pathway, with SAHA up‐regulating p21waf1/cip1 expression via promoter histone acetylation; this, in turn, blocked the Rb‐E2F1 pathway. We conclude that combination therapy with SAHA and S‐1 in lung cancer may be promising due to its potential to overcome S‐1 resistance via modulation of 5‐FU/S‐1 sensitivity‐associated biomarker (TS) by HDAC inhibitor. (Cancer Sci 2010)
Even though a wide range of anticancer agents have been developed, many patients with advanced solid tumors still exhibit poor prognosis. With respect to treatment of advanced lung cancer, there are many anticancer agents in clinical use, such as cisplatin (CDDP), carboplatin (CBDCA), docetaxel, paclitaxel, vinorelbine, gemicitabine, 5‐fluorouracil (5‐FU) derivatives, camptotecin‐11 (CPT‐11), etc. A number of combination therapy regimens employing platinum compounds have proven to be effective and are widely applied in the initial treatment for inoperative non‐small‐cell lung cancer (NSCLC).( 1 , 2 ) In addition, docetaxel, pemetrexed, and epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors have been reported to be effective in the context of second‐line chemotherapy for NSCLC.( 3 , 4 , 5 ) However, the effect of these therapies on improving patient survival remains far from satisfactory at present.( 1 , 2 , 3 , 4 , 5 ) It is consequently desirable to find more appropriate therapeutic opportunities for this disease.
Recently, molecular‐targeted therapy in the treatment of cancer has made substantial progress. Histone deacetylase (HDAC) inhibitors have been shown to acetylate the nucleosomal histones of condensed chromatin, and cause the reactivation of genes silenced by hyperacetylated histones.( 6 ) Histone deacetylase inhibitors, including suberoylanilide hydroxamic acid (SAHA), have demonstrated therapeutic benefit as monotherapy in hematologic, breast, and bladder malignancies, as well as mesothelioma and NSCLC, without evidence of severe adverse events.( 7 , 8 , 9 , 10 ) We previously examined the sensitivity of 16 NSCLC cell lines to HDAC inhibitors via an MTT assay.( 11 ) Histone deacetylase inhibitors displayed strong antitumor activity against a subgroup of the NSCLC cell lines, suggesting the need for the use of predictive markers to select patients receiving this treatment. We have also analyzed gene expression profiles in these cell lines using cDNA arrays to identify transcripts that correlate with sensitivity to HDAC inhibitor treatment.( 11 ) We related the cytotoxic activity of particular agents to the corresponding expression pattern in each of the cell lines using a modified National Cancer Institute (NCI) program.( 11 ) We found that nine genes can distinguish sensitive from resistant cell lines.( 11 ) Histone deacetylase inhibitors may also be promising new agents for the treatment of NSCLC. Recently, it has been demonstrated that combinations of HDAC inhibitors with well‐established chemotherapeutics can provide synergistic antitumor effects.( 12 , 13 , 14 )
S‐1 (composed of tegafur, 2‐chloro‐2,4‐dihydroxypyridine[CDHP], and potassium oxonate in the molar ratio 1:0.4:1( 15 )), which inhibits dihydropyrimidine dehydrogenase (DPD) activity, has been developed for cancer chemotherapy including the treatment of NSCLC (Taiho Pharmaceutical, Tokyo, Japan).( 15 ) Many studies have demonstrated that intratumoral DPD activity affects resistance to 5‐FU.( 16 , 17 , 18 ) In particular, NSCLCs may be considered to be a tumor type that commonly displays a high level of DPD expression, which in turn is associated with decreased 5‐FU activity.( 19 ) S‐1 mediates its antitumor effect via the blockage of DPD by CDHP.( 15 , 17 ) Tegafur is a prodrug that generates 5‐FU in the blood largely as a result of metabolism by cytochrome p450 enzymes in the liver. The active metabolite of 5‐FU, FdUMP, forms a covalent complex with 5,10‐methylenetrahydrofolate and thymidylate synthase (TS), resulting in inhibition of DNA synthesis.( 20 ) It is well known that TS is frequently overexpressed in cancer cells compared with normal tissues, and that tumors with high TS expression are generally resistant to 5‐FU.( 14 , 15 , 17 , 21 , 22 ) Thymidylate synthase expression seems to be one of the key molecules connected with sensitivity to S‐1, DPD blocking agent. In a clinical trial of S‐1 in advanced NSCLC patients (not having received prior chemotherapy) carried out in Japan, the response rate was 22% with the median survival time being 10.2 months.( 23 ) Furthermore, a phase II trial of S‐1 + CDDP combination revealed a 47% response rate and acceptable safety profile for NSCLC patients.( 24 ) Thus, combining other agents with S‐1 therapy may be a promising strategy for NSCLC patients.
In this study, we examined the antitumor effects of 5‐FU/S‐1 in combination with SAHA against lung cancer cells using an MTT assay. Combined treatment with low dosage SAHA enhanced 5‐FU/S‐1 cytotoxicity, as well as provided a synergistic effect in 5‐FU‐resistant cells. Using Cox multivariate regression analysis, Nakano et al. reported that TS, orotate phosphoribosyltransferase (OPRT), and DPD status to be significant prognostic factors in respect to determining the survival of resected NSCLC patients that were post‐operatively treated with a combination of uracil and ftorafur (UFT).( 23 ) In our study, we investigated the status and modulation after treatment of these three molecules. The mechanism underlying this synergy seemed to be related to down‐regulation of TS mRNA and protein by SAHA.
Materials and Methods
Cell lines. Three lung cancer cell lines and one breast cancer cell line were used in this study, as follows: A549, PC9, PC9/f14, and MCF7. The A549 and MCF7 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). The PC9 cell line was obtained from IBL (Gunma, Japan). PC9/f14 (A. Gemma, K. Takenaka) is a highly metastatic subline of PC9 cells established at Nippon Medical School Medical School using an artificial metastasis method.( 25 )
Drugs and growth‐inhibition assay. Suberoylanilide hydroxamic acid was purchased from Sigma. 5‐Fluorouracil was provided by Kyowa Hakko (Tokyo, Japan). CDHP was provided by Taiho Pharmaceutical. They were dissolved in dimethyl sulfoxide (DMSO) for in vitro studies.
We used the colorimetric MTT assay to examine the activity of 5‐FU monotherapy or SAHA monotherapy against all three lung cancer cell lines, as well as MCF7 cells which are known to be highly sensitive to 5‐FU.( 26 ) In addition, combination therapy against these lines was also examined. For these studies, 5‐FU, 5‐FU with SAHA, S‐1 (5‐FU + CDHP 1 μM), and S‐1 with SAHA, were incubated with cells for 72 h. Cell suspensions (200 μL, 5 × 103 cells/mL) were seeded into 96‐well microtiter plates and 10 μL of drug solution added, at various concentrations. After incubation for 72 h at 37°C, 20 μL of MTT solution (5 mg/mL in phosphate‐buffered saline [PBS]) was added to each well and incubation then continued for a further 4 h at 37°C. The IC50 value was defined as the concentration of drug(s) needed for a 50% reduction in absorbance (560 nm) based on cell growth curves.
Real‐time RT‐PCR analysis. Real‐time quantitative RT‐PCR assessment of TS, DPD, and OPRT gene expression was performed using the ABI Prism 7700 Sequence Detector system (Perkin Elmer/Applied Biosystems, Foster City, CA, USA). The PCR primers were designed using the Primer Express software program (Perkin Elmer/Applied Biosystems). The relevant oligonucleotide sequences were as follows:
TS, sense primer: tctggaagggtgttttggag, antisense primer: cctccactggaagccataaa; DPD, sense primer: atggaggagtgtctgggaca, antisense primer: ttgaggccagtgcagtagtc; OPRT, sense primer: tggcatcagtgaccttcaagccctcct, antisense primer: tagtgttttggaaac‐tgttgaggtt; GAPDH, sense primer: tggtatcgtggaaggactcatgac, antisense primer: atgccagtgacttcccgttcagc.
Total RNA was extracted from cultured cells and reverse transcribed using the RevaTra Ace Kit, with a random hexamer being used as primer (Toyobo, Osaka, Japan). A portion of the resulting cDNA was used for quantitative PCR in a 25 μL final volume, together with the desired primers, and SYBR Green PCR Master Mix (Applied Biosystems). The initial thermal cycling conditions were 95°C for 10 min, as recommended by the manufacturer, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The level of gene expression was expressed as ratio of the relevant mRNA in a particular sample to the level of GAPDH mRNA in that sample. Each sample was measured at least three times.( 25 )
Western blot analysis. Western blot analysis was performed as previously described.( 27 ) The blotted membranes were first incubated overnight at 4°C with primary antibodies specific for the following antigens. Antibodies to TS, DPD, and OPRT were provided by Taiho Pharmaceutical. Antibodies against E2F1, phosphorylated Rb, p53, p27, p16, acetylated H3, and acetylated H4 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against p21waf1/cip1 and poly‐(ADP‐ribose) polymerase (PARP) were purchased from Biosource International (Camarillo, CA, USA) while those against β‐actin was purchased from Cell Signaling Technology (Beverly, MA, USA). The membranes were then incubated with peroxidase‐conjugated secondary antibodies and protein was detected with enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham, Buckinghamshire, UK).
Chromatin immunoprecipitation (ChIP) assay. The ChIP‐IT Express kit (Active Motif, Carlsbad, CA, USA) was used to perform chromatin immunoprecipitation assays.( 28 ) PC9/f14 cells were treated with 10 μM SAHA for 24 h. Cells were treated with 1% formaldehyde, followed by glycine stop‐solution. Cells were then collected, centrifuged at 4°C, and resuspended in lysis buffer. After an additional 30 min of lysis and 10 strokes in a homogenizer, nuclei were collected by 10 min of centrifugation at 2400g and resuspended in shearing buffer.
Enzymatic shearing cocktail was added to digest DNA, facilitating preparation of DNA fragments ranging in size from 200 to 500 bp. After centrifugation, 10 μL of the supernatants (containing sheared chromatin) were utilized as assay input. Immunoprecipitation was carried out overnight at 4°C using an incubation mixture containing 10 μL sheared chromatin, protein G magnetic beads, and 3 μg of antibodies (acetylated H3, acetylated H4; Santa Cruz Biotechnology). After centrifugation, washing, and elution, the samples were reverse cross‐linked and treated with RNase A and proteinase K. DNA (both immunoprecipitated samples and input) was recovered by phenol/chloroform extraction and ethanol precipitation and then analyzed by PCR.
The primers were designed as follows: p21waf1/cip1 promoter p1 (285 bp), forward primer: 5′‐ACCAACGCAGGCGAGGGACT‐3′, reverse primer: 5′‐CCGGCTCCACAAGGAACTGA‐3′; p21waf1/cip1 promoter p2 (291 bp), forward primer: 5′‐CGT‐GGTGGTGGTGTGCTAGA‐3′, reverse primer: 5′‐CTGTCTG‐CACCTTCGCTCCT‐3′.
The conditions used were 95°C for 5 min, 35 cycles at 96°C for 1 min, annealing at 60°C for 30 s, 72°C for 1 min, and a final extension at 72°C for 10 min.
Statistical analysis. Data are presented as mean ± SE of at least three independent experiments. Statistical analysis was performed using an unpaired Student’s t ‐test. P‐values <0.05 were considered statistically significant.
Results
Effect of 5‐FU, S‐1 compound (5‐FU + CDHP), SAHA, and combination treatments on cell growth in vitro. The activity of 5‐FU against a panel of lung cancer cell lines, as well as MCF7 cells, was determined by MTT assay (Fig. 1). Clinical trials with 5‐FU/S‐1 have previously shown that serum levels in treated patients reached 1–3 μM.( 29 ) PC9 cells were moderately resistant to 5‐FU, as compared to MCF7 cells (which had previously been reported to be sensitive to 5‐FU).( 26 ) A549 cells were also shown to be quite resistant to 5‐FU (Fig. 1a). PC9/f14 cells were ∼4.5‐fold more resistant than their parental cell line, PC9 (Fig. 1a). Next, we examined the efficacy of SAHA against the same cell lines. Clinical trials with SAHA (Vorinostat) showed previously that serum levels in treated patients reached 0.43–2.98 μM.( 7 , 8 ) In a similar vein to 5‐FU response data, PC9/f14 and A549 cells were found to be resistant to SAHA, while MCF7 and PC9 were comparatively sensitive to this agent (Fig. 1b). For the purpose of determining a clinical setting for a SAHA and S‐1 combination, we examined the growth‐inhibitory potential offered by 5‐FU monotherapy, S‐1 compounds (5‐FU + 1 μM CDHP) with or without low dose (1 μM) SAHA. One μM CDHP was within the serum concentration range found in clinical dosage within S‐1‐treated patients. Combination with SAHA significantly enhanced the growth‐inhibitory effect of 5‐FU, as compared to 5‐FU monotherapy, in all examined cell lines. Similarly, S‐1 compound (combination with CDHP) significantly enhanced the growth‐inhibitory effect of 5‐FU, as compared to 5‐FU monotherapy, in PC9 and PC9/f14 cells (P < 0.05) (Fig. 2a). The combination of S‐1 compounds (5‐FU + CDHP) with SAHA significantly promoted the growth‐inhibitory effect, as compared to S‐1 compound (5‐FU + CDHP) monotherapy, in A549 and PC9/f14 cells (P < 0.05) (Fig. 2a). These results indicated that the combination of S‐1 and SAHA had a synergistic effect in certain cell types, especially in 5‐FU‐resistant cells. In PC9/f14 cells, each result was expressed as a ratio of MTT reduction in treated samples compared with the untreated sample (100%) for 5‐FU alone or compared with the 1 μM SAHA‐treated sample (92.4% and 87.0% of untreated samples [see Fig. S1]) for 5‐FU therapy after SAHA 1 μM treatment, and concurrent therapy with SAHA 1 μM (see Fig. S1, Fig. 2b). The IC50 values of 5‐FU therapy after SAHA 1 μM treatment and concurrent 5‐FU therapy with SAHA 1 μM were 31.7 ± 8.42 μM and 30.4 ± 5.27 μM, whereas that of 5‐FU monotherapy was 99.7 ± 26.7 μM.
Figure 1.
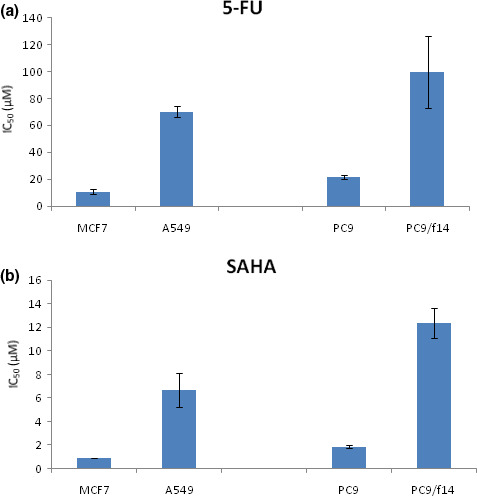
Effect of 5‐fluorouracil (5‐FU) and suberoylanilide hydroxamic acid (SAHA) on cell growth in vitro. An MTT assay was used to investigate effects on cell viability mediated by 5‐FU and SAHA. Cells were seeded into 96‐well plates and treated with various doses of 5‐FU and SAHA for 72 h, and then incubated with MTT reagent for 4 h. Cell viabilities were determined by measuring absorbance at 560 nm. The IC50 value was defined as the concentration of drug(s) needed for a 50% reduction in absorbance (560 nm) based on cell growth curves. Points, mean of at least three independent experiments; bars, SE. Note: The PC9/f14 cell line is a highly metastatic derivative of PC9 cells established at Nippon Medical School using an artificial metastasis method( 26 ).
Figure 2.
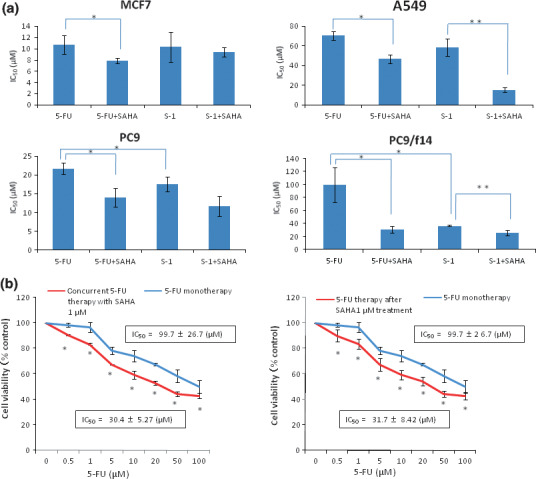
Effect of combination therapy with 5‐fluorouracil (5‐FU)/S‐1 and suberoylanilide hydroxamic acid (SAHA) on cell growth in vitro. (a) Indicated non‐small‐cell lung cancer (NSCLC) cells were treated with 5‐FU or S‐1, a compound composed of 5‐FU and 2‐chloro‐2,4‐dihydro‐xypyridine (CDHP) (1 μM), in combination with SAHA (1 μM). MTT assay was performed 72 h after addition of indicated drugs. IC50 was defined as the drug concentration required to inhibit cell proliferation by 50% compared with that of untreated control cells. Data represent the mean value of IC50 ± SE of three independent experiments in triplicate. *P < 0.05 compared to 5‐FU alone. **P < 0.05 compared to S‐1 alone. (b) 5‐Fluorouracil therapy for 72 h after low‐dose SAHA treatment (1 μM) for 24 h, and concurrent therapy for 72 h, was examined in PC9/f14 cells. Each result is expressed as cell viability in treated samples compared with the untreated sample (100%) for 5‐FU alone or compared with the 1 μM SAHA‐treated sample (92.4% and 87.0% of the untreated sample; Fig. S1) for 5‐FU therapy after 1 μM SAHA treatment, and concurrent therapy with 1 μM SAHA treatment. Data represent the mean value of cell viability ± SE of three independent experiments in triplicate; *P < 0.05 compared to 5‐FU mono‐therapy.
These combination therapies significantly enhanced 5‐FU sensitivity approximately threefold, as compared to 5‐FU monotherapy, in PC9/f14 5‐FU‐resistant cells (Fig. 2b). These results indicate that the combination of 5‐FU with SAHA could have a synergistic growth‐inhibitory effect against PC9/f14 cells.
5‐Fluorouracil‐resistant lung cancer cells display high levels of TS mRNA and protein expression. The expression level of TS, OPRT, and DPD mRNA in the lung cancer cell lines and MCF7 cells was quantified by real‐time RT‐PCR analysis (Fig. 3a). Thymidylate synthase mRNA expression in PC9/f14 cells, which are 5‐FU resistant, was about 45 times the level of that seen with MCF7 cells, which are 5‐FU sensitive. MCF7 cells have wild‐type p53 status, while PC9 and PC9/f14 cells contain mutated p53( 30 , 31 ) (Fig. 3b‐1). By western blot analysis, the expression of TS protein also increased inversely in relationship to 5‐FU sensitivity (Fig. 3b). Expression of OPRT and DPD protein did not correlate with 5‐FU sensitivity (Fig. 3b).
Figure 3.
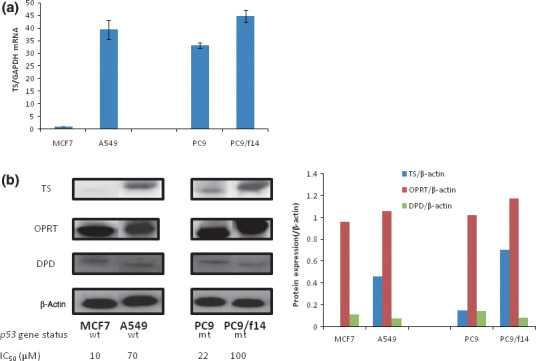
Correlation of 5‐fluorouracil (5‐FU) sensitivity with expression of factors related to 5‐FU metabolism in cancer cell lines. The expression of thymidylate synthase (TS), orotate phospho‐ribosyltransferase (OPRT), and dihydropyrimidine dehydrogenase (DPD) mRNA and protein was determined by real‐time RT‐PCR and western blot analysis, respectively, in lung cancer cell lines, as well as MCF7 cells. (a) The level of gene expression was expressed as ratio of the relevant mRNA in a particular sample to the level of GAPDH mRNA in that sample. The ratio was compared to the corresponding expression level observed in MCF7 cells. Points, mean of at least three independent experiments; bars, SE. (b) Expression of TS, OPRT, and DPD proteins were determined by western blot analyses. Quantitative analyses of protein expression ([TS, OPRT, and DPD]/β‐actin) was examined using NIH image software.
Suberoylanilide hydroxamic acid down‐regulates TS mRNA and protein expression in lung cancer cells. We examined the levels of TS, OPRT, and DPD mRNA and protein expression following SAHA treatment of PC9/f14 cells in order to identify key mechanism(s) underlying the synergistic growth‐inhibitory effect mediated by combination of S‐1 compounds + SAHA. From this analysis, it was observed that SAHA treatment reduced TS mRNA expression in dose‐dependent manner (Fig. 4a‐1). In addition, TS protein expression was found to be reduced in dose‐dependent manner, similar to that observed at the mRNA level, following SAHA treatment (Fig. 4a). Again, OPRT and DPD protein expression did not change in response to treatment with SAHA (Fig. 4a‐2).
Figure 4.
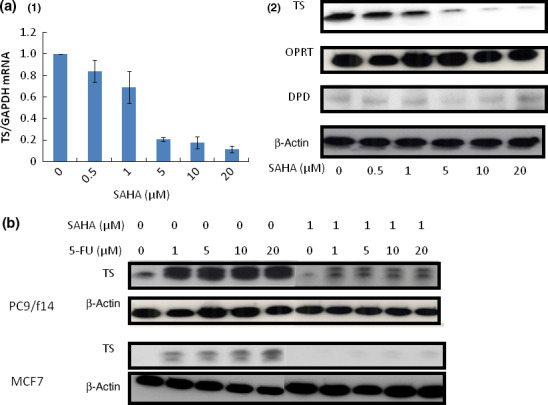
Effect of suberoylanilide hydroxamic acid (SAHA) treatment on expression of the factors related to 5‐fluorouracil (5‐FU) metabolism and the expression of thymidylate synthase (TS) in cancer cell lines after 5‐FU monotherapy and SAHA combination therapy. (a) The effect of SAHA treatment for 24 h on TS, orotate phosphoribosyltransferase (OPRT), and dihydropyrimidine dehydrogenase (DPD) mRNA and protein expression in PC9/f14 cells was examined by real‐time RT‐PCR and western blot analyses, respectively. (1) For the different doses of SAHA used, the level of gene expression was expressed as a ratio of the relevant mRNA to the level of GAPDH mRNA. Points, mean of at least three independent experiments; bars, SE. (2) Expression of TS, OPRT, and DPD proteins, following exposure to different doses of SAHA, were determined by western blot analyses. (b) PC9/f14 and MCF7 cells were incubated with various doses of 5‐FU and SAHA (1 μM) for 24 h. Thymidylate synthase expression was determined by western blot analysis. A rapid increase in TS protein expression was observed after the exposure to 5‐FU. Suberoylanilide hydroxamic acid suppressed 5‐FU‐mediated induction of TS protein.
Suberoylanilide hydroxamic acid suppresses 5‐FU‐mediated induction of TS protein. A rapid increase in TS protein expression, after exposure to 5‐FU, was shown in 5‐FU‐sensitive and ‐resistant cells (Fig. 4b). Use of a low dose of SAHA (1 μM) suppressed this induction of TS protein expression by 5‐FU in all examined cell types (Fig. 4b).
Rb‐E2F1 pathway modulation by SAHA. We examined the modulation of E2F1‐Rb activity by SAHA treatment. Moreover, we examined the effect of SAHA treatment on p27 and p16, which block the cyclin D–CDK4/6 complex, and p21waf1/cip1 which blocks the cyclin E–CDK2 complex in addition to E2F1 and Rb.
p21waf1/cip1 expression increased in a SAHA dose‐dependent manner, while phosphorylated Rb and E2F1 expression decreased (Fig. 5a). p27 and p16 expression did not change in response to SAHA treatment (Fig. 5a). Suberoylanilide hydroxamic acid monotherapy also induced PARP cleavage in a dose‐dependent manner when used at >5 μM.
Figure 5.
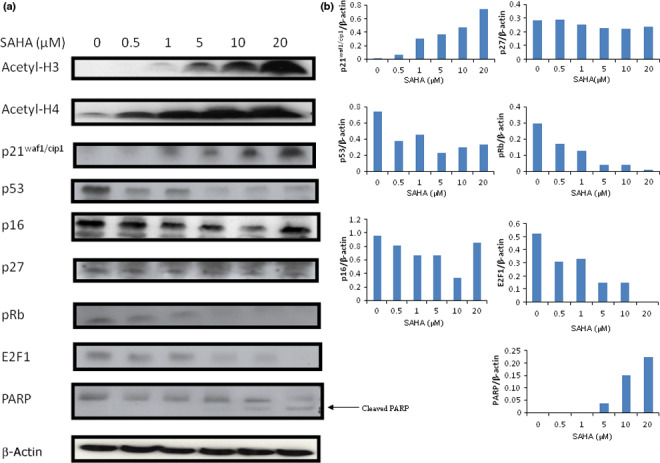
Mechanistic studies relating to down‐regulation of thymidylate synthase (TS) transcription by suberoylanilide hydroxamic acid (SAHA) treatment. (a) PC9/f14 cells were incubated with various doses of SAHA for 24 h. Protein expression levels were determined by western blot analyses. (b) Quantitative analyses of protein expression (E2F1, p53, pRb, p16, p21waf1/cip1, p27, and cleaved poly‐(ADP‐ribose)polymerase/β‐actin) were examined using NIH image software.
Histone deacetylase inhibitors, such as SAHA, activate gene transcription by increasing the acetylation of histones in particular promoter regions. Both acetylated H3 and H4 were dramatically increased in a SAHA dose‐dependent manner (Fig. 5a). Chromatin fragments from PC9/f14 cells, cultured with or without SAHA for 24 h, were immunoprecipitated with an antibody against acetylated H3 and H4. The level of H3 and H4 acetylation in the p21waf1/cip1 promoter region was increased after SAHA treatment (Fig. 6a). These results suggested that SAHA induced TS down‐regulation, decreasing E2F1 and phosphorylated Rb via p21waf1/cip1 up‐regulation based on its promoter acetylation (Fig. 6).
Figure 6.
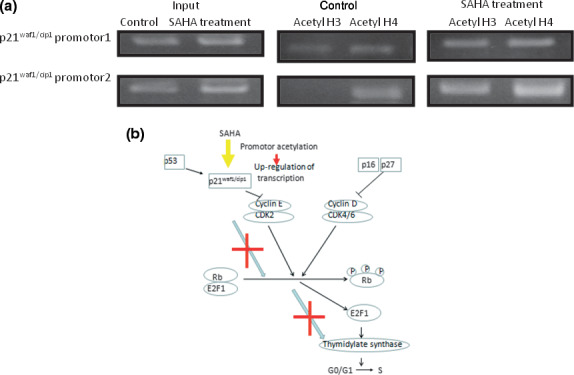
Summary of the proposed mechanism underlying down‐regulation of thymidylate synthase (TS) transcription by suberoylanilide hydroxamic acid (SAHA) treatment. (a) Acetylation of the p21waf1/cip1 promoter was determined by a ChIP assay. Soluble chromatin was immunoprecipitated with anti‐H3 and H4 antibodies from untreated cells and 1 μM SAHA. Polymerase chain reaction primers for two regions of the p21waf1/cip1 gene were used to amplify the DNA isolated from immunoprecipitated chromatin. (b) The mechanism underlying the down‐regulation of TS transcription by SAHA treatment is summarized.
Discussion
In our study, we demonstrated that low‐dose SAHA, which paralleled serum levels in treated patients with little evidence of adverse effects, mediated down‐regulation of TS mRNA expression and protein levels, as well as led to enhanced 5‐FU/S‐1 cytotoxity, especially in 5‐FU‐resistant lung cancer cells. We found that S‐1 (or 5‐FU) and SAHA exerted a synergistic antiproliferative effect against NSCLC cells, as determined by MTT assay.
We also demonstrated the ability of SAHA to down‐regulate TS mRNA and protein expression in dose‐dependent manner (Fig. 4a). The rapid induction of TS in response to exposure to 5‐FU was shown in all cell types. Thymidylate synthase, as an RNA binding protein, regulates its own synthesis by impairing the translation of its mRNA, and the binding to a specific inhibitor leads to up‐regulation of TS protein.( 32 , 33 ) Repeated 5‐FU treatment may result in sustained TS expression and eventually lead to 5‐FU resistance.( 32 , 33 )
In addition, SAHA led to decreased E2F1 and phosphorylated Rb expression via p21waf1/cip1 upr‐egulation due to acetylation of its promoter (Fig. 6). p21waf1/cip1 expression may be transcriptionally enhanced, accompanied by an accumulation of acetylated histones H3 and H4, which are associated with the p21waf1/cip1 gene promoter. Thus, we have supported the results of other studies in this context (5, 6).( 34 , 35 ) Suberoylanilide hydroxamic acid up‐regulated p21waf1/cip1 and inhibited the activity of cyclin E–CDk2 complex, resulting in down‐regulation of both E2F1 and phosphorylated Rb in PC9/f14 cells (5, 6). G1 cyclin/kinase activity, including cyclins D and E, led to unphosphorylated Rb and the release of E2F1 from the E2F1–Rb complex (Fig. 6).
Several reports have shown E2F1 overexpression correlates with TS expression and tumor proliferation.( 36 , 37 , 38 ) E2F1 regulates transcription of genes that encode S‐phase‐activating proteins that are required for DNA synthesis, as such thymidine kinase, DHFR, TS, and ribonucleotide reductase.( 39 ) Free E2F1 has been shown to transactivate target genes such as the TS gene.( 39 , 40 ) E2F1‐transfected cells, which showed up‐regulation of TS, were also reported to be more resistant to 5‐FU than non‐transfected cells.( 39 ) Peter et al. reported that, after 5‐FU treatment, TS bound by 5‐fluoro‐deoxyuridine monophosphate (FdUMP) derepressed the autoregulation of TS mRNA transcription, leading to more TS mRNA and, subsequently, increased synthesis of TS protein. Subsequently, the amount of E2F1 increased which generally induced transcription, leading to increased TS mRNA, encoding for more TS protein.( 20 ) Taken collectively, transcriptional regulation may attributed to Rb‐E2F1 pathway modulation by p21waf1/cip1 up‐regulation via its promoter histone acetylation by SAHA.
p53 expression was down‐regulated in response to SAHA treatment in PC9/f14 (p53 mutant) cells (Fig. 5a). These results supported the report by Di Gennaro et al. regarding the down‐regulation of p53 expression by SAHA in cells containing mutated, nonfunctional forms of p53.( 41 ) p21waf1/cip1 expression may be transcriptionally enhanced via its promoter histone acetylation by SAHA (Fig. 6). These results suggested that the decreases observed in relation to p53 levels may be due, in part, to a negative feedback mechanism; however, additional investigations will be needed to clarify this suggestion.
In conclusion, SAHA may overcome 5‐FU resistance by down‐regulating TS expression and up‐regulating p21waf1/cip1 expression via histone acetylation at its promoter, which in turn blocks the Rb–E2F1 pathway. This is the first report that SAHA enhanced 5‐FU/S‐1 sensitivity via the modulation of 5‐FU metabolism in lung cancer cells. Our preclinical results will facilitate future clinical investigations of combination chemotherapy with S‐1 and SAHA in patients with NSCLC.
Disclosure Statement
Akihiko Gemma and Akinobu Yoshimura serve as in advisory roles to Taiho Pharmaceutical.
Supporting information
Fig. S1.The cell viability of the 5‐fluorouracil (5‐FU) control (0 μM for 5‐FU for 72 h) after suberoylanilide hydroxamic acid (SAHA) treatment for 24 h, and 5‐FU control (0 μM for 5‐FU) concurrent with SAHA treatment for 72 h, are expressed compared to untreated cells (92.4 ± 3.76% and 87.0 ± 0.51% of the untreated sample).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This study was supported by a Grant‐in‐Aid from the Ministry of Education, Science, Sports and Culture of Japan (to A.G. and S.K.). We thank Kyowa Hakko and Taiho Pharmaceutical for providing 5‐FU, CDHP, and antibodies to TS, DPD, and OPRT. We thank Tsuneo Shimokawa, Hideaki Mizutani, and Hidehiko Kuribayashi for their advice in respect to study design and technical support.
References
- 1. Non‐small Cell Lung Cancer Collaborative Group . Chemotherapy in non‐small cell lung cancer: a meta‐analysis using updated data on individual patients from 52 randomized clinical trials. Br Med J 1995; 311: 899–909. [PMC free article] [PubMed] [Google Scholar]
- 2. Sandler AB, Nemunaitis J, Denham C et al. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non‐small‐cell lung cancer. J Clin Oncol 2000; 18: 122–30. [DOI] [PubMed] [Google Scholar]
- 3. Shepherd FA, Dancy J, Ramlau R et al. Prospective randomized trial of docetaxel versus best supportive care in patients with non‐small‐cell lung cancer previously treated with platinum‐based chemotherapy. J Clin Oncol 2000; 18: 2095–103. [DOI] [PubMed] [Google Scholar]
- 4. Hanna N, Shepherd FA, Fossella FV et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non‐small‐cell lung cancer previously treated with chemotherapy. J Clin Oncol 2004; 22: 1589–97. [DOI] [PubMed] [Google Scholar]
- 5. Shephered FA, Rodrigues Pereira J, Ciuleanu T et al. National Cancer Institute of Canada Clinical Trials Group. Erlotinib in previously treated non‐small‐cell lung cancer. N Engl J Med 2005; 353: 123–32. [DOI] [PubMed] [Google Scholar]
- 6. Johnstone RW. Histone‐deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002; 1: 287–99. [DOI] [PubMed] [Google Scholar]
- 7. Kelly WK, Richon VM, O’Connor O et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res 2003; 9: 3578–88. [PubMed] [Google Scholar]
- 8. Kelly WK, O’Connor OA, Krug LM et al. Phase I Study of an Oral Histone deacetylase inhibitor, Suberoylanilide hydroxamic acid, in Patients with advanced cancer. J Clin Oncol 2005; 23: 3923–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rubin EH, Agrawal NG, Friedman EJ et al. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin Cancer Res 2006; 12: 7039–45. [DOI] [PubMed] [Google Scholar]
- 10. Duvic M, Talpur R, Ni X et al. Phase II trial of oral vorinostat (SAHA) for refractory cutaneous T‐cell lymphoma (CTCL). Blood 2007; 1: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyanaga A, Gemma A, Noro R et al. Anti‐tumor activity of Histone deacetylase inhibitors in non‐small cell lung cancer cells: development of a molecular predictive model. Mol Cancer Ther 2008; 7(6): 1923–30. [DOI] [PubMed] [Google Scholar]
- 12. Kim MS, Black M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anti‐cancer drugs targeting DNA. Cancer Res 2003; 63: 7291–300. [PubMed] [Google Scholar]
- 13. Jang ER, Lim SJ, Lee ES et al. The histone deacetylase inhibitor inhibitor trichostatin A sentitizes estrogen receptor α‐negative breast cancer cells to tamoxifen. Oncogene 2003; 23: 1–13. [DOI] [PubMed] [Google Scholar]
- 14. Marchion DC, Bieaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence‐specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem 2004; 92: 223–37. [DOI] [PubMed] [Google Scholar]
- 15. Shirasaka T, Shimamoto Y, Fukushima M. Inhibition by oxonic acid of gastrointestinal toxicity of 5‐fluorouracil without loss of its antitumor activity in rats. Cancer Res 1993; 5: 4004–9. [PubMed] [Google Scholar]
- 16. Nakano J, Huang C, Liu D et al. Evaluation of biomarkers associated with 5‐FU sensitivity for non‐small‐cell lung cancer patients postoperatively treated with UFT. Br J Cancer 2006; 95: 607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Inoue K, Takao M, Watanabe F et al. Role of dihydropyrimidine dehydrogenase inhibitory fluoropyrimidine against non‐small‐cell lung cancer‐incorrelation with the tumoral expression of thymidylate synthase and dihydropyrmidine. Lung Cancer 2005; 49(1): 47–54. [DOI] [PubMed] [Google Scholar]
- 18. Peters GJ, Van Groeningen CJ, Laurensse EJ, Piendo HM. Sensitivity of human, murine, and rat cells to 5‐FU and 57‐deoxy‐5‐fluorouridine in relation to drug‐metabolizing enzymes. Cancer Res 1986; 46: 20–8. [PubMed] [Google Scholar]
- 19. Huang CL, Yokomise H, Fukushima M, Kinoshita M. Tailor‐maid chemotherapy for non‐small cell lung cancer patients. Future Oncol 2006; 2(2): 289–99. [DOI] [PubMed] [Google Scholar]
- 20. Peters GJ, Van Der Wilt CL, Van Triest B et al. Thymidylate Synthase and drug resistance. Eur J Cancer 1995; 31(A): 1299–305. [DOI] [PubMed] [Google Scholar]
- 21. Rustum YM, Harstrick A, Cao S et al. Thymidylate synthase inhibitors in cancer therapy: direct and indirect inhibitors. J Clin Oncol 1997; 15: 389–400. [DOI] [PubMed] [Google Scholar]
- 22. Saga Y, Suzuki M, Mizukami H et al. Overexpression of Thymidylate synthase mediates desensitization for 5‐fluorourai of tumor cells. Int J Cancer 2003; 106: 324–6. [DOI] [PubMed] [Google Scholar]
- 23. Kawahara M, Furuse K, Segawa Y et al. S‐1 Cooperative Study Group (Lung Cancer Working Group). Phase II study S‐1, a novel oral fluorouracil, in advanced non‐small‐cell lung cancer. Br J Cancer 2001; 85: 939–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ichinose Y, Yoshimori K, Sakai H et al. S‐1 plus cisplatin combination chemotherapy in patients with advanced non‐small‐cell lung cancer: a multi‐institutionl Phase II trial. Clin Cancer Res 2004; 10: 7860–4. [DOI] [PubMed] [Google Scholar]
- 25. Gemma A, Takenaka K, Hosoya Y et al. Altered expression of several genes in highly metastatic subpopulations of a human pulmonary adenocarcinoma cell line. Eur J Cancer 2001; 37(12): 1554–61. [DOI] [PubMed] [Google Scholar]
- 26. Wang W, Cassidy J, O’Brien V, Ryan KM, Collie‐Duguid E. Mechanistic and predictive profiling of 5‐FU resistance in human cancer cells. Cancer Res 2004; 64: 8167–76. [DOI] [PubMed] [Google Scholar]
- 27. Noro R, Gemma A, Kosaihira S et al. Gefitinib (IRESSA) sensitive lung cancer cell lines show phosphorylation of Akt without ligand stimulation. BMC Cancer 2006; 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McMillan RE, Sikes ML. Differential activation of dual promoters alters Dβ2 germline transcription during thymocyte development. J Immunol 2008; 180: 3218–28. [DOI] [PubMed] [Google Scholar]
- 29. Van Groeningen Cees J., Peters Godefridus J., Schornagel Jan H. et al. Phase I clinical and pharmacokinetic study of oral S‐1 in patients with advanced solid tumors. J Clin Oncol 2000: 18: 2772–9. [DOI] [PubMed] [Google Scholar]
- 30. Okano T, Gemma A, Hosoya Y et al. Alterations in novel candidate tumor suppressor genes, ING1 and ING2 in human lung cancer. Oncol Rep 2006; 15: 545–9. [PubMed] [Google Scholar]
- 31. Lu X, Errington J, Curtin NJ, Lunec J, Newell DR. The impact of p53 status on cellular sensitivity to antifolate drugs. Clin Cancer Res 2001; 7: 2114–23. [PubMed] [Google Scholar]
- 32. Chu E, Callender MA, Farell MP, Schmitz JC. Thymidylate synthase inhibitors as anticancer agents: from bench to bed side. Cancer Chemother Pharmacol 2003; 52(Suppl): S80–9. [DOI] [PubMed] [Google Scholar]
- 33. Welsh SJ, Titley J, Brunton L et al. Comparison of Thymidylate synthase (TS) protein up‐regulation after TS inhibitor in normal and tumor cell lines and tissues. Clin Cancer Res 2006; 6: 2538–46. [PubMed] [Google Scholar]
- 34. Takai N, Kawamata N, Gui D, Said JW, Miyakawa I, Koeffler HP. Human Ovarian Carcinoma cells: histone deacetylase inhibitors exhibit anti‐proliferative activity and potently induce apoptosis. Cancer 2004; 101(12): 2760–70. [DOI] [PubMed] [Google Scholar]
- 35. Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21 expression and gene‐associated histone acetylation. Pro Natl Acad Sci 2000; 97(18): 10014–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Banerjee D, Gorlick R, Liefshitz A et al. Levels of E2F1 expression are higher in lung metastasis of colon cancer as compared with hepatic metastasis and correlate with levels of thymidylate synthase. Cancer Res 2000; 60: 2365–7. [PubMed] [Google Scholar]
- 37. Kasahara M, Takahashi Y, Nagata T et al. Thymidylate synthase expression correlates closely with E2F1 expression in colon cancer. Clin Cancer Res 2000; 6: 2707–11. [PubMed] [Google Scholar]
- 38. Huang CL, Liu D, Nakano J et al. E2F1 overexpression correlates with thymidylate synthase and surviving gene expressions and tumor proliferation in non‐small‐cell lung cancer. Clin Cancer Res 2007; 13(23): 6938–46. [DOI] [PubMed] [Google Scholar]
- 39. Banerjee D, Schnieders B, Fu JZ, Adhikari D, Zhao SC, Bertino JR. Role of E2F1 in Chemosensitivity. Cancer Res 1998; 58: 4292–6. [PubMed] [Google Scholar]
- 40. DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis and G1/S regulatory genes. Mol Cell Biol 1995; 15: 4215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Di Gennaro E, Bruzzese F, Pepe S et al. Modulation of thymidylate synthase and p53 expression by HDAC inhibitor vorinostat resulted in synergistic antitumor effect in combination with 5‐FU or raltitrexed. Cancer Biol Ther 2009; 8(9): 782–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1.The cell viability of the 5‐fluorouracil (5‐FU) control (0 μM for 5‐FU for 72 h) after suberoylanilide hydroxamic acid (SAHA) treatment for 24 h, and 5‐FU control (0 μM for 5‐FU) concurrent with SAHA treatment for 72 h, are expressed compared to untreated cells (92.4 ± 3.76% and 87.0 ± 0.51% of the untreated sample).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item