

Abstract
The development of gastric cancer is closely associated with Helicobacter pylori (H. pylori) infection. The expression of cylooxigenase‐2 (COX‐2), a rate‐limiting enzyme for prostaglandin biosynthesis, is induced in H. pylori‐associated chronic gastritis, which thus results in the induction of proinflammatory prostaglandin, PGE2. The COX‐2/PGE2 pathway plays a key role in gastric tumorigenesis. On the other hand, several oncogenic pathways have been shown to trigger gastric tumorigenesis. The activation of Wnt/β‐catenin signaling is found in 30–50% of gastric cancers, thus suggesting that Wnt signaling plays a causal role in gastric cancer development. Mutations in the bone morphogenetic protein (BMP) signaling pathway are responsible for the subset of juvenile polyposis syndrome (JPS) that develops hamartomas in the gastrointestinal tract. BMP suppression appears to contribute to gastric cancer development because gastric cancer risk is increased in JPS. Wnt signaling is important for the maintenance of gastrointestinal stem cells, while BMP promotes epithelial cell differentiation. Accordingly, it is possible that both Wnt activation and BMP suppression can cause gastric tumorigenesis through enhancement of the undifferentiated status of epithelial cells. Recent mouse model studies have indicated that induction of the PGE2 pathway is required for the development of both gastric adenocarcinoma and hamartoma in the Wnt‐activated and BMP‐suppressed gastric mucosa, respectively. This article reviews the involvement of the PGE2, Wnt, and BMP pathways in the development of gastric cancer, and gastric phenotypes that are found in transgenic mouse models of PGE2 induction, Wnt activation, BMP suppression, or a combination of these pathways. (Cancer Sci 2009; 100: 1779–1785)
Epidemiological studies indicate that the regular use of nonsteroidal anti‐inflammatory drugs (NSAIDs) lowers the mortality rate of gastrointestinal cancer.( 1 ) The major target of NSAIDs is cyclooxygenases (COXs), COX‐1 and COX‐2, which are rate‐limiting enzymes for prostaglandin biosynthesis (Fig. 1). COX‐1 is constitutively expressed in most tissues and it is considered to be responsible for physiological levels of prostaglandin.( 2 ) In contrast, COX‐2 is induced in inflammation by various stimuli including cytokines and growth factors.( 3 , 4 , 5 ) The induction of COX‐2 expression is also found in a variety of cancer tissues. Mouse genetic studies have demonstrated that disruption of the Ptgs2 gene encoding COX‐2 results in the suppression of tumor development in the intestine and skin.( 6 , 7 ) Moreover, various animal studies have confirmed that treatment with NSAIDs or COX‐2 selective inhibitors (COXIBs) suppressed chemically induced tumor formation and xenografted tumor growth.( 8 ) These results, taken together, indicate that the COX‐2 pathway plays an essential role in cancer development.
Figure 1.
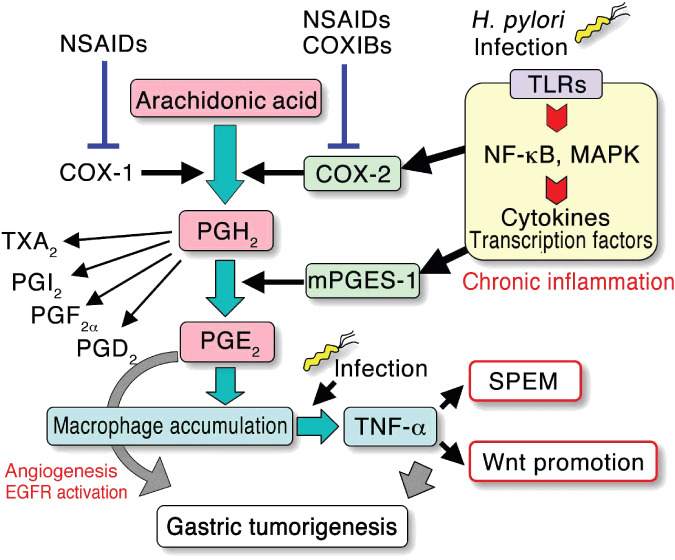
Schematic presentation of arachidonic acid metabolism in the context of gastric tumorigenesis. The expression of cyclooxygenase (COX)‐2 and microsomal PGE synthase‐1 (mPGES‐1) is induced by Helicobacter pylori (H. pylori)‐associated inflammatory responses. The simultaneous expression of both COX‐2 and mPGES‐1 leads to induction of the prostaglandin PGE2 pathway, which results in macrophage accumulation. These macrophages are activated by infectious stimuli, resulting in the induction of tumor necrosis factor (TNF)‐α‐dependent SPEM development and the promotion of Wnt signaling, which may contribute to gastric tumorigenesis. The induction of angiogenesis and activation of epidermal growth factor receptor (EGFR) signaling are also possible mechanisms of PGE2 in tumorigenesis. COXIBs, COX‐2 selective inhibitors; NF‐κB, nuclear factor‐κB; NSAIDS, nonsteroidal anti‐inflammatory drugs; SPEM, spasmolytic polypeptide/TFF2‐expressing metaplasia; TLRs, Toll‐like receptors.
However, the mechanism of the COX‐2 pathway underlying gastric tumorigenesis has not yet been fully elucidated. To investigate the possible crosstalk between the COX‐2 pathway and oncogenic activation in gastric carcinogenesis, a series of mouse models have been constructed and examined as discussed in this review.
Induction of the COX‐2/PGE2 pathway in gastric cancer
Regular use of NSAIDs is associated with a decreased incidence of gastric cancer.( 9 , 10 , 11 , 12 ) Induction of COX‐2 is found in approximately 70% of gastric cancer, whereas the expression of COX‐1 is not elevated.( 13 , 14 ) Gastric cancer can be divided into two histological subtypes: intestinal and diffuse types, and the expression of COX‐2 is found predominantly in the intestinal‐type gastric cancer.( 15 ) These results suggest that the COX‐2 pathway plays a role in the development of intestinal‐type gastric cancer.
Infection with Helicobacter pylori (H. pylori) causes chronic gastritis, which is associated with gastric carcinogenesis.( 16 ) The expression of COX‐2 is significantly induced in the H. pylori‐infected gastric mucosa, and that COX‐2 expression is suppressed by the eradication of H. pylori.( 17 ) Although the molecular mechanism for COX‐2 induction in tumors has not been elucidated, it is possible that the cytokine network is activated by infection and induces the expression of COX‐2. H. pylori can stimulate Toll‐like receptors (TLRs), leading to activation of the nuclear factor‐κB (NF‐κB) pathway that induces the expression of COX‐2 (Fig. 1).( 18 , 19 ) Moreover, TLR2/TLR9 signaling by H. pylori activates mitogen activated protein kinases (MAPK) including p38, resulting in the activation of CRE and AP‐1 elements on the COX‐2 gene promoter.( 19 , 20 )
Microsomal PGE synthase‐1 (mPGES‐1), a PGE2 converting enzyme is functionally coupled with COX‐2.( 21 ) Simultaneous induction of COX‐2 and mPGES‐1 is observed in gastric cancer tissues suggesting induction of the PGE2 pathway in gastric tumors (Fig. 1).( 22 , 23 ) The level of mPGES‐1 also decreases after the eradication of H. pylori,( 24 ) thus indicating that H. pylori infection induces the PGE2 pathway through induction of both COX‐2 and mPGES‐1. The PGE2 level significantly increases in gastric cancer,( 25 ) and the level is associated with the H. pylori infection status.( 26 )
Gastric tumor development in mouse and rat models induced by chemical carcinogens or Helicobacter infection is suppressed by treatment with NSAIDs or COXIBs.( 27 , 28 , 29 ) H. pylori infection in Mongolian gerbils induces gastric tumorigenesis, which is quite similar to the course of human gastric carcinogenesis. Importantly, treatment with H. pylori‐infected and chemical carcinogen‐treated Mongolian gerbils with a COXIB suppressed gastric carcinogenesis.( 30 , 31 ) These animal studies suggest that the COX‐2 pathway thus plays an essential role in H. pylori infection–associated gastric tumorigenesis.
K19‐C2mE transgenic mice: A model for PGE2 induction in the stomach
K19‐C2mE transgenic mice express both COX‐2 and mPGES‐1 in gastric epithelial cells, which results in induction of the PGE2 pathway in the stomach (Fig. 2).( 32 ) K19‐C2mE mice develop hyperplastic lesions in the glandular stomach. Histologically, the major cell type of hyperplasia is the mucous cell, which is similar to that found in the spasmolytic polypeptide/TFF2‐expressing metaplasia (SPEM; Fig. 2).( 33 ) The development of SPEM is associated with an H. pylori infection and gastric adenocarcinoma, thus suggesting that SPEM is an H. pylori‐induced precancerous lesion.( 34 ) It is thus possible that the PGE2 pathway induced by H. pylori infection is responsible for the development of SPEM. Treatment with N‐methyl‐N‐nitrosourea (MNU) to the H. pylori‐infected mice causes gastric tumor development. Notably, the multiplicity of gastric tumors induced by H. pylori infection and MNU treatment was significantly higher in K19‐C2mE mice compared with wild‐type mice.( 35 ) These results suggest that PGE2‐induced metaplastic hyperplasia is a precursor for chemical carcinogen‐induced gastric tumor.
Figure 2.
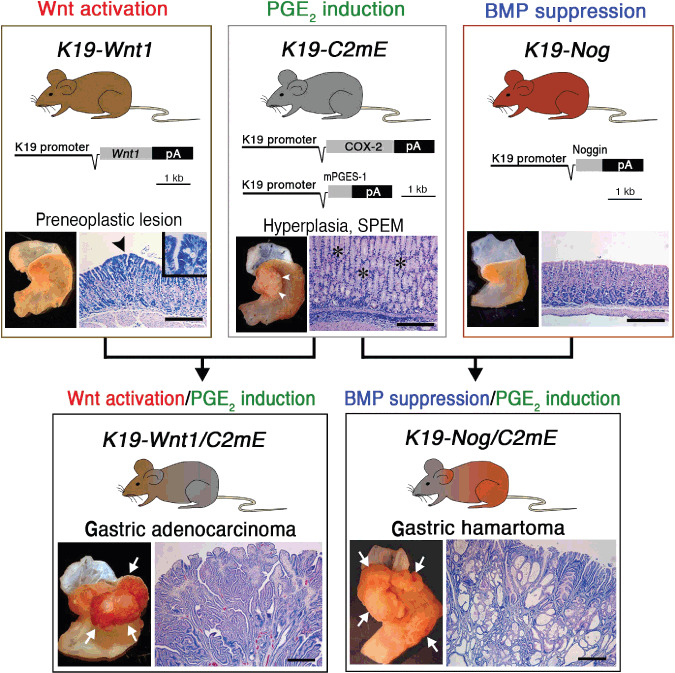
Transgenic mouse models of gastric tumorigenesis. Transgenic vector construction(s) and representative macroscopic and microscopic photographs of the stomach are shown for each line. K19‐Wnt1/C2mE and K19‐Nog/C2mE are compound transgenic mice of K19‐Wnt1 and K19‐C2mE, and K19‐Nog and K19‐C2mE, respectively. The arrowhead in the K19‐Wnt1 mouse stomach indicates a preneoplastic lesion. The arrowheads and asterisks in the K19‐C2mE mouse stomach indicate gastric hyperplasia and mucous metaplasia (SPEM), respectively. The arrows in K19‐Wnt1/C2mE and K19‐Nog/C2mE indicate gastric tumors. Note that the histology of the K19‐Wnt1/C2mE mouse shows dysplastic adenocarcinoma, while that of the K19‐Nog/C2mE mouse shows hamartoma with dilated cystic structure. Bars indicate 100 µm. (Reproduced from Oshima et al. Cancer Res, 69: 2729–33, 2009.) BMP, bone morphogenetic protein; COX‐2, cyclooxygenase‐2; SPEM, spasmolytic polypeptide/TFF2‐expressing metaplasia.
Notably, macrophages infiltrate and are activated in the gastric mucosa of K19‐C2mE mice.( 32 ) The activation of these macrophages is suppressed by treatment with antibiotics, thus indicating that infectious stimuli activate the accumulated macrophages. Importantly, the development of SPEM is also suppressed by antibiotic treatment, thus suggesting that bacterial infection activates macrophages, which is required for SPEM development. Consistently, disruption of the tumor necrosis factor (TNF)‐α gene in K19‐C2mE mice results in the suppression of SPEM development, thus suggesting that TNF‐α derived from activated macrophages plays an essential role in SPEM formation.( 33 ) Moreover, it is conceivable that the induction of PGE2 signaling is the primary cause for the mucosal macrophage accumulation, because the treatment of K19‐C2mE mice with a COXIB, but not with antibiotics, inhibits macrophage infiltration. A possible mechanism for the processes from H. pylori infection to SPEM development through PGE2 induction and macrophage activation is depicted in Figure 1.
Activation of Wnt signaling in gastric cancer
Canonical Wnt signaling (Wnt/β‐catenin signaling) is a critical pathway in the regulation of development as well as in tumorigenesis.( 36 ) In the absence of the Wnt ligand, cytoplasmic β‐catenin is phosphorylated by GSK‐3β within a complex containing adenomatous polyposis coli (APC) and Axin, thus resulting in the degradation of β‐catenin through the ubiquitin proteasome pathway.( 37 ) When Wnt ligands bind Frizzled receptors, phosphorylation of β‐catenin is suppressed, leading to stabilization and nuclear translocation of β‐catenin (Fig. 3). Nuclear β‐catenin interacts with T‐cell factor/lymphocyte enhancer factor (TCF/LEF) to induce transcription of Wnt target genes. APC or β‐catenin mutation causes tumor development by activation of the canonical Wnt signaling.
Figure 3.
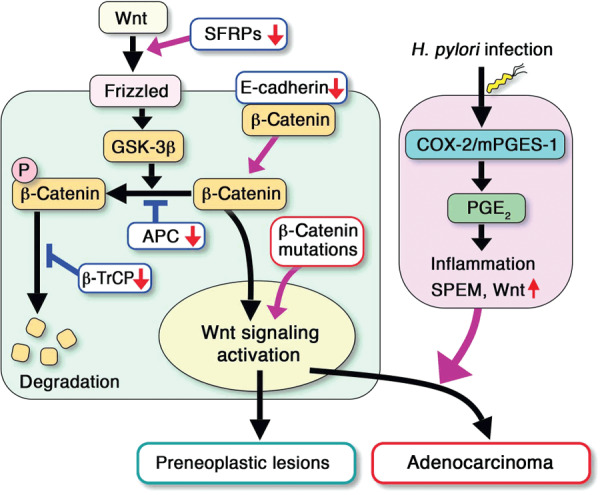
Schematic presentation of the canonical Wnt signaling and cyclooxygenase (COX)‐2/prostaglandin PGE2 pathway in gastric tumor development. β‐Catenin mutations, SFRPs methylation, and downregulation of E‐cadherin or β‐TrCP can activate Wnt signaling in gastric cancer. Cooperation of the Helicobacter pylori (H. pylori)‐induced COX‐2/PGE2 pathway with Wnt activation leads to the development of gastric adenocarcinoma. Without the induction of the PGE2 pathway, Wnt activation alone does not cause gastric cancer development. mPGES‐1, microsomal PGE synthase‐1.
Patients with germ‐line mutations in the APC gene have an increased risk of gastric cancer.( 38 ) Moreover, β‐catenin accumulation, a hallmark of Wnt activation, is found in 30–50% of gastric cancers.( 39 , 40 ) These results suggest that Wnt activation is one of the major causes of gastric cancer development. In gastric cancer, mutations in β‐catenin are reported, while APC mutations are rarely detected.( 41 , 42 , 43 ) However, the incidence of β‐catenin mutations is less than 30% in the Wnt‐activated gastric cancers,( 39 ) thus suggesting mechanism(s) other than APC or β‐catenin mutation for activation of the Wnt pathway. It has been suggested that the cytoplasmic β‐catenin level is increased by E‐cadherin downregulation or β‐TrCP mutation through decrease of E‐cadherin‐bound membrane β‐catenin or inhibition of β‐catenin ubiquitination, respectively (Fig. 3).( 44 , 45 ) These mechanisms may thus contribute to the activation of Wnt signaling in gastric tumorigenesis. On the other hand, the expression of the SFRP1, ‐2, and ‐5 genes are silenced by promoter methylation in gastric cancer cells.( 46 ) SFRPs are secreted endogenous antagonist of the Wnt ligands. Accordingly, it is possible that SFRP methylation is also an important mechanism for Wnt activation in gastric tumorigenesis (Fig. 3).( 46 )
K19‐Wnt1 transgenic mice: A model for Wnt activation in the stomach
K19‐Wnt1 transgenic mice express Wnt1, one of the canonical Wnt ligands, in the gastric epithelial cells, which results in the activation of Wnt signaling in the stomach (Fig. 2).( 40 ) The number of undifferentiated epithelial cells increases in the K19‐Wnt1 mouse stomach, thus suggesting that Wnt signaling keeps gastric epithelial cells in an undifferentiated status. Small preneoplastic lesions spontaneously develop in the gastric mucosa of K19‐Wnt1 mice, which consist of dysplastic epithelial cells (Fig. 2). However, gastric tumors do not develop in K19‐Wnt1 mice. It is thus possible that the activation of Wnt signaling can trigger tumorigenesis and form small preneoplastic lesions; however, Wnt activation alone is not sufficient for tumor development (Fig. 3).
To examine the effect of the PGE2 pathway in the Wnt‐activated gastric mucosa, K19‐Wnt1 mice were crossed with K19‐C2mE to construct compound transgenic mice (K19‐Wnt1/C2mE mice), in which both the Wnt and PGE2 pathways were activated in the stomach simultaneously. Importantly, K19‐Wnt1/C2mE mice developed gastric adenocarcinoma (Fig. 2).( 40 ) The tumors consisted of dysplastic epithelial cells, which sometimes invade the smooth muscle layers. These results clearly indicate that the simultaneous activation of the Wnt and PGE2 pathways is responsible for the development of gastric adenocarcinomas. Importantly, the gene expression profile of K19‐Wnt1/C2mE mouse gastric tumors is similar to that of human intestinal‐type gastric cancer (Hiraku Itadani et al., submitted manuscript, 2009). Therefore, K19‐Wnt1/C2mE mice recapitulate human intestinal‐type gastric cancer not only in the molecular etiology, but also in the pathological and molecular characteristics of tumors.
These results of mouse model studies suggest the following possible scenario for gastric tumorigenesis (Fig. 3): H. pylori infection causes the induction of the COX‐2/PGE2 pathway, which thus leads to SPEM development. The activation of Wnt signaling in the normal gastric mucosa causes small preneoplastic lesions, but is not sufficient for tumor formation. When Wnt signaling is activated in the PGE2 induction–associated inflamed mucosa, gastric adenocarcinomas develop through cooperation of the Wnt and PGE2 pathways.
Suppression of BMP signaling in gastric tumors
Juvenile polyposis syndrome (JPS) is characterized by hereditary gastrointestinal hamartomatous polyposis,( 47 ) a subset of which is caused by germline mutations in the BMP receptor type IA gene (BMPR1A).( 48 ) BMP ligands bind to a complex of the BMP receptor type II and I, leading to phosphorylation of Smad1,5,8, thereby allowing them to form a complex with Smad4.( 49 , 50 ) These Smad complexes translocate to the nuclei and function as transcription enhancers. BMP signaling inhibits epithelial proliferation and promotes differentiation.( 51 , 52 ) The suppression of BMP signaling in the mouse intestine results in hamartomatous polyp development,( 52 , 53 ) elongated villi, and crypt fission.( 54 ) These results suggest that the suppression of BMP signaling causes tumorigenesis by the inhibition of epithelial cell differentiation. Although the main affected site in JPS patients is the intestine, gastric polyps also develop in JPS, and the cancer risk in JPS patients increases both in the colon and stomach.( 55 , 56 ) Moreover, the expression of BMP‐2 is suppressed by promoter methylation in gastric cancer cells,( 57 ) and stimulation of gastric cancer cells with BMP‐2 suppresses proliferation.( 58 ) These results suggest that the inhibition of BMP signaling contributes to gastric tumorigenesis through the suppression of differentiation.
K19‐Nog transgenic mice: A model for BMP suppression in the stomach
K19‐Nog mice express noggin, an endogenous BMP antagonist, in the gastric epithelial cells, thus resulting in the inhibition of BMP signaling in the stomach (Fig. 2).( 59 ) Noggin is a polypeptide that inhibits BMP signaling by binding the BMP ligands.( 50 ) In the K19‐Nog mice, the phosphorylation of Smad1,5,8 is suppressed in the gastric gland by BMP inhibition. However, K19‐Nog mice do not develop gastric lesions, and the histology of the gastric mucosa is normal (Fig. 2). To examine the effect of cooperation of BMP suppression and PGE2 induction, K19‐Nog mice were crossed with K19‐C2mE to construct compound transgenic mice (K19‐Nog/C2mE mice), in which BMP signaling is suppressed and the PGE2 pathway is induced in the gastric mucosa. Importantly, K19‐Nog/C2mE mice develop large tumors in the glandular stomach (Fig. 2). These results indicate that the suppression of BMP signaling is insufficient for gastric tumorigenesis; however, the induction of the PGE2 pathway does promote tumor formation in the BMP‐suppressed gastric mucosa.
Histologically, K19‐Nog/C2mE mouse tumors are not dysplastic, but consist of irregular branching of epithelial cell layers, combined with dilated cysts (Fig. 2). Such histological characteristics are distinct from adenocarcinomas of K19‐Wnt1/C2mE mice, but are typical of the hamartomas of JPS patients.( 55 , 56 , 60 ) These results indicate that the suppression of BMP signaling associated with PGE2 induction causes gastric hamartoma development. Accordingly, it is possible that types of genetic alterations determine the histological types of tumors, e.g. adenocarcinoma by Wnt activation or hamartoma by BMP suppression. Furthermore, the induction of the PGE2 pathway promotes tumor formation regardless of the histological types (Fig. 2). Accordingly, it is thus possible that H. pylori infection contributes to development of both types of gastric tumors through PGE2 induction.
It has been reported that BMP signaling negatively regulates Wnt signaling in the intestinal crypt.( 53 ) Namely, suppression of BMP signaling enhances Wnt activity through the activation of PI3K/Akt pathway. However, the β‐catenin level in the K19‐Nog/C2mE hamartomas is the same as that in the wild‐type mouse stomach, while it is markedly elevated in K19‐Wnt1/C2mE gastric tumors.( 59 ) These results indicate that gastric hamartomas develop in K19‐Nog/C2mE mice due to a Wnt‐activation independent mechanism.
Possible mechanisms of the PGE2 pathway in gastric tumorigenesis
There are four G protein‐coupled receptors for PGE2, EP1–EP4. Among these receptors, the expression of EP4 increased significantly in gastric tumors of both K19‐Wnt1/C2mE mice and K19‐Nog/C2mE mice.( 59 ) It is thus possible that PGE2 signaling through EP4 plays a role in the development of adenocarcinoma and hamartoma in the Wnt‐activated and BMP‐suppressed gastric mucosa, respectively.
The promotion of the Wnt signaling activity beyond the basal activation level may be important for malignant progression.( 61 ) For example, increased accumulation of β‐catenin is found in the invasion front of colon cancer, suggesting that the increased Wnt activation level contributes to tumor invasion.( 62 ) Hepatocyte growth factor (HGF) and platelet‐derived growth factor (PDGF) increase the Wnt signaling activity in colon cancer cells, suggesting that these factors function as Wnt promoters.( 63 , 64 ) Notably, the level of Wnt signaling activity in gastric cancer cells significantly increases when the cells are stimulated with a conditioned medium from activated macrophages (Fig. 4a).( 65 ) Moreover, TNF‐α, but not other proinflammatory cytokines, caused an increase in the Wnt signaling activity in gastric cancer cells (Fig. 4b).( 65 ) These results suggest the hypothesis that Wnt activation, due to either genetic or epigenetic alterations in normal epithelial cells, is not sufficient for gastric tumor development. However, the Wnt activation level increases further in the inflamed mucosa by macrophage‐derived TNF‐α, and such Wnt promotion contributes to gastric cancer development (Fig. 4c). The induction of the PGE2 pathway leads to macrophage accumulation in the gastric mucosa. It is therefore conceivable that the induction of Wnt promotion is one of the important mechanisms of the PGE2 pathway in gastric tumorigenesis (Fig. 1).
Figure 4.

Promotion of Wnt signaling by macrophage‐derived tumor necrosis factor (TNF)‐α in gastric cancer cells. (a) Representative FACS analyses of Wnt‐reporter gastric cancer cells, AGS‐GFP, in which GFP expression is regulated by β‐catenin/TCF. GFP intensity increased significantly when cells were treated with conditioned medium (CM) from activated macrophages. (b) GFP intensity of reporter cells treated with indicated cytokines are shown in the bar graph. Note that Wnt activity is elevated by treatment with TNF‐α in a dose‐dependent manner. (c) Hypothesis for gastric tumor development. The level of Wnt signaling activated by genetic/epigenetic alteration is not sufficient for tumorigenesis. However, Helicobacter pylori (H. pylori) infection‐induced inflammation promotes the Wnt activation level through macrophage‐derived TNF‐α, which contributes to gastric tumorigenesis. (a and b, reproduced from Oguma et al. EMBO J, 27: 1671–81, 2008, with permission from the Nature Publishing Group.)
Conclusions
Studies with mouse models have elucidated the roles of the PGE2 pathway in gastric tumorigenesis in the Wnt‐activated and BMP‐suppressed gastric mucosa. Alterations in morphogen signals, such as the Wnt and BMP pathways, can therefore trigger gastric tumorigenesis by the suppression of epithelial differentiation. However, alterations of these signals in the non‐inflamed stomach do not cause gastric tumor formation. In contrast, alterations of these signals in the inflamed gastric mucosa lead to the development of gastric tumors through cooperation with the PGE2 pathway. Moreover, mouse studies show the possible mechanisms of PGE2 in gastric tumorigenesis, i.e. macrophage accumulation and activation, subsequent SPEM formation, and Wnt signaling promotion. Considering the multifunctional nature of PGE2, it is possible that other mechanisms triggered by the PGE2 pathway may also contribute to gastric tumorigenesis, which should be further elucidated using mouse models in the future. These studies will provide a rationale for the inhibition of the PGE2 pathway as a possible preventative strategy against gastric tumorigenesis.
Acknowledgment
We thank Manami Watanabe for her helpful work with the papers that we cited in this review article.
References
- 1. Thun MJ, Namboodiri MM, Heath CW Jr. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med 1991; 325: 1593–6. [DOI] [PubMed] [Google Scholar]
- 2. Dewitt DL, Smith WL. Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc Natl Acad Sci USA 1998; 85: 1412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen‐responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci USA 1991; 88: 2692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fletcher BS, Kujubu DA, Perrin DM, Herschman HR. Structure of the mitogen‐inducible TIS10 gene and demonstration that the TIS10‐encoded protein is a functional prostaglandin G/H synthase. J Biol Chem 1992; 267: 4338–44. [PubMed] [Google Scholar]
- 5. Hla T, Neilson K. Human cylooxygenase‐2 cDNA. Proc Natl Acad Sci USA 1992; 89: 7384–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oshima M, Dinchuk JE, Kargman SL et al . Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX‐2). Cell 1996; 87: 803–9. [DOI] [PubMed] [Google Scholar]
- 7. Chulada PC, Thompson MB, Mahler JF et al . Genetic disruption of Ptgs‐1, as well as Ptgs‐2, reduces intestinal tumorigenesis in Min mice. Cancer Res 2000; 60: 4705–8. [PubMed] [Google Scholar]
- 8. Oshima M, Taketo MM. COX selectivity and animal models for colon cancer. Curr Pharm Des 2002; 8: 1021–34. [DOI] [PubMed] [Google Scholar]
- 9. Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW Jr. Aspirin use and risk of fatal cancer. Cancer Res 1993; 53: 1322–7. [PubMed] [Google Scholar]
- 10. Schreinemachers DM, Everson RB. Aspirin use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology 1994; 5: 138–46. [DOI] [PubMed] [Google Scholar]
- 11. Zaridze D, Borisova E, Maximovitch D, Chkhikvadze V. Aspirin protects against gastric cancer: results of a case‐control study from Moscow, Russia. Int J Cancer 1999; 82: 473–6. [DOI] [PubMed] [Google Scholar]
- 12. Akre K, Ekstrom AM, Signorello LB, Hansson LE, Nyren O. Aspirin and risk for gastric cancer: a population‐based case‐control study in Sweden. Br J Cancer 2001; 84: 965–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ristimäki A, Honkanen N, Jänkälä H, Sipponen P, Härkönen M. Expression of cyclooxygenase‐2 in human gastric carcinoma. Cancer Res 1997; 57: 1276–80. [PubMed] [Google Scholar]
- 14. Saukkonen K, Rintahaka J, Sivula A et al . Cyclooxygenase‐2 and gastric carcinogenesis. APMIS 2003; 111: 915–25. [DOI] [PubMed] [Google Scholar]
- 15. Saukkonen K, Nieminen O, Van Rees B et al . Expression of cyclooxygenase‐2 in dysplasia of the stomach and in intestinal‐type gastric adenocarcinoma. Clin Cancer Res 2001; 7: 1923–31. [PubMed] [Google Scholar]
- 16. Correa P. Helicobacter pylori infection and gastric cancer. Cancer Epidemiol Biomarkers Prev 2003; 12: 238s–41s. [PubMed] [Google Scholar]
- 17. McCarthy CJ, Crofford LJ, Greenson J, Scheiman JM. Cyclooxygenase‐2 expression in gastric antral mucosa before and after eradication of Helicobacter pylori infection. Am J Gastroenterol 1999; 94: 1218–23. [DOI] [PubMed] [Google Scholar]
- 18. Smith MF Jr, Mitchell A, Li G et al . Toll‐like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori‐induced NF‐κB activation and chemokine expression by epithelial cells. J Biol Chem 2003; 278: 32 552–60. [DOI] [PubMed] [Google Scholar]
- 19. Chang YJ, Wu MS, Lin JT et al . Induction of cyclooxygenase‐2 overexpression in human gastric epithelial cells by Helicobacter pylori involves TLR2/TLR9 and c‐Src‐dependent nuclear factor‐κB activation. Mol Pharmacol 2004; 66: 1465–77. [DOI] [PubMed] [Google Scholar]
- 20. Chang YJ, Wu MS, Lin JT, Chen CC. Helicobacter pylori‐induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase‐2 induction through TLR2/TLR9 and promoter regulation. J Immunol 2005; 175: 8242–52. [DOI] [PubMed] [Google Scholar]
- 21. Murakami M, Naraba H, Tanioka T et al . Regulation of prostaglandin E2 biosynthesis by inducible membrane‐associated prostaglandin E2 synthase that acts in concert with cyclooxygenase‐2. J Biol Chem 2000; 275: 32783–92. [DOI] [PubMed] [Google Scholar]
- 22. Van Rees BP, Sivula A, Thorén S et al . Expression of microsomal prostaglandin E synthase‐1 in intestinal type gastric adenocarcinoma and in gastric cancer cell lines. Int J Cancer 2003; 107: 551–6. [DOI] [PubMed] [Google Scholar]
- 23. Jang TJ. Expression of proteins related to prostaglandin E2 biosynthesis is increased in human gastric cancer and during gastric carcinogenesis. Virchows Arch 2004; 445: 564–71. [DOI] [PubMed] [Google Scholar]
- 24. Nardone G, Rocco A, Vaira D et al . Expression of COX‐2, mPGE‐synthase1, MDR‐1 (P‐gp), and Bcl‐XL: a molecular pathway of H. pylori‐related gastric carcinogenesis. J Pathol 2004; 202: 305–12. [DOI] [PubMed] [Google Scholar]
- 25. Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase‐2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clin Cancer Res 2000; 6: 135–8. [PubMed] [Google Scholar]
- 26. Al‐Marhoon MS, Nunn S, Soames RW. CagA+Helicobacter pylori induces greater levels of prostaglandin E2 than cagA‐strains. Prostaglandins Other Lipid Med 2004; 73: 181–9. [DOI] [PubMed] [Google Scholar]
- 27. Hu PJ, Yu J, Zeng ZR et al . Chemoprevention of gastric cancer by celecoxib in rats. Gut 2004; 53: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nam KT, Hahm KB, Oh SY et al . The selective cyclooxygenase‐2 inhibitor nimesulide prevents Helicobacter pylori‐associated gastric cancer development in a mouse model. Clin Cancer Res 2004; 10: 8105–13. [DOI] [PubMed] [Google Scholar]
- 29. Xiao F, Furuta T, Takashima M, Shirai N, Hanai H. Involvement of cyclooxygenase‐2 in hyperplastic gastritis induced by Helicobacter pylori infection in C57BL/6 mice. Aliment Pharmacol Ther 2001; 15: 875–86. [DOI] [PubMed] [Google Scholar]
- 30. Magari H, Shimizu Y, Inada K et al . Inhibitory effects of etodolac, a selective cyclooxygenase‐2 inhibitor, on stomach carcinogenesis in Helicobacter pylori‐infected mongolian gerbils. Biochem Biophys Res Commun 2005; 334: 606–12. [DOI] [PubMed] [Google Scholar]
- 31. Futagami S, Suzuki K, Hiratsuka T et al . Celecoxib inhibits Cdx2 expression and prevents gastric cancer in Helicobacter pylori‐infected mongolian gerbils. Digestion 2006; 74: 187–98. [DOI] [PubMed] [Google Scholar]
- 32. Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX‐2 /mPGES‐1 transgenic mice. EMBO J 2004; 23: 1669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oshima M, Oshima H, Matsunaga A, Taketo MM. Hyperplastic gastric tumors with spasmolytic polypeptide‐expressing metaplasia caused by tumor necrosis factor‐α‐dependent inflammation in cyclooxygenase‐2/microsomal prostaglandin E synthase‐1 transgenic mice. Cancer Res 2005; 65: 9147–51. [DOI] [PubMed] [Google Scholar]
- 34. Nomura S, Baxter T, Yamaguchi H et al . Spasmolytic polypeptide expressing metaplasia to preneoplasia in H. felis‐infected mice. Gastroenterology 2004; 127: 582–94. [DOI] [PubMed] [Google Scholar]
- 35. Takasu S, Tsukamoto T, Cao X‐Y et al . Role of cyclooxygenase‐2 and microsomal prostaglandin E synthase‐1 expression and β‐catenin activation in gastric carcinogenesis in N‐merhyl‐N‐nitrosourea‐treated K19‐C2mE transgenic mice. Cancer Sci 2008; 99: 2356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taketo MM. Wnt signaling and gastrointestinal tumorigenesis in mouse models. Oncogene 2006; 25: 7522–30. [DOI] [PubMed] [Google Scholar]
- 37. Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev 2005; 19: 877–90. [DOI] [PubMed] [Google Scholar]
- 38. Offerhaus GJ, Giardiello FM, Krush AJ et al . The risk of upper gastrointestinal cancer in familial adenomatous polyposis. Gastroenterology 1992; 102: 1980–2. [DOI] [PubMed] [Google Scholar]
- 39. Clements WM, Wang J, Sarnaik A et al . β‐catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res 2002; 62: 3503–6. [PubMed] [Google Scholar]
- 40. Oshima H, Matsunaga A, Fujimura T, Tsukamoto T, Taketo MM, Oshima M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 2006; 131: 1086–95. [DOI] [PubMed] [Google Scholar]
- 41. Park WS, Oh RR, Park JY et al . Frequent somatic mutations of the β‐catenin gene in intestinal‐type gastric cancer. Cancer Res 1999; 59: 4257–60. [PubMed] [Google Scholar]
- 42. Woo DK, Kim HS, Lee HS, Kang YH, Yang HK, Kim WH. Altered expression and mutation of β‐catenin gene in gastric carcinomas and cell lines. Int J Cancer 2001; 95: 108–13. [DOI] [PubMed] [Google Scholar]
- 43. Ebert MPA, Fei G, Kahmann S et al . Increased β‐catenin mRNA levels and mutational alterations of the APC and β‐catenin gene are present in intestinal‐type gastric cancer. Carcinogenesis 2002; 23: 87–91. [DOI] [PubMed] [Google Scholar]
- 44. Cheng XX, Wang ZC, Chen XY et al . Correlation of Wnt‐2 expression and β‐catenin intracellular accumulation in Chinese gastric cancers: relevance with tumour dissemination. Cancer Lett 2005; 223: 339–47. [DOI] [PubMed] [Google Scholar]
- 45. Kim CJ, Song JH, Cho YG et al . Somatic mutations of the β‐TrCP gene in gastric cancer. APMIS 2007; 115: 127–33. [DOI] [PubMed] [Google Scholar]
- 46. Nojima M, Suzuki H, Toyota M et al . Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 2007; 26: 4699–713. [DOI] [PubMed] [Google Scholar]
- 47. Entius MM, Westerman AM, Van Velthuysen ML et al . Molecular and phenotypic markers of hamartomatous polyposis syndromes in the gastrointestinal tract. Hepatogastroenterology 1999; 46: 661–6. [PubMed] [Google Scholar]
- 48. Howe JR, Bair JL, Sayed MG et al . Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001; 28: 184–7. [DOI] [PubMed] [Google Scholar]
- 49. Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross‐talk. Cytokine Growth Factor Rev 2005; 16: 251–63. [DOI] [PubMed] [Google Scholar]
- 50. Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors 2004; 22: 233–41. [DOI] [PubMed] [Google Scholar]
- 51. Hardwick JCH, Van Den Brink GR, Bleuming SA et al . Bone morphogenetic protein 2 is expressed by, and acts upon, mature epithelial cells in the colon. Gastroenterology 2004; 126: 111–21. [DOI] [PubMed] [Google Scholar]
- 52. Haramis A‐PG, Begthel H, Van Den Born M et al . De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 2004; 303: 1684–6. [DOI] [PubMed] [Google Scholar]
- 53. He XC, Zhang J, Tong W‐G et al . BMP signaling inhibits intestinal stem cell self‐renewal through suppression of Wnt‐β‐catenin signaling. Nat Genet 2004; 36: 1117–21. [DOI] [PubMed] [Google Scholar]
- 54. Auclair BA, Benoit YD, Rivard N, Mishina Y, Perreault N. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology 2007; 133: 887–96. [DOI] [PubMed] [Google Scholar]
- 55. Chow E, Macrae F. Review of juvenile polyposis syndrome. J Gastroenterol Hepatol 2005; 20: 1634–40. [DOI] [PubMed] [Google Scholar]
- 56. Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol 2005; 100: 476–90. [DOI] [PubMed] [Google Scholar]
- 57. Wen XZ, Miyake S, Akiyama Y, Yuasa Y. BMP‐2 modulates the proliferation and differentiation of normal and cancerous gastric cells. Biochem Biophys Res Commun 2004; 316: 100–6. [DOI] [PubMed] [Google Scholar]
- 58. Wen XZ, Akiyama Y, Baylin S, Yuasa Y. Frequent epigenetic silencing of the bone morphogenetic protein 2 gene through methylation in gastric carcinomas. Oncogene 2006; 25: 2666–73. [DOI] [PubMed] [Google Scholar]
- 59. Oshima H, Itadani H, Kotani H, Taketo MM, Oshima M. Induction of prostaglandin E2 pathway promotes gastric hamartoma development with suppression of bone morphogenetic protein signaling. Cancer Res 2009; 69: 2729–33. [DOI] [PubMed] [Google Scholar]
- 60. Covarrubias DJ, Huprich JE. Best cases from the AFIP. Juvenile polyposis of the stomach. Radiographics 2002; 22: 415–20. [DOI] [PubMed] [Google Scholar]
- 61. Fodde R, Brabletz T. Wnt/β‐catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol 2007; 19: 150–8. [DOI] [PubMed] [Google Scholar]
- 62. Brabletz T, Jung A, Hermann K, Günther K, Hohenberger W, Kirchner T. Nuclear overexpression of the oncoprotein β‐catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract 1998; 194: 701–4. [DOI] [PubMed] [Google Scholar]
- 63. Rasola A, Fassetta M, De Bacco F et al . A positive feedback loop between hepatocyte growth factor receptor and β‐catenin sustains colorectal cancer cell invasive growth. Oncogene 2006; 26: 1078–87. [DOI] [PubMed] [Google Scholar]
- 64. Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF‐induced epithelial mesenchymal transition by displacing Axin from β‐catenin. Cell 2006; 127: 139–55. [DOI] [PubMed] [Google Scholar]
- 65. Oguma K, Oshima H, Aoki M et al . Activated macrophages promote Wnt signaling through tumor necrosis factor‐α in gastric tumor cell. EMBO J 2008; 27: 1671–81. [DOI] [PMC free article] [PubMed] [Google Scholar]