

Abstract
In a previous study, we developed a novel mouse model for colitis‐related carcinogenesis, utilizing a single dose of azoxymethane (AOM) followed by dextran sodium sulfate (DSS) in drinking water. In the present study, we investigated whether colonic neoplasms can be developed in mice initiated with a single injection of another genotoxic colonic carcinogen 1,2‐dimethylhydrazine (DMH), instead of AOM and followed by exposure of DSS in drinking water. Male crj: CD‐1 (ICR) mice were given a single intraperitoneal administration (10, 20 or 40 mg/kg body weight) of DMH and 1‐week oral exposure (2% in drinking water) of a non‐genotoxic carcinogen, DSS. All animals were killed at week 20, histological alterations and immunohistochemical expression of β‐catenin, cyclooxygenase (COX‐2) and inducible nitric oxide synthase (iNOS) were examined in induced colonic epithelial lesions (colonic dysplasias and neoplasms). Also, the β‐catenin gene mutations in paraffin‐embedded colonic adenocarcinomas were analyzed by the single strand conformation polymorphism method, restriction enzyme fragment length polymorphism and direct sequencing. The incidences of colonic neoplasms with dysplastic lesions developed were 100% with 2.29 ± 0.95 multiplicity, and 100% with 10.38 ± 4.00 multiplicity in mice given DMH at doses of 10 mg/kg or 20 mg/kg and 2%DSS, respectively. Although approximately half of the mice given DMH at a dose of 40 mg/kg bodyweight were dead after 2–3 days after the injection, mice who received DMH 40 mg/kg and 2%DSS had 100% incidence of colonic neoplasms with 9.75 ± 6.29 multiplicity. Immunohistochemical investigation revealed that adnocarcinomas, induced by DMH at all doses and 2%DSS, showed positive reactivities against β‐catenin, COX‐2 and iNOS. In DMH/DSS‐induced adenocarcinomas, 10 of 11 (90.9%) adenocacrcinomas had β‐catenin gene mutations. Half of the mutations were detected at codon 37 or 41, encoding serine and threonine that are direct targets for phosphorylation by glycogen synthase kinase‐3β. The present results suggests that, as in the previously reported model (AOM/DSS) our experimental protocol, DMH initiation followed by DSS, may provide a novel and useful mouse model for investigating inflammation‐related colon carcinogenesis and for identifying xenobiotics with modifying effects. (Cancer Sci 2005; 96: 69–76)
Colorectal cancer (CRC) is one of the most common non‐smoking related cancers. The risk for CRC is associated with extent and duration of inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn's disease. 1 , 2 The etiopathogenesis of IBD remains uncertain, although it is generally assumed that chronic inflammation is the primary driving force. (3) To understand the pathogenesis of IBD and IBD‐related CRC, several animal models were reported. The chemically induced and genetic models of colonic inflammation do not completely mimic the disease situation found in UC patients, (4) although they are more readily available, reproducible and conductive to therapeutic and mechanistic studies. Most used is an animal model with dextran sodium sulfate (DSS) administration through the diet, or drinking fluid. A non‐genotoxic carcinogen, DSS (5) induces colonic inflammation in rodents with clinical and histopathological similarity to human UC. (6) However, the colitis model using DSS needs a long period or cycle administration of DSS to induce colitis and colitis‐related CRC, and the incidence and/or multiplicity of induced tumors are relatively low. (7) Recently, we developed a novel mouse model for inflammation‐related colon carcinogenesis utilizing a single and low dose of azoxymethane (AOM), a metabolite of 1,2‐dimethylhydrazine (DMH), followed by a strong tumor‐promoter DSS in drinking water. (8) This combined treatment with AOM and DSS resulted in a high incidence and greater multiplicity of colonic neoplasms within 20 weeks. Moreover, the first colonic malignancy was observed as early as 12 weeks of the experimental schedule. This model can be used for detecting the chemicals with weak colonic carcinogenicity in mice within a short‐term period and for analyzing gene mutations in induced colonic neoplasms. The colon carcinogen DMH has been widely used to study chemically‐induced colon cancer in rodents. Regardless of the mode of administration, DMH specifically induces colorectal tumors. (9) DMH‐induced colon tumors in rodents are very close to human colon cancer with regard to morphology, pattern of growth and clinical manifestations. (10) Colorectal adenocarcinomas, induced by DMH in mice, often invade into the submucosa and muscular layer, but those induced by AOM and methylazoxymethanol did not show such biological and histological natures. 10 , 11 However, the major weakness of the model, using DMH, is that multiple injections of DMH and long‐term experimental period are required to induce colon tumors in laboratory animals. β‐Catenin, acting as a structural protein at cell–cell adherens junctions and as a transcriptional activator mediating Wnt signal transduction, (12) participates in a large cytoplasmic protein complex, which contains the tumor suppressor gene product of adenomatous polyposis coli (APC), glycogen synthase kinase‐3β (GSK‐3β) and axin/conductin. (13) Frequent mutation of the β‐catenin gene was found in chemically induced colonic neoplasms in rodents. 14 , 15 For example, β‐catenin mutations were frequently observed in AOM‐induced colon tumors in rats and mice. 15 , 16 In rats, 32% of colonic adenocarcinomas, induced by DMH, possessed β‐catenin gene mutations. (17) Mutation of the APC gene is known to repress the degradation and result in accumulation of β‐catenin. (18) About 80% of colorectal neoplasms harbor mutations in the APC gene and half of the reminder have β‐catenin mutation. 19 , 20 , 21 In the colonic neoplasms (adenomas and adenocarcinomas), β‐catenin was universally localized to the cytoplasm and/or nucleus. (22) In addition, altered expression of β‐catenin was reported in inflammation‐related colonic cancer in rodents 8 , 23 and humans. (24) These findings suggest that the mutation of β‐catenin gene plays an important role in the development of colon carcinogenesis in rodents as well as in humans. In the current study, we tried to induce colonic neoplasms in mice with a single administration of DMH at three dose levels followed by a 1‐week exposure of DSS in drinking water. In addition, we analyzed mutations of the β‐catenin gene in induced colonic adenocarcinomas and compared with those found in colonic malignancies induced by AOM and DSS. (8)
Materials and Methods
Animals, chemicals and diets. Male Crj: CD‐1 (ICR) mice (Charles River Japan Inc., Tokyo, Japan) aged 5 weeks were used. They were maintained at the Animal Facility of Kanazawa Medical University according to the Institutional Animal Care Guidelines. All animals were housed in plastic cages (four or five mice/cage) with free access to drinking water and a pelleted basal diet (CRF‐1; Oriental Yeast Co., Ltd, Tokyo, Japan), under controlled conditions of humidity (50 ± 10%), light (12:12 h light : dark cycle) and temperature (23 ± 2°C). After 7‐days of quarantine, they were randomized by body weight into experimental and control groups. DMH was purchased from Wako Pure Chemical Ind. Ltd. (Osaka, Japan). DSS with a molecular weight of 36 000–50 000 was obtained from ICN Biochemicals, Inc. (Aurora, OH, USA [Cat no. 160110]).
Experimental procedure. A total of 43 male ICR mice were divided into seven experimental and control groups. DMH was dissolved in 0.9% saline and the pH adjusted to 6.5 using 0.25 M NaOH. Groups 1 (seven mice), 2 (eight mice) and 3 (eight mice) were given a single intraperitoneal (i.p.) injection of DMH at a dose of 10, 20 or 40 mg/kg body weight, respectively. Starting 1 week after the injection, animals in groups 1–3 were given 2% (w/v) DSS in drinking water for 7 days, and then followed without any further treatment for 18 weeks. Groups 4 (five mice) and 5 (five mice) were given DMH 20 and 40 mg/kg body weight alone, respectively. Group 6 (five mice) was given 2% DSS alone. Group 7 (five mice) was an untreated control. All animals were killed at week 20 by ether overdose. At the termination of the study, all organs, including small and large intestines, in the mice were carefully inspected for macroscopic pathological lesions. The large bowels were flushed with saline, excised, their length measured (from ileocecal junction to the anal verge), cut open longitudinally along the main axis, and then washed with saline. Macroscopic inspection on the large bowels was carefully carried out and they were cut and fixed in 10% buffered formalin for at least 24 h. Formalin‐fixed colonic tissues were routinely processed for histological examination. Histological diagnosis was performed on hematoxylin–eosin (HE) stained section. Eleven colonic tumors, histologically diagnosed as adenocarcinoma, were stored in a deep‐freezer at −80°C for analyzing β‐catenin mutation. On HE‐stained sections, histological alterations, such as mucosal ulceration, dysplasia and colonic neoplasms, were examined. Colitis with or without ulceration was scored on HE‐stained sections, according to the following morphological criteria described by Cooper et al.: (25) grade 0, normal colonic mucosa; grade 1, shortening and loss of the basal one‐third of the actual crypts with mild inflammation in the mucosa: grade 2, loss of the basal two‐thirds of the crypts with moderate inflammation in the mucosa; grade 3, loss of the entire crypts with severe inflammation in the mucosa and submucosa, but with retainment of the surface epithelium; and grade 4, presence of mucosal ulcer with severe inflammation (neutrophil, lymphocyte, and plasma cell infiltration) in the mucosa, submucosa, musculaaris propria and/or subserosa. High‐ or low‐grade of dysplasia of colonic mucosa was diagnosed according to the criteria described by Riddell et al. (26) and Pascal. (27) Colonic neoplasms were diagnosed according to the description by Ward. (28) Histopathological examination was also carried out in other organs.
Immunohistochemistry. As in our previous study, (8) immunohistochemistry for β‐catenin, cyclooxygenase (COX)‐2 and nitric oxide synthase (iNOS), was performed on 3‐µm‐thick paraffin‐embedded sections from colons of mice in all groups, utilizing the labeled streptavidin–biotin method using a LSAB Kit (DAKO, Glostrup, Denmark) with microwave accentuation. The paraffin‐embedded sections were heated for 30 min at 65°C, deparaffinized in xylene, and rehydrated through graded ethanols at room temperature. A 0.05 M Tris HCl buffer (pH 7.6) was used to prepare solutions and for washes between various steps. Incubations were performed in a humidified chamber. Sections were treated for 40 min at room temperature with 2% bovine serum albumin, and incubated overnight at 4°C with primary antibodies, such as anti‐β‐catenin mouse monoclonal antibody (diluted 1:1000; Transduction Laboratories, Lexington, KY, USA), anti‐COX‐2 mouse monoclonal antibody (diluted 1:200; Transduction Laboratories), and anti‐iNOS mouse monoclonal antibody (cat. no. N32020‐150; diluted 1:250, Transduction Laboratories). To reduce the non‐specific staining of mouse tissue by the mouse antibodies, a mouse on mouse immunoglobulin G blocking reagent (Vector Laboratories Inc., Burlingame, CA, USA) was applied for 1 h. Horseradish peroxidase activity was visualized by treatment with H2O2 and 3,3′‐diaminobenzidine for 5 min. At the last step, the sections were weakly counterstained with Mayer's hematoxylin (Merck Ltd, Tokyo, Japan). For each case, negative controls were performed on serial sections. On the control sections, incubation with the primary antibodies was omitted. Intensity and localization of immunoreactivities against all primary antibodies used were examined on all sections using a microscope (Olympus BX41, Olympus Optical Co., Ltd, Tokyo, Japan) and recorded.
DNA extraction. For analysis of β‐catenin mutations, 11 colonic adenocarcinomas developed in DMH (10 or 20 mg/kg body weight)/DSS‐treated mice were used. DNA was extracted from frozen tissue using Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA).
Polymerase chain reaction‐single strand conformation polymorphism analysis. DNA from colonic adenocarcinomas was polymerase chain reaction (PCR)‐amplified with primers (5′‐primer, GCTGACCTGATGGAGTTGGA; 3′‐primer, GCTACTTGCTCTTGCGTGAA), which were designed to amplify exon 3 of the β‐catenin gene containing the consensus sequence for GSK‐3β phosphorylation. (15) The length of the PCR product with these primers is 227 bp. The primers were purchased from Sigma‐Aldrich Japan K.K. (Tokyo, Japan). PCR for non‐radioisotopic single strand conformation polymorphism (SSCP) was performed in 50 µL of reaction mixture consisting of 0.5 µM of each primer, 1 × PCR buffer (Takara Bio, Otsu, Japan), 250 µM each dNTP, 2.5 U TaKaRa Ex Taq (Takara Bio) and 1 µL of template DNA. The mixture was heated at 94°C for 1 min and subjected to 30 cycles of denaturation (94°C, 0.5 min), annealing (55°C, 0.5 min) and extension (72°C, 1 min) using a using a TaKaRa PCR Thermal Cycler Dice (Takara Bio). The amplified PCR product was analyzed for its mobility‐shifted bands using a GenePhor (Amersham Biosciences Corp., NJ, USA) with a GeneGel Clean (Amersham Biosciences Corp.) according to the manufacturer's protocol. Electrophoresis was carried out at 90 V for 25 min and then 500 V for 50 min at 20°C, and the gels were soaked in 10% trichloroacetic acid and in 50% methanol for 10 min each. DNA bands were detected by silver staining using 2D Silver Staining Solution II (Daiichi Chemical DNA Co., Tokyo, Japan).
Restriction fragment length polymorphism assay for PCR products of β‐catenin. To detect β‐catenin mutations at codons 32, 33 and 34, PCR products were treated with a restriction enzyme HinfI (Wako Pure Chemical Industries, Tokyo, Japan) and electrophoresed on 5% agarose gels. Recognition sequences of HinfI are GANTC. The PCR product of 227 bp is digested by HinfI to 82, 7 and 138 bp in the case of the wild‐type, to 89 and 138 bp with mutations at the first or second bases of codons 32 or 33, and to 82 and 145 bp with mutations at the second or third bases of codons 34 or 35.
Direct DNA sequencing. The PCR products were purified and concentrated to 20 µL using Microcon 100 (Amicon Inc., Beverley, MA, USA). With 2 µL of the purified PCR products and 5′ or 3′ PCR primers, cycle sequencing reactions were carried out using a BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and purified by isopropanol precipitation. The sequences were determined with an ABI PRISM 310 Genetic Analyzer (PerkinElmer, Wellesley, MA, USA).
Statistical analysis. All measurements were compared by Student's t‐test, Welch's t‐test, chi‐squared test or Fisher's exact probability test for multiple group comparisons.
Results
General observations. Approximately half of the mice injected 40 mg/kg body weight of DMH (four mice of group 3, and two mice of group 5) died of hepatotoxicity of DMH 2–3 days after the injection. This was confirmed by histological examination of liver. Also bloody stool was found during and soon after of DSS exposure (days 12–21) in a few mice who received 2% DSS in drinking water, and their body weight gains were slightly decreased (data not shown). Thereafter, however, no such clinical symptoms were observed. The body and liver weights, and lengths of large bowel of mice in all groups at the end of the study (week 20) are listed in Table 1. There were no significant differences among the groups in these measurements.
Table 1.
Body weights, liver weights, and lengths of large bowel in each group
| Group no. | Treatment (no. mice examined) | Body weight (g) | Liver weight (g) | Length of large bowel (cm) |
|---|---|---|---|---|
| 1 | DMH 10 mg/kg→2%DSS (7) | 42.6 ± 2.7 † | 2.47 ± 0.29 | 13.8 ± 1.3 |
| 2 | DMH 20 mg/kg→2%DSS (8) | 43.3 ± 3.0 | 2.63 ± 0.35 | 13.5 ± 1.5 |
| 3 | DMH 40 mg/kg→2%DSS (4) | 44.3 ± 3.9 | 2.64 ± 0.21 | 13.9 ± 1.2 |
| 4 | DMH 20 mg/kg (5) | 45.9 ± 3.6 | 2.81 ± 0.41 | 14.5 ± 1.1 |
| 5 | DMH 40 mg/kg (3) | 43.2 ± 1.9 | 2.86 ± 0.46 | 14.9 ± 0.1 |
| 6 | 2%DSS (5) | 42.1 ± 5.5 | 2.82 ± 0.31 | 13.9 ± 1.0 |
| 7 | None (5) | 44.6 ± 3.2 | 2.32 ± 0.45 | 15.0 ± 0.9 |
Mean ± standard deviation. DMH, 1,2‐Dimethylhydrazine; DSS, dextran sodium sulfate.
Pathological findings. Macroscopically, nodular, polypoid or flat‐type colonic tumors were observed in the middle and distal colon of all mice in groups 1–3 (Fig. 1), but not in the small intestine. Their histopathology was well‐ or moderately‐differentiated tubular adenocarcinoma (Fig. 2a) or tubular adenoma (Fig. 2b). Histologically, there were no tumors in any organs other than the large bowel in these groups. The incidences̀ and multiplicities of large bowel adenoma, adenocarcinoma and total tumors (adenoma + adenocarcinoma) are summarized in Table 2. The incidences of total tumors and adenocarcinoma in mice of given DMH/DSS (groups 1–3) were 100%. The multiplicities of total tumors, adenoma and adenocarcinoma in groups 2 and 3 were significantly higher than those of group 1 (P ≤ 0.001, P ≤ 0.01 or P ≤ 0.05, respectively). In mice of groups 4–7, no neoplasms developed in any organs including large bowel. Besides colonic neoplasms, all mice in groups 1–3 had colonic dysplasia (Fig. 2c). Their multiplicities were 4.71 ± 2.29, 7.13 ± 1.27 and 8.25 ± 3.10 in groups 1, 2 and 3, respecively (Table 3). The multiplicities of total dysplasia and high‐grade dysplasia in groups 2 and 3 were significantly greater than those of group 1 (P ≤ 0.02, P ≤ 0.01 or P ≤ 0.05, respectively). There were no such dysplatic lesions in mice of groups 4–7. In addition, colonic mucosal ulceration (grade 1) was found in the distal colon of mice in groups 1, 2, 3 and 5 (Table 3).
Figure 1.
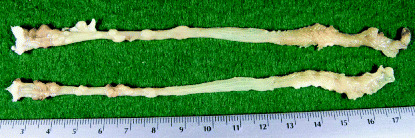
Representative macroscopic view of the colonc from group 2 (1,2‐dimethylhydrazine [DMH] 20 mg/kg body weight →2% dextran sodium sulfate [2%DSS]). Note the numerous polypoid tumors in the colon.
Figure 2.

Histopathology of colonic neoplasms developed in mice. (a) adenocarcinoma (b) adenoma, and (c) dysplasia. Hematoxylin–eosion stain. Original magnification (a) ×4, (b) ×10, and (c) ×20.
Table 2.
Incidence of large bowel neoplasms in mice treated with 1,2‐dimethylhydrazine and dextran sodium sulfate
| Group no. | Treatment (no. mice examined) | No. mice with large bowel neoplasms | ||
|---|---|---|---|---|
| Total (%) (multiplicity) | Adenoma (%) (multiplicity) | Adenocarcinoma (%) (multiplicity) | ||
| 1 | DMH 10 mg/kg→2%DSS (7) | 100 (2.29 ± 0.95) | 57.1 (1.00 ± 1.15) | 100 (1.29 ± 0.50) |
| 2 | DMH 20 mg/kg→2%DSS (8) | 100 (10.38 ± 4.00)* | 87.5 (4.63 ± 3.29)** | 100 (5.75 ± 1.83)* |
| 3 | DMH 40 mg/kg→2%DSS (4) | 100 (9.75 ± 6.29)** | 100 (5.25 ± 4.65)*** | 100 (4.50 ± 1.73)* |
| 4 | DMH 20 mg/kg (5) | 0 | 0 | 0 |
| 5 | DMH 40 mg/kg (3) | 0 | 0 | 0 |
| 6 | 2%DSS (5) | 0 | 0 | 0 |
| 7 | None (5) | 0 | 0 | 0 |
Significantly different from group 1 by Student's t‐test (*P < 0.001, **P < 0.01, and ***P < 0.05). DMH, 1,2‐Dimethylhydrazine; DSS, dextran sodium sulfate. Numbers in parentheses are multiplicity (mean ± standard deviation) of large bowel tumors.
Table 3.
Incidence of large bowel ulceration and dysplasia in mice treated with 1,2‐dimethylhydrazine and dextran sodium sulfate
| Group no. | Treatment (no. mice examined) | Incidence of mucosal ulcer (%) (multiplicity) | Incidence of colonic dysplasia (multiplicity) | ||
|---|---|---|---|---|---|
| Total | Low‐grade | High‐grade | |||
| 1 | DMH 10 mg/kg→2%DSS (7) | 100 (2.43 ± 1.40) | 100 (4.71 ± 2.29) | 100 (3.00 ± 1.29) | 100% (1.71 ± 1.11) |
| 2 | DMH 20 mg/kg→2%DSS (8) | 100 (1.86 ± 0.4) | 100 (7.13 ± 1.25)* | 100 (2.88 ± 1.25) | 100% (4.25 ± 1.58)** |
| 3 | DMH 40 mg/kg→2%DSS (4) | 100 (2.50 ± 1.29) | 100 (8.25 ± 3.10) | 100 (4.00 ± 1.41) | 100% (4.25 ± 2.22)*** |
| 4 | DMH 20 mg/kg (5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| 5 | DMH 40 mg/kg (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| 6 | 2%DSS (5) | 40 (0.51 ± 0.32) | 0 (0) | 0 (0) | 0 (0) |
| 7 | None (5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Significantly different from group 1 by Student's t‐test (*P < 0.02, **P < 0.01, and ***P < 0.05). DMH, 1,2‐Dimethylhydrazine; DSS, dextran sodium sulfate. Numbers in parentheses are multiplicity (mean ± standard deviation) of large bowel tumors.
Immunohistochemical findings. The immunoreactivities against β‐catenin, COX‐2 and iNOS were noted in all colonic lesions, including neoplasms and dysplastic lesions. The immunoreactivity showed dark brown reaction products with slight variation in the intensity and distribution. Strong β‐catenin expression was seen in the nucleus and cytoplasm of adenocarcinoma cells (Fig. 3a). Although the intensity was relatively weaker than carcinoma cells, adenoma cells showed positivity for β‐catenin in their cytoplasm and cell membrane. β‐Catenin immunoreactivity was also found in the cell membrane and cytoplasm of dysplastic cells, but intensity was weaker than adenoma cells. Non‐lesional cryptal cells showed weak positivity of β‐catenin in their cell membrane. In addition, positive reaction against β‐catenin antibody was found in the cytoplasm of vascular endothelium, infiltrated inflammatory cells, and ganglion cells in myenteric (Auerbach's) plexus. Strong COX‐2 immunoreactivity was found in adenocarcinoma cytoplasm (Fig. 3b). Adenoma cells also were found in their cytoplasm, and the intensity was weaker than adenocarcinoma cells. Dysplastic cells showed weak positivity for COX‐2 when compared to neoplastic cells. Non‐lesional cryptal cells at lower part of crypts were weakly positive for COX‐2, and the stainability was lower than dysplastic crypts. Strongly positive reaction of COX‐2 was also seen in the endothelium of small blood vessels, and inflammatory cells infiltrated in the lamina propria. Smooth muscle cells and fibroblasts in the wall of the large bowel showed weak reaction of COX‐2. iNOS‐immunohistochemistry showed strong immunoreactivity in the cytoplasm of adenocarcinoma (Fig. 3c) and adenoma cells; the intensity was greater in carcinoma cells when compared to adenoma cells. Also, dysplastic cells were positive for iNOS in their cytoplasm, but the intensity was weaker than adenoma cells. The faint positive reaction was found in the cytoplasm of non‐lesional cryptal cells. Immunohistochemical iNOS expression was strong in the endothelial cells of small blood vessels and inflammatory cells in the lamina propria. COX‐2‐ and iNOS‐stained inflammatory cells were also frequently observed in areas of mucosal ulceration in groups 1, 2, 3 and 6. The results of immunoreactivities against β‐catenin, COX‐2, and iNOS are summarized in Table 4.
Figure 3.

Immunohistochemistry of (a) β‐catenin, (b) cyclooxygenase (COX‐2), and (c) nitric oxide synthase (iNOS), and immunofluorescent staining of β‐catenin in colonic adenocarcinoma in mice. Original magnification (a–c) ×10.
Table 4.
Expression of β‐catenin, nitric oxide synthase and cyclooxygenase in 1,2‐dimethylhydrazine/dextran sodium sulfate‐induced mouse colon lesions
| Protein | Normal mucosa | Dysplasia | Adenoma | Adenocarcinoma |
|---|---|---|---|---|
| β‐Catenin | ± ∼ + | + ∼ ++ | ++ | +++ |
| (M † ) | (M, C ‡ ) | (M, C) | (C, N § ) | |
| COX‐2 | – ∼ ± | + | ++ | +++ |
| (C) | (C) | (C) | (C) | |
| iNOS | – ∼ ± | + | ++ | +++ |
| (C) | (C) | (C) | (C) |
† Cell membrane; ‡ cytoplasm, § nucleus. –, No staining; ±, faint and partial staining; +, weak staing; ++, moderate staining, +++, strong staining. COX‐2, cyclooxygenase; iNOS, nitric oxide synthase.
Mutation in β‐catenin gene. In this study, we analyzed the status of the β‐catenin gene in the histological sections of DMH/DSS‐induced colon adenocarcinomas. We detected β‐catenin gene mutations in 10 out of 11 colonic adenocarcinomas induced by DMH (10 or 20 mg/kg bodyweight) and 2% DSS (4, 5). All mutations detected in colon adenocarcinomas converged at codons 32, 34, 37 and 41, all being functionally important codons for β‐catenin degradation: five were located at the second base of codon 34, three at the second base of codon 41, two at the first base of codon 37 and one at the first base of codon 32. Except for mutation (A : T to G : C) at the first base of codon 37, all were G : C to A : T transitions (Table 5).
Figure 4.

Polymerase chain reaction–single strand conformation polymorphism (PCR‐SSCP) analysis of the β‐catenin gene in mouse colon adenocarcinomas. 1,2‐Dimethylhydrazine/dextran sodium sulfate (DMH/DSS)‐induced mouse colon adenocarcinomas (lanes 1–11). Lanes 1–3: DMH (10 mg/kg body weight)/DSS‐induced mouse colon adenocarcinoma samples. Lanes 4–11: DMH (20 mg/kg body weight)/DSS‐induced mouse colon adenocarcinoma samples. Lane ND: DMH (20 mg/kg body weight)‐induced mouse colon mucosa sample. Lane NC: negative control mouse colon mucosa sample. Arrowheads indicate tumor‐specific bands.
Figure 5.

Restriction fragment length polymorphism (RFLP) analysis of the β‐catenin gene in mouse colon adenocarcinomas. Lanes 1–11: 1,2‐dimethylhydrazine/dextran sodium sulfate (DMH/DSS)‐induced mouse colon adenocarcinomas samples. Lanes 1–3: DMH (10 mg/kg bodyweight)/DSS‐induced mouse colon adenocarcinoma samples. Lanes 4–11: DMH (20 mg/kg body weight)/DSS‐induced mouse colon adenocarcinoma samples. Lane M: DNA size markers. Lane N: Negative control mouse colon mucosa sample. Arrowheads indicate tumor‐specific bands.
Table 5.
Mutations in exon 3 of the β‐catenin gene in 1,2‐dimethylhydrazine/dextran sodium sulfate‐induced mouse colonic adenocarcinomas
| Sample | β‐catenin status | Amino acid substitution | |
|---|---|---|---|
| DMH 10/DSS‐1 | Codon 37 | TCT→CCT | Ser→Pro |
| DMH 10/DSS‐2 | Codon 32 | GAT→AAT | Asp→Asn |
| DMH 10/DSS‐3 | Codon 37 | TCT→CCT | Ser→Pro |
| DMH 20/DSS‐4 | Wild type | – | |
| DMH 20/DSS‐5 | Codon 34 | GGA→GAA | Gly→Glu |
| DMH 20/DSS‐6 | Codon 34 | GGA→GAA | Gly→Glu |
| DMH 20/DSS‐7 | Codon 41 | ACC→ATC | Thr→Ile |
| DMH 20/DSS‐8 | Codon 34 | GGA→GAA | Gly→Glu |
| Codon 41 | ACC→ATC | Thr→Ile | |
| DMH 20/DSS‐9 | Codon 34 | GGA→GAA | Gly→Glu |
| DMH 20/DSS‐10 | Codon 41 | ACC→ATC | Thr→Ile |
| DMH 20/DSS‐11 | Codon 34 | GGA→GAA | Gly→Glu |
DMH, 1,2‐Dimethylhydrazine; DSS, dextran sodium sulfate.
Discussion
In the current study, a single i.p. injection of DMH (10, 20 or 40 mg/kg body weight) followed by a 1‐week exposure of 2% DSS in drinking water, could produced colonic adenocarcinomas with 100% incidence in male ICR mice within 20 weeks. All of the DMH/DSS‐induced colonic adenocarcinomas were immunohistochemically positive for β‐catenin, COX‐2 and iNOS. Moreover, 10 (91%) out of 11 colonic adenocarcinomas had β‐catenin mutations. However, no colonic neoplasms were found in mice treated with DMH alone or DSS alone. These findings indicated a powerful tumor‐promoting ability of DSS on DMH‐initiated colon carcinogenesis in male ICR mice, as found in our previous experiment using AOM as a carcinogen. (8) In the current study, dosing of 40 mg/kg body weight of DMH was lethal in almost half of the mice. This was caused by hepatotoxicity (necrosis and bleeding in the liver) of DMH at the dose. Therefore, appropriate dose of DMH was considered to be 10 or 20 mg/kg body weight in this model. β‐Catenin is a multifunctional molecule involved in the cadherin‐mediated cell–cell adhesion and Wnt‐APC signal transduction. (29) Regulation of membrane, cytoplasmic and nuclear pools of β‐catenin is crucial for modulating its adhesion and signaling functions. (30) Normally, β‐catenin is localized in cell–cell junctions with very low levels of β‐catenin in the cytoplasm and nucleus. Accumulation of β‐catenin in the cytoplasm or nucleus as a consequence of mutant APC, β‐catenin or Axin, is associated with colon carcinogenesis. (31) β‐Catenin accumulation moves from the cytoplasm to the nucleus when the β‐catenin or APC genes are mutated or the Wnt signaling pathway is activated. (32) Immunohistochemically aberrant expression of β‐catenin was reported in colonic neoplasms and dysplasia in a DSS‐induced mouse colitis model, (23) and human colitis‐related dysplasia and neoplasms. 33 , 34 The observed frequent mutations in the GSK‐3β phosphorylation consensus motif of the β‐catenin gene appear to be associated with alteration of the cellular localization and functional site of the protein, as shown by immnohistochemical staining in the present study. Our results are comparable to those in a recent report describing altered distribution of β‐catenin in UC‐related CRC. (35) Although, the β‐catenin gene is frequently mutated at codons 33, 41 and 45 of the GSK‐3β phosphorylation motif in human colon cancers without APC mutations, (36) the mutations of the gene in chemically induced rat colon tumors is found at codons 32, 33 and 34. 15 , 17 In the present study, we detected β‐catenin gene mutation of mouse colon adenocarcinomas, induced by DMH/DSS, at codon 32, 34, 37 and 41. The location was slightly different from a report documenting that β‐catenin gene mutations of mouse colon tumors, induced by AOM, were present at codons 33, 34, 37 and 41, but not at codon 32. (16) Recently, we detected β‐catenin gene mutations of mouse colon adenocarcinomas, induced by AOM/DSS, at codons 32, 33 and 34. (37) However, in the current protocol, half of the mutations caused by DMH/DSS treatment were at codon 37 and 41, which are important serine and threonine sites for GSK‐3β phosphorylation. Koesters et al. (17) reported that the different mutational spectra, observed in Ctnnb1, directly relates to the particular carcinogenic treatment. They demonstrated that the β‐catenin mutations at codons 37 and 41 possess higher oncogenic potential. (17) Therefore, it may be speculated that DSS exposure caused a shift in the mutation sites induced by DMH alone treatment. Since mutation of β‐catenin is reported to be an early event of colorectal carcinogenesis, (38) molecular analysis at early stage of colon carcinogenesis should be carried out in this model. Although the mutations of the APC and β‐catenin are rare in UC‐related CRC, as compared with sporadic CRC, nuclear β‐catenin expression is related to UC‐related CRC development. (39) Since the data on β‐catenin mutation in UC‐related CRC are limited, more studies are required to determine the role of the β‐catenin mutation in UC‐related CRC. Also, expression of c‐myc, cyclinD1 and c‐jun, which are targets of the β‐catenin/APC pathway, 17 , 40 , 41 may also influence colon carcinogenesis in the present model. Such analysis is underway in our laboratory. Nitric oxide (NO) and prostaglandin, as the main inflammatory mediators, take part in the pathogenesis of IBD, with enhanced expression of iNOS and COX‐2 in the morbid colonic mucosa. 42 , 43 Moreover, expression of the iNOS and COX‐2 is increased in human colorectal tumors with inflammation, (44) and in carcinogen‐induced colon tumors in rodents. (45) There exist various proposed pathways for NO‐induced regulation of COX‐2 expression and for modulation of cancer development. (46) They include cross–talk or interactions between endogenous NO and COX‐2, and between β‐catenin/APC pathway and COX‐2. (47) β‐Catenin/APC is reported to play a critical role in NO induction of COX‐2 in colon epithelial cells. (48) Furthermore, Howe et al. (49) demonstrated that β‐catenin/Tcf‐4 complex transactivates the expression of PEA3, a transcription factor of Ets family, and stimulates COX‐2 expression. More recently, it has been reported that NO increases PEA3 expression through β‐catenin/APC pathway and directly augments the COX‐2 promoter activity of the PEA3/p300 in YAMC cells. (50) In the present study, all colonic neoplasms were immunohistochemically positive for iNOS and COX‐2, which was in accordance with the previous reports. (8) Furthermore, strong β‐catenin expression was found in the nucleus and cytoplasm of adenocarcinoma cells. It may be possible that the increased expression of iNOS may be related to the altered localization of β‐catenin, and/or increased expression of COX‐2 in the colonic tumor formation and/or progression. Our recent work suggest involvement of oxidative/nitrosative stress in tumor‐promoting effect of DSS on AOM‐induced colon carcinogenesis in mice. (51)
In conclusion, the results in the current study indicate that a single dose of DMH followed by DSS resulted in a high incidence of colonic epithelial malignancies with β‐catenin mutations within 20 weeks. Furthermore, half of the β‐catenin mutations, detected in adenocarcinomas, were at codon 37 and 41, and strong β‐catenin expression was seen in their nucleus and cytoplasm. Also, our findings suggest the importance of inflammation caused by DSS exposure in mouse colon carcinogenesis. The experimental protocol described here could be applied to investigate detailed molecular mechanism of inflammation‐related CRC, as is the AOM/DSS model. (8)
Acknowledgments
This research was supported in part by the Grant‐in‐Aid (13–15) for Cancer Research from the Ministry of Health, Labour and Welfare of Japan; the Grant‐in‐Aid for the 3rd Term for a Comprehensive 10‐year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan; the Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the grant (C2004‐4) for the Collaborative Research from Kanazawa Medical University; and the grant (H2004‐6) for the Project Research from the High‐Technology Center of Kanazawa Medical University.
References
- 1. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta‐analysis. Gut 2001; 48: 526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Van Hogezand RA, Eichhorn RF, Choudry A, Veenendaal RA, Lamers BHW. Malignancies in inflammatory bowel disease: Fact or fiction? Scand J Gastroenterol 2002; 235: (Suppl.) 48–53. [DOI] [PubMed] [Google Scholar]
- 3. Weitzman SA, Gordon LI. Inflammation and cancer: role of phagocyte‐generated oxidants in carcinogenesis. Blood 1990; 76: 655–63. [PubMed] [Google Scholar]
- 4. Warren BF, Watkins PE. Animal models of inflammatory bowel disease. J Pathol 1994; 172: 313–6. [DOI] [PubMed] [Google Scholar]
- 5. Mori H, Ohbayashi F, Hirono I, Shimada T, Williams GM. Absence of genotoxicity of the carcinogenic sulfated polysaccharides carrageenan and dextran sulfate in mammalian DNA repair and bacterial mutagenicity assays. Nutr Cancer 1984; 6: 92–7. [DOI] [PubMed] [Google Scholar]
- 6. Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. Novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990; 98: 694–702. [DOI] [PubMed] [Google Scholar]
- 7. Okayasu I, Yamada M, Mikami T, Yoshida T, Kanno J, Ohkusa T. Dysplasia and carcinoma development in a repeated dextran sulfate sodium‐induced colitis model. J Gastroenterol Hepatol 2002; 17: 1078–83. [DOI] [PubMed] [Google Scholar]
- 8. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation‐related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 2003; 94: 965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rogers AE, Nauss KM. Rodent models for carcinoma of the colon. Dig Dis Sci 1985; 30: 87S–102S. [DOI] [PubMed] [Google Scholar]
- 10. Pozharisski KM, Likhachev AJ, Klimashevski VF, Shaposhnikov JD. Experimental intestinal cancer research with special reference to human pathology. Adv Cancer Res 1979; 30: 165–237. [DOI] [PubMed] [Google Scholar]
- 11. Ross JS. Experimental large intestinal adenocarcinoma induced by hydrazines and human colorectal cancer: a comparative study. In: Malt RA, Williamson RCN, eds. Colonic Carcinogenesis. MTP Press, Hingham, 1982; 187–207. [Google Scholar]
- 12. Miller JR, Moon RT. Signal transduction through β‐catenin and specification of cell fate during embryogenesis. Genes Dev 1996; 10: 2527–39. [DOI] [PubMed] [Google Scholar]
- 13. Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S, Kikuchi A. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of β‐catenin. J Biol Chem 1998; 273: 10823–6. [DOI] [PubMed] [Google Scholar]
- 14. Dashwood RH, Suzui M, Nakagama H, Sugimura T, Nagao M. High frequency of β‐catenin (Ctnnb1) mutations in the colon tumors induced by two heterocyclic amines in the F344 rat. Cancer Res 1998; 58: 1127–9. [PubMed] [Google Scholar]
- 15. Takahashi M, Fukuda K, Sugimura T, Wakabayashi K. β‐Catenin is frequently mutated and demonstrates altered cellular location in azoxymethane‐induced rat colon tumors. Cancer Res 1998; 58: 42–6. [PubMed] [Google Scholar]
- 16. Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequent mutations of the β‐catenin gene in mouse colon tumors induced by azoxymethane. Carcinogenesis 2000; 21: 1117–20. [PubMed] [Google Scholar]
- 17. Koesters R, Hans MA, Benner A, Prosst R, Boehm J, Gahlen J, Doeberitz MK. Predominant mutation of codon 41 of the β‐catenin proto‐oncogene in rat colon tumors induced by 1,2‐dimethylhydrazine using a complete carcinogenic protocol. Carcinogenesis 2001; 22: 1885–90. [DOI] [PubMed] [Google Scholar]
- 18. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β‐catenin‐Tcf signaling in colon cancer by mutations in β‐catenin or APC. Science 1997; 275: 1787–90. [DOI] [PubMed] [Google Scholar]
- 19. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992; 1: 229–33. [DOI] [PubMed] [Google Scholar]
- 20. Powell SM, Zilz N, Beazer‐Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature 1992; 359: 235–7. [DOI] [PubMed] [Google Scholar]
- 21. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β‐catenin/Tcf pathway in colorectal cancer. Cancer Res 1998; 58: 1130–4. [PubMed] [Google Scholar]
- 22. Iwamoto M, Ahnen DJ, Franklin WA, Maltzman TH. Expression of β‐catenin and full‐length APC protein in normal and neoplastic colonic tissues. Carcinogenesis 2000; 21: 1935–40. [DOI] [PubMed] [Google Scholar]
- 23. Cooper HS, Murthy S, Kido K, Yoshitake H, Flanigan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis‐associated neoplasia in the human. a study of histopathology, β‐catenin and p53 expression and the role of inflammation. Carcinogenesis 2000; 21: 757–68. [DOI] [PubMed] [Google Scholar]
- 24. Hill KA, Wang KL, Stryker SJ, Gupta R, Weinrach DM, Rao MS. Comparative analysis of cell adhesion molecules, cell cycle regulatory proteins, mismatch repair genes, cyclooxygenase‐2, and DPC4 in carcinomas arising in inflammatory bowel disease and sporadic colon cancer. Oncol Report 2004; 11: 951–6. [PubMed] [Google Scholar]
- 25. Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Laboratory Invest 1993; 69: 238–49. [PubMed] [Google Scholar]
- 26. Riddell RH, Goldman H, Ransohof DF, Appleman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC, Sommers SC, Yardley JH. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical application. Human Pathol 1983; 14: 931–68. [DOI] [PubMed] [Google Scholar]
- 27. Pascal RR. Dysplasia and early carcinoma in inflammatory bowel disease and colorectal carcinomas. Human Pathol 1994; 25: 1160–71. [DOI] [PubMed] [Google Scholar]
- 28. Ward JM. Morphogenesis of chemically induced neoplasms of the colon and small intestine in rats. Laboratory Invest 1974; 30: 505–13. [PubMed] [Google Scholar]
- 29. Bullions LC, Levine AJ. The role of β‐catenin in cell adhesion, signal transduction, and cancer. Curr Opin Oncol 1998; 10: 81–7. [DOI] [PubMed] [Google Scholar]
- 30. Morin PJ. β‐Catenin signaling and cancer. Bioessays 1999; 21: 1021–30. [DOI] [PubMed] [Google Scholar]
- 31. Brabletz T, Jung A, Kirchner T. β‐Catenin and the morphogenesis of colorectal cancer. Virchows Arch 2002; 441: 1–11. [DOI] [PubMed] [Google Scholar]
- 32. Hao XP, Pretlow TG, Rao JS, Pretlow TP. β‐Catenin expression is altered in human colonic aberrant crypt foci. Cancer Res 2001; 61: 8085–8. [PubMed] [Google Scholar]
- 33. Walsh SV, Loda M, Torres CM, Antonioli D, Odze R. P53 and β‐catenin expression in chronic ulcerative colitis − associated polypoid dysplasia and sporadic adenomas: an immunohistochemical study. Am J Surg Pathol 1999; 23: 963–9. [DOI] [PubMed] [Google Scholar]
- 34. Mikami T, Mitomi H, Hara A, Yanagisawa N, Yoshida T, Tsuruta O, Okayasu I. Decreased expression of CD44, α‐catenin, and deleted colon carcinoma and altered expression of β‐catenin in ulcerative colitis‐associated dysplasia and carcinoma, as compared with sporadic colon neoplasms. Cancer 2000; 89: 733–40. [DOI] [PubMed] [Google Scholar]
- 35. Aust DE, Terdiman JP, Willenbucher RF, Chew K, Ferrell L, Florendo C, Molinaro‐Clark A, Baretton GB, Lohrs U, Waldman FM. Altered distribution of β‐catenin, and its binding proteins E‐cadherin and APC, in ulcerative colitis‐related colorectal cancers. Mod Pathol 2001; 14: 29–39. [DOI] [PubMed] [Google Scholar]
- 36. Erdman SH, Wu HD, Hixson LJ, Ahnen DJ, Gerner EW. Assessment of mutations in Ki‐ras and p53 in colon cancers from azoxymethane‐ and dimethylhydrazine‐treated rats. Mol Carcinog 1997; 19: 137–44. [DOI] [PubMed] [Google Scholar]
- 37. Tanaka T, Suzuki R, Kohno H, Sugie S, Takahashi M, Wakabayashi K. Colonic adenocarcinomas rapidly induced by the combined treatment with 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine and dextran sodium sulfate in male ICR mice possess β‐catenin gene mutations and increase immunoreactivity for β‐catenin, cyclooxygenase‐2, and inducible nitric oxide synthase. Carcinogenesis 2005; 26: 229–38. [DOI] [PubMed] [Google Scholar]
- 38. Takahashi M, Mutoh M, Kawamori T, Sugimura T, Wakabayashi K. Altered expression of β‐catenin, inducible nitric oxide synthase and cyclooxygenase‐2 in azoxymethane‐induced rat colon carcinogenesis. Carcinogenesis 2000; 21: 1319–27. [PubMed] [Google Scholar]
- 39. Chiang JM, Chou YH, Chen TC, Ng KF, Lin JL. Nuclear β‐catenin expression is closely related to ulcerative growth of colorectal carcinoma. Br J Cancer 2002; 86: 1124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tetsu O, McCormick F. β‐Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398: 422–6. [DOI] [PubMed] [Google Scholar]
- 41. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, Da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c‐MYC as a target of the APC pathway. Science 1998; 281: 1509–12. [DOI] [PubMed] [Google Scholar]
- 42. Sakamoto C. Roles of COX‐1 and COX‐2 in gastrointestinal pathophysiology. J Gastroenterol 1998; 33: 618–24. [DOI] [PubMed] [Google Scholar]
- 43. Kankuri E, Asmawi MZ, Korpela R, Vapaatalo H, Moilanen E. Induction of iNOS in a rat model of acute colitis. Inflammation 1999; 23: 141–52. [DOI] [PubMed] [Google Scholar]
- 44. Ambs S, Merriam WG, Bennett WP, Felley‐Bosco E, Ogunfusika MO, Oser SM, Klein S, Shields PG, Billiar TR, Harris CC. Frequent nitric oxide synthase‐2 expression in human colon adenomas: implication for tumor angiogenesis and colon cancer progression. Cancer Res 1998; 58: 334–41. [PubMed] [Google Scholar]
- 45. Takahashi M, Fukuda K, Ohata T, Sugimura T, Wakabayashi K. Increased expression of inducible and endothelial constitutive nitric oxide synthases in rat colon tumors induced by azoxymethane. Cancer Res 1997; 57: 1233–7. [PubMed] [Google Scholar]
- 46. Takahashi M, Wakabayashi K. Gene mutations and altered gene expression in azoxymethane‐induced colon carcinogenesis in rodents. Cancer Sci 2004; 95: 475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Araki Y, Okamura S, Hussain SP, Nagashima M, He P, Shiseki M, Miura K, Harris CC. Regulation of cyclooxygenase‐2 expression by the Wnt and ras pathways. Cancer Res 2003; 63: 728–34. [PubMed] [Google Scholar]
- 48. Mei JM, Hord NG, Winterstein DF, Donald SP, Phang JM. Expression of prostaglandin endoperoxide H synthase‐2 induced by nitric oxide in conditionally immortalized murine colonic epithelial cells. FASEB J 2000; 14: 1188–201. [DOI] [PubMed] [Google Scholar]
- 49. Howe LR, Crawford HC, Subbaramaiah K, Hassell JA, Dannenberg AJ, Brown AM. PEA3 is up‐regulated in response to Wnt1 and activates the expression of cyclooxygenase‐2. J Biol Chem 2001; 276: 20108–15. [DOI] [PubMed] [Google Scholar]
- 50. Liu Y, Borchert GL, Phang JM. Polyoma enhancer activator 3, an Ets transcription factor, mediates the induction of cyclooxygenase‐2 by nitric oxide in colorectal cancer cells. J Biol Chem 2004; 279: 18694–700. [DOI] [PubMed] [Google Scholar]
- 51. Suzuki R, Kohno H, Sugie S, Tanaka T. Sequential observations on the occurrence of preneoplastic and neoplastic lesions in the mouse colon treated with azoxymethane and dextrane sodium sulfate. Cancer Sci 2004; 95: 721–7. [DOI] [PMC free article] [PubMed] [Google Scholar]