

Abstract
Cancer is a disease of genetic and epigenetic alterations, which are emphasized as the central mechanisms of tumor progression in the multistepwise model. Discovery of rare subpopulations of cancer stem cells (CSCs) has created a new focus in cancer research. The heterogeneity of tumors can be explained with the help of CSCs supported by antiapoptotic signaling. CSCs mimic normal adult stem cells by demonstrating resistance to toxic injuries and chemoradiation therapy. Moreover, they might be responsible for tumor relapse following apparent beneficial treatments. Compared with hematopoietic malignancies, conventional therapy regimes in solid tumors have improved the overall survival marginally, illustrating the profound impact of treatment resistance. This implies that the present therapies, which follow total elimination of rapidly dividing and differentiated tumor cells, need to be modified to target CSCs that repopulate the tumor. In this review article, we report on recent findings regarding the involvement of CSCs in chemoradiation resistance and provide new insights into their therapeutic implications in cancer. (Cancer Sci 2008; 99: 1871–1877)
The past few decades have seen an increase in evidence that suggests that cancer is a genomic disease and is associated with genetic and epigenetic alterations. Accumulation of these alterations during carcinogenesis activates proto‐oncogenes and inactivates tumor suppressors.( 1 , 2 ) The events occur in a stepwise manner, with mono‐ or oligo‐clonal cancer initiating cells acquiring a few genetic and epigenetic alterations in the early stages to numerous alterations in the advanced stages, which leads to aggressive characteristics such as invasions and clinical metastasis. Classical tumor suppressors including RB1 and TP53, and oncogenes including RAS and MYC, have been extensively studied.( 3 , 4 ) Epigenetic changes are associated with modifications in the spatial arrangement of the three‐dimensional structure of DNA resulting from interdigitation of DNA binding proteins such as histones and their modifiers, the Polycomb/Trithorax proteins, the DNA methyltransferases, and histone deacetylase enzymes,( 5 ) which lead to somatic cell‐related developmental disorders.( 5 )
Recent progress in stem cell research has greatly contributed to the field of oncology with the identification and characterization of cancer stem cells (CSCs) from a variety of tumors.( 6 , 7 ) The discovery of rare subpopulations of CSCs has shifted the focus of cancer research. CSCs are believed to mimic to normal adult stem cells, which are undifferentiated, have self‐renewal ability, and can give rise to differentiated cells.( 6 , 7 ) CSCs are believed to be more resistant to toxic injuries and chemoradiation therapy than differentiated daughter cells.( 6 , 7 ) It is suggested that normal stem or progenitor cells can eventually become malignant because of the occurrence of genetic and epigenetic alterations in CSCs, which are responsible for the development and maintenance of the tumor mass.( 6 , 7 ) Therapy‐resistant CSCs might regrow and lead to relapse of cancer following an apparently beneficial treatment.( 8 ) Although the CSC hypothesis is supported by some data, there are many uncertainties regarding the theoretical and interpretational aspects of the data supporting it. If correct, the CSC hypothesis could have profound implications on the field of cancer medicine. Here we discuss the recent progress made in the study of CSCs, which can enhance our understanding of the mechanisms of therapy resistance. Selective targeting of malignant CSCs could lead to improved preventive diagnosis and the development of novel therapeutics for cancer.
Rare subpopulations of CSCs in bulk tumors
CSCs can explain the biological heterogeneity of tumors.( 7 , 9 ) CSCs are a subpopulation of cancer cells with characteristics similar to normal stem cells such as: (1) the capability of self‐renewal; (2) the potential to divide and differentiate to generate all functional elements of a particular tissue; and (3) strict control over stem cell numbers.( 6 , 10 ) CSCs are defined as cells within tumors with tumor‐initiating properties.( 6 , 7 ) They are identified by inoculating cells in immune‐deficient animals, which have no control over cell growth in serial transplantations.( 6 ) CSCs, which are present in small numbers in whole tumors, are believed to be tumorigenic in contrast to the bulk of cancer cells, which are considered to be non‐tumorigenic.( 6 ) It has been hypothesized that CSCs persist in tumors as a distinct population and might cause relapse and metastasis by giving rise to new tumors. Thus, development of therapeutics that target CSCs holds promise for the improvement of survival and quality of life of cancer patients, especially for those with clinical metastasis. The clinicopathological process is driven by the mechanism of cell death, growth, and differentiation, which are altered in the bulk of cancer cells.
A small population of cancer‐initiating CSCs is potentially important, since they may play a role in the resistance to chemotherapy and radiation therapy and appear to be responsible for cancer recurrence after the cancer treatments, even when most of the cancer cells appear to be killed.( 11 ) Since the identification of rare CSCs in leukemia,( 12 , 13 , 14 ) molecular markers for detection of CSCs have been reported in solid tumors of the head, neck( 15 ) gastrointestinal system,( 16 ) colon,( 17 , 18 ) breast,( 19 ) and brain.( 20 , 21 )
Tumor heterogeneity
The CSC model can be used to explain the so‐called biological heterogeneity of tumors.( 7 , 9 ) Although genomic alterations occur in CSCs and are transmitted to daughter cells, occurrence of stochastic changes in numerous progenies is a matter of debate.( 7 , 9 ) The genomic alterations that are detected frequently in the chromosomal regions( 1 , 22 ) suggest that the events happen non‐randomly. The accumulation of genomic changes not only plays a role in carcinogenesis, but also in variations of the cancer cell genome and the corresponding biological behaviors, such as the interaction of the stroma and vessels of the host in the surrounding tissue.( 23 ) The extreme biological heterogeneity caused by the CSCs can lead to a lack of consistency in treatment planning, since similar cases under a clinico‐pathological point of view may differ widely in prognosis in cancers, for example those in the gastrointestinal tract.( 24 )
Alterations of survival signaling in stem cells
Adult stem cells and their malignant counterparts share almost all the intrinsic and extrinsic factors for regulation of self‐renewal, differentiation, and proliferation pathways.( 8 ) The following molecular markers characterizing the stem cells and transit amplifying populations of solid tumors such as the gastrointestinal tract are reported.( 25 ) For example, at present, candidate markers are proposed, including musashi‐1,( 26 ) hairy and enhancer of split homolog‐1 (Hes‐1),( 27 ) CD133 (prominin‐1, PROM1),( 28 , 29 ) EpCAM,( 30 ) Claudin‐7,( 30 ) CD44 variant isoforms,( 30 ) Lgr5,( 31 ) Hedgehog (Hh),( 32 ) bone morphogenic protein (Bmp),( 33 , 34 ) Notch,( 35 ) and Wnt.( 25 )
The mammalian gastrointestinal tract develops from the embryonic gut. It consists of an endodermally derived epithelium surrounded by cells of mesodermal origin.( 25 ) Cell signaling between these two tissue layers plays a critical role in coordinating pattern formation and organogenesis of the gut and its derivatives.( 25 ) Many studies have revealed that the critical signal transduction pathways include Bmp/Tgf‐β, Hh, fibroblast growth factor (Fgf), Wnt, Notch, Akt/PKB, and Bcl‐2. These constitute the stem cell signaling network and play a key role in a variety of processes, such as embryogenesis, maintenance of adult tissue homeostasis, tissue repair during chronic persistent inflammation, and carcinogenesis.( 25 , 36 , 37 ) In gastrointestinal stem cells, Wnt signaling is the most dominant force in controlling cell proliferation, differentiation, and apoptosis along the crypt–villus axis.( 25 ) Hh signaling is frequently activated in esophageal, gastric, and pancreatic cancers due to transcriptional up‐regulation of Hh ligands and epigenetic silencing of inhibitory molecule, Hhip1. Hh signaling is rarely activated in colorectal cancer owing to the negative regulation by the Wnt signaling pathway.( 36 ) Moreover, gastric mucosal repair and parietal cell proliferation during chronic Helicobacter pylori infection is associated with sonic Hh signaling.( 38 )
Hh. The Hh family of signaling proteins, hedgehog ligands, sonic (Shh), Indian (Ihh), and desert (Dhh), are secretory proteins which signal through autocrine and paracrine mechanisms to control cell proliferation, differentiation, and morphology.( 32 ) The Hh proteins exert their function by binding to a 12‐pass transmembrane protein called patched (Ptch),( 39 ) which relieves the inhibitory affect of Ptch on a serpentine protein called Smoothened (Smo), leading to hyper‐phosphorylation of Smo.( 40 , 41 ) The signal pathway induces activation and repression of target genes through the Gli family of transcription factors, Gli‐1, ‐2, ‐3, which regulate the transcription of target genes.
Bmp. Bmps, members of the Tgf‐β family of signaling proteins, are secretory ligands that signal via autocrine and paracrine mechanisms to regulate cell proliferation and differentiation.( 33 , 34 ) Bmp ligands bind to cell surface–associated proteins called Bmp receptors type I and type II.( 33 , 34 ) Bmp proteins have also been associated with epithelial to mesenchymal transition (EMT), as described below.
Notch. Notch signaling is critical for cell–cell communication and regulates a broad spectrum of cell fate decisions during embryonic development and in the adult organism via stem cell proliferation, differentiation, and cell death.( 35 ) Notch is instrumental in regulating processes such as neurogenesis, somatogenesis, and angiogenesis.( 42 ) Notch proteins are members of the conserved transmembrane receptor family including four members, Notch1–4.( 35 ) The Notch genes encode transmembrane receptors, which contain a large extracellular domain, composed of a variable number of epidermal growth factor (Egf)–like repeats and an intracellular signaling domain, which consists of six ankyrin/cdc10 motifs and nuclear localization signals.( 43 ) Notch receptors interact through their extracellular domain with other membrane‐associated ligands, the Delta and Serrate/Jagged families, which are composed of five proteins, Jagged1 and 2, and Delta‐like 1, 3, and 4.( 43 ) Notch signaling is activated by ligand–receptor interactions and triggers proteolytic cleavage by the gamma‐secretase complex, which releases the Notch intracellular domain (NICD) into the nucleus. The Notch intracellular domain binds to the CBF1 DNA binding protein of the transcriptional activator complex, the activation of which can lead to the expression of target genes, such as Hes family genes, involved in cell growth and differentiation. Notch signals are transduced to the canonical pathway (CSL‐NICD‐Mastermind signaling cascade) or the non‐canonical pathway (NF‐κB‐NICD and CSL‐NICD‐Deltex signaling cascades) based on the expression profile of the Notch ligands, receptors, and signaling modifiers.( 44 )
EMT
Recent studies indicate that embryonic pathways affect the survival of tumor‐initiating stem cells and orchestrate a complex microenvironment or niche that promotes tumor survival and progression.( 45 ) Increased motility and invasiveness of certain cancer cells is associated with the EMT process, which occurs during embryonic development.( 46 ) EMT is an important change in cell phenotype, which allows the escape of epithelial cells from the structural constraints imposed by tissue architecture.( 47 ) Although the morphology of various tumors has indicated epithelial and mesenchymal components of metaplasia,( 48 , 49 ) the EMT phenomenon was recognized by Elizabeth Hay in the 1980s to be a central process in early embryonic morphogenesis.( 46 ) The phrase EMT first appeared in 1980s with reference to a study of cellular change elicited by extracellular matrix,( 50 ) and this phenomenon was further characterized by Hay.( 46 ) The EMT process can be affected by Shh,( 51 ) Bmp2,( 52 ) and Notch signaling.( 53 )
EMT and stem cell formation are important processes in development, and are characterized by aberrant nuclear expression of β‐catenin. The unusual combination of EMT with stem cell competence might result in migration of CSCs, which initiates tumor invasion and metastasis.( 54 ) Although radiation resistance remains to be fully understood, recent studies have shown that induction of EMT might contribute to the decreased efficacy of therapy in chemoresistant cancer cells as the tumor cells switch from a proliferative to invasive phenotype.( 55 , 56 , 57 , 58 ) The involvement of EMT was demonstrated by the chemoresistance to paclitaxel,( 55 , 58 ) oxaliplatin,( 56 ) and erlotinib.( 57 ) Twist transcriptionally up‐regulates Akt2 in cancer cells leading to resistance to paclitaxel.( 55 ) Further understanding of the mechanisms of chemoresistance will enable improvements in chemotherapy for solid tumors.
Adenosine triphosphate (ATP)–binding cassette (ABC) drug transporters
Multidrug resistance (MDR) in which tumor cells simultaneously possess intrinsic or acquired cross‐resistance to diverse chemotherapeutic agents, including taxanes and anthracyclines, hampers the effective treatment of cancer.( 59 ) Several proteins associated with MDR have been identified, including P‐glycoprotein (P‐gp), multidrug resistance–associated protein (MRP1), lung resistance protein (LRP), breast cancer resistance protein (BCRP), all members of the ABC superfamily of efflux transporters with a wide variety of substrates.( 59 , 60 ) These transmembrane proteins cause MDR either by decreasing the total intracellular retention of drugs or by redistributing the intracellular accumulation of drugs away from target organelles.( 59 ) Some of the characteristics of CSCs exhibit the functional ATP‐dependent drug efflux and elevated tumorigenic potential.
Certain stem cell types are known to have highly efficient pumps for the dye Hoechst 33342.( 61 ) Stem cells isolated by their ability to efflux Hoechst 33342 dye, are referred to as the ‘side population’ (SP).( 61 , 62 ) Hoechst 33342 is a DNA‐binding dye is that binds preferentially to A‐T rich regions of DNA. The dye is excited by UV wavelengths. However, the emission wavelength shifts to the red end of the spectrum in apoptosis and also when the dye concentration is high. Although it is able to enter live cells, and it can also be actively pumped out by the ABC transporters which include p‐glycoprotein and ABCG2 (another name for BCRP). Agents such as verapamil and reserpinel are specific inhibitors of these transporters.( 63 ) The presence of a rare, drug‐effluxing stem cell‐like SP of cells contain a subpopulation of CSC‐like cells, which resemble previously characterized tissue stem cells with individual cell quiescence. The existence of ATP transporter–dependent CSCs might be relatively common, particularly within established tumors.( 64 ) Drug resistance has been reported in glioma,( 65 ) ovarian,( 66 ) lung,( 67 ) and gastrointestinal cancers.( 16 , 68 )
The study of SP in lung cancer cells demonstrates elevated expression of ABCG2 and other ATP‐binding cassette transporters, resistance to multiple chemotherapeutic drugs, and high expression of human telomerase reverse transcriptase, suggesting that this fraction may represent a reservoir with unlimited proliferation; and low expression of minichromosome maintenance 7 (MCM7), a member of the MCM family of proteins critical to the DNA replication complex, suggesting that a majority of the SP fraction was in the G0 quiescent state.( 67 , 69 ) Although SP is enriched with tumorigenic stem‐like cancer cells, the study of glioma and breast and prostate cancers indicates that ABCG2 expression primarily identifies fast‐cycling tumor progenitors. The ABCG2‐negative population predominantly consists of primitive stem‐like cancer cells, which preferentially express several ‘stemness’ genes, including Notch1 and β‐catenin,( 70 ) suggesting that SP might be a heterogeneous population and might possess distinct characteristics depending on the tumor types.
Verapamil (VER), a potent calcium channel blocker, can overcome P‐gp‐mediated MDR and may increase sensitivity to cytotoxic anticancer drugs in refractory cancer, although efficacy of VER for treating solid tumors is still a matter for debate.( 71 ) A recent study shows that ABCG2 expression in breast cancer MCF7 cells is regulated during EMT, and that the EMT effect reflects post‐translational regulation of ABCG2 function by E‐cadherin as well as transcriptional repression of the ABCG2 gene, indicating that TGFβ‐directed EMT associates with ABCG2 expression and SP abundance.( 72 )
Checkpoint response and DNA damage repair
The human genome is exposed to a variety of genotoxins, which can elicit unscheduled replication. After DNA is damaged or replication is perturbed, cells respond by activation of evolutionarily conserved signal transduction pathways that delay cell cycle progression and induce DNA repair. These signal transduction pathways include protein sensors that recognize aberrant DNA structures and activate kinases, thereby inducing phosphorylation cascades that ultimately result in cell cycle arrest and DNA repair.( 73 , 74 , 75 , 76 ) Failure of this cell cycle surveillance mechanism can cause genomic instability that eventually leads to cancer formation in mammals.( 77 , 78 ) Cell cycle checkpoints are signal transduction pathways that maintain the proper order of cell cycle events. As has been shown previously, γ‐H2ax is an important marker for detection of breaks in double‐strands,( 79 ) and is used for evaluating DNA repair dynamics.( 80 ) In mammalian cells, the signal initiated by sensors, 2‐phosphatidylinositol 3‐kinase‐related kinases (Pikk), ataxia‐telangiectasia mutated (Atm), and Atm‐ and Rad3‐related (Atr), play a central role in the checkpoint signaling pathways.( 81 ) Atm and Atr are activated by genotoxins and phosphorylate downstream signaling proteins, including Chk1 and Chk2, two protein kinases that regulate checkpoint responses.( 82 , 83 , 84 )
Although there is increasing evidence that a rare CSC population of undifferentiated cells is responsible for tumor formation and maintenance, it remains to be explored in case of colorectal cancer. Recently, high‐density CD133+ population was isolated from tumorigenic cells in colon cancer, which accounts for about 2.5% of the tumor cells.( 17 ) The study suggests that subcutaneous injection of colon cancer CD133+ cells but not CD133− cells readily reproduced the original tumor in immunodeficient mice. Moreover, serial transplantation for several generations of CD133+ colon cancer cells results in exponential growing tumors for more than a year and CD133+ cells reproduce the same morphological and antigenic patterns of the original tumor,( 17 ) thereby suggesting the candidacy of CD133 as a marker of CSCs. The data show that colorectal cancer is created and propagated by a small number of undifferentiated tumorigenic CD133+ cells, which possess high proliferation potential (Fig. 1).( 17 , 85 ) Previous studies of CD133+ cells suggest an association of the mechanism of DNA damage repair,( 21 ) hypoxia,( 86 ) and chemokine SDF‐1‐CXCR4 axis.( 87 ) CD133+ cells were found to be significantly resistant to chemotherapeutic agents including temozolomide, carboplatin, paclitaxel (Taxol), and etoposide (VP16), compared to autologous CD133− cells.( 88 )
Figure 1.
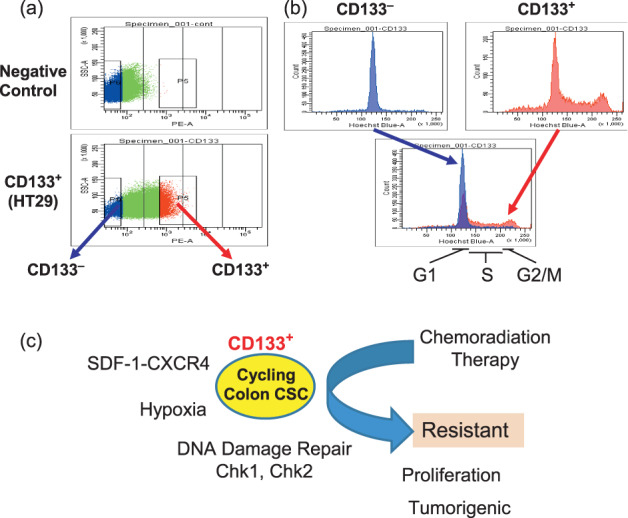
CD133+ cancer stem cells (CSCs) from colon cancer. (a) CD133+ and CD133− colon cancer cells were isolated by FACS sorting. (b) Cell cycle analysis of CD133+ and CD133− colon cancer cells. CD133+ fraction exhibits cell cycling prolifration, compared with CD133− fraction. (c) Schema represents CD133+ colon CSCs are resistant to therapy.
The study of CD133+ glioma stem cells indicated that the radioresistance of CD133+ CSCs can be reversed with a specific inhibitor of the Chk1 and Chk2 checkpoint kinases,( 21 ) indicating that CD133+ cells confer glioma radioresistance and could be the source of tumor recurrence after radiation treatment. It is known that the Chk1 and Chk2 proteins play roles in the execution of checkpoint response to delay or arrest the cell cycle, which elicits repair of the DNA damage response.( 89 ) It is suggested that the activity might be involved in aberrant repairs of damaged cells to enable their survival and might contribute to the accumulation of further genomic alterations during carcinogenesis. These observations together with the increase in proliferation of CD133+ colon cancer cells( 85 ) suggest that the uncoupling of repair and cell cycle control might be a critical feature of CD133+ CSCs. Elucidation of DNA damage response in isolated CSCs would enable targeting DNA damage checkpoint response in CSCs. This would open new avenues for overcoming the current chemotherapy or radiation therapy resistance, and provide a new model for cancer medicine.
The response of CD24(–/low)/CD44+ breast CSCs to radiation indicates that the levels of reactive oxygen species increase in both mammospheres and monolayer cultures after irradiation but are lower in mammospheres than in monolayer cultures.( 90 ) H2ax phosphorylation increased in irradiated monolayer cultures, but no change was observed in mammospheres, suggesting that rapid, altered repairs occur in CSCs. The process might be involved in the activation of Notch1.( 90 ) The percentage of CSCs in the nonadherent cell population of monolayer cultures indicate that breast CSCs are a relatively radioresistant subpopulation and increase in numbers after short courses of fractionated irradiation. These findings offer a possible mechanism for the accelerated repopulation of tumor cells observed during gaps in radiotherapy.
The oncogenic function of Notch1 and Notch4 is shown by studies of the mammary epithelial cell system.( 91 , 92 , 93 ) Transgenic overexpression of the Notch intracellular domain of Notch1 and Notch3 resulted in the development of mammary tumors.( 94 ) Knockdown of Notch proteins resulted in sensitization to genotoxic deionizing radiation (Fig. 2). The effect was more apparent in CD44+ breast‐cancer‐initiating cells than in CD44− cells, suggesting a possible role of the Notch family as survival factors of breast‐cancer‐initiating cells, i.e. the protective role of cancer‐initiating cells from the induction of cell death, which could lead to the survival and accumulation of damaged cells carrying deleterious mutations. Considering recent study,( 95 ) the Notch pathway may be a site for promising candidates for molecular targets to elicit the sensitivity of Her2‐negative cancer‐initiating cells as a therapeutic approach.
Figure 2.
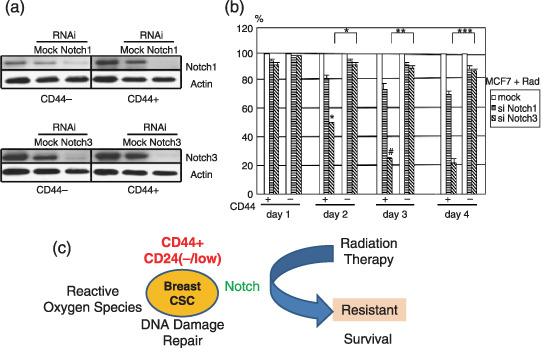
Notch pathway as a candidate therapeutic target in breast tumors. CD44‐positive and negative MCF7 cells were studied by FACS analysis and immunoblot with anti‐Notch1, Notch3, and actin antibodies. (a) siRNAs knockdown of Notch1 and Notch3. siRNAs of Notch1 (144334; NM_017617) and Notch3 (143322; NM_000435) were used (Ambion‐Applied Biosystems). (b) MTS proliferation assay (Promega, San Luis Obispo, CA, USA) of CD44+‐ and CD44− breast cancer MCF7 cells with knockdown of Notch1 and Notch3. Cells (2 × 103 per well) were seeded and grown in 96‐well plat bottom plates for 24 hours. After cells were treated by radiation exposure at 4 Gy, cells were allowed to grow in a 37°C incubator for indicated periods and the absorbance at 490 nm was measured. The average data was calculated in comparison with the control result and the percent inhibition is shown. Each character (* and #) indicates statistical significance (P < 0.05). (c) Schema represents CD44+ CD24(–/low) breast cancer stem cells (CSCs) resistant to therapy.
Perspectives
Similar to the skin, where cancers are common, the gastrointestinal epithelium possesses a high rate of cell turnover.( 96 ) There is a small subset of cancer cells, the CSCs, which constitute a reservoir of self‐sustaining cells with the exclusive ability to self‐renew and maintain tumors.( 9 ) The CSC hypothesis requires us to rethink the methods of tumor diagnosis and treatment. For effective treatment, we may have to focus on the minority CSC population that possesses dormancy and leads to tumor growth, requiring a shift from attempts to eliminate the bulk of rapidly dividing and terminally differentiated cells to targeting CSCs within a tumor.( 9 ) Our current understanding is that CSCs are either dormant or in a proliferative phase. Dormant CSCs may confer drug resistance through damage response, the ABC transporter, and niche signaling, whereas CSC proliferation would give rise to tumor volume. In tumors, there could be at least two possible defects: the transition from dormancy to proliferation and the regulation of cell cycling.( 97 ) Recent findings securely fasten the rationale for several different therapeutic approaches for cancer (Fig. 3), including targeting Hh, Notch, Wnt, and chemokine CXCR4;( 98 , 99 , 100 ) ABC transporter; CD33;( 101 ) tyrosine kinase bcr‐abl;( 102 ) Chk1 and Chk2;( 21 ) and the retinoic acid pathway.( 103 ) While the role of somatic mutations has been extensively documented in determining tumor phenotype,( 104 ) undoubtedly the detection and biological regulation of CSCs during carcinogenesis could be a critical issue for the development of effective prevention, diagnostic, and novel therapeutic approaches to gastrointestinal cancers.
Figure 3.
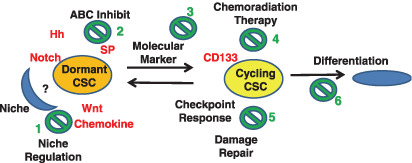
Cancer stem cells (CSCs) and chemoradiation therapy. In the schema, dormant CSCs are supported by a niche and resistant therapy, whereas cycling CSCs give rise to the volume of tumors and are sensitive to the genotoxic chemoradiation therapy. If so, the remaining CSCs followed by apparent chemoradiation therapy would contribute to the regrowth and metastasis of tumors. The possible therapeutic approaches include: (1) targeting niche regulation through inhibition of stem cell Hh, Notch, and Wnt signaling and chemokine, which inhibit CSCs through the interference of self‐renewal; (2) inhibition of ABC transporters such as SP; (3) targeting molecular markers such as CD33 and imatinib for tyrosine kinase bcr‐abl; (4) chemoradiation therapy; (5) control of checkpoints, such as inhibition of Chk1 and Chk2; and (6) induction of differentiation, such as retinoic acid.
During the preparation of manuscript, a direct link between the EMT and the gain of epithelial CSC properties has been proposed, which is often activated during cancer invasion and metastasis.( 105 ) The transient induction of an EMT in tumors may make possible the generation of relatively unlimited numbers of CSCs and their daughter cells.( 105 ) It is suggested that the use of pharmacogenetic techniques to transiently induce or inhibit an EMT in large populations of epithelial cells may provide a means of control of normal epithelial and CSCs.
Acknowledgments
Grant support: Ministry of Education, Culture, Sports, Science, and Technology of Japan, the 21st Century COE Program ‘integrative life sciences – postgenome research of systemic life sciences’, and GCOE program ‘cell‐fate decision: function and dysfunction in homeostasis’ at Kyushu University.
References
- 1. Nowell PC. Chromosomes and cancer: the evolution of an idea. Adv Cancer Res 1993; 62: 1–17. [DOI] [PubMed] [Google Scholar]
- 2. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet 1993; 9: 138–41. [DOI] [PubMed] [Google Scholar]
- 3. Hickman ES, Moroni MC, Helin K. The role of p53 and pRB in apoptosis and cancer. Curr Opin Genet Dev 2002; 12: 60–6. [DOI] [PubMed] [Google Scholar]
- 4. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer 2003; 3: 459–65. [DOI] [PubMed] [Google Scholar]
- 5. Waterland RA. Epigenetic mechanisms and gastrointestinal development. J Pediatr 2006; 149: S137–42. [DOI] [PubMed] [Google Scholar]
- 6. Sagar J, Chaib B, Sales K, Winslet M, Seifalian A. Role of stem cells in cancer therapy and cancer stem cells: a review. Cancer Cell Int 2007; 7: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414: 105–11. [DOI] [PubMed] [Google Scholar]
- 8. Giordano A, Fucito A, Romano G, Marino IR. Carcinogenesis and environment. the cancer stem cell hypothesis and implications for the development of novel therapeutics and diagnostics. Front Biosci 2007; 12: 3475–82. [DOI] [PubMed] [Google Scholar]
- 9. Clarke MDJ, Dirks PB, Eaves CJ et al . Cancer stem cells – perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66: 9339–44. [DOI] [PubMed] [Google Scholar]
- 10. Bixby S, Kruger GM, Mosher JT, Joseph NM, Morrison SJ. Cell‐intrinsic differences between stem cells from different regions of the peripheral nervous system regulate the generation of neural diversity. Neuron 2002; 35: 643–56. [DOI] [PubMed] [Google Scholar]
- 11. Tan BT, Park CY, Ailles LE, Weissman IL. The cancer stem cell hypothesis: a work in progress. Lab Invest 2006; 86: 1203–7. [DOI] [PubMed] [Google Scholar]
- 12. Wulf GG, Wang RY, Kuehnle I et al . A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood 2001; 98: 1166–73. [DOI] [PubMed] [Google Scholar]
- 13. Lapidot T, Sirard C, Vormoor J et al . A cell initiating human acute myeloid leukemia after transplantation into SCID mice. Nature 1994; 367: 645–8. [DOI] [PubMed] [Google Scholar]
- 14. Bonnet D, Dick JE. Human acute leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730–7. [DOI] [PubMed] [Google Scholar]
- 15. Prince ME, Sivanandan R, Kaczorowski A et al . Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA 2007; 104: 973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haraguchi N, Utsunomiya T, Inoue H et al . Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells 2006; 24: 506–13. [DOI] [PubMed] [Google Scholar]
- 17. Ricci‐Vitiani L, Lombardi DG, Pilozzi E et al . Identification and expansion of human colon‐cancer‐initiating cells. Nature 2007; 445: 111–15. [DOI] [PubMed] [Google Scholar]
- 18. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445: 106–10. [DOI] [PubMed] [Google Scholar]
- 19. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piccirillo SG, Reynolds BA, Zanetti N et al . Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour‐initiating cells. Nature 2006; 444: 761–5. [DOI] [PubMed] [Google Scholar]
- 21. Bao S, Wu Q, McLendon RE, Hao Y et al . Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444: 756–60. [DOI] [PubMed] [Google Scholar]
- 22. Cavenee W, Dryja TP, Phillips RA et al . Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983; 305: 779–84. [DOI] [PubMed] [Google Scholar]
- 23. Weinberg R. Tumor suppressor genes. Science 1991; 254: 1138–46. [DOI] [PubMed] [Google Scholar]
- 24. Almadori G, Bussu F, Paludetti G. Should there be more molecular staging of head and neck cancer to improve the choice of treatments and thereby improve survival? Curr Opin Otolaryngol Head Neck Surg 2008; 16: 117–26. [DOI] [PubMed] [Google Scholar]
- 25. Yen TH, Wright NA. The gastrointestinal tract stem cell niche. Stem Cell Rev 2006; 2: 203–12. [DOI] [PubMed] [Google Scholar]
- 26. Nakamura M, Okano H, Blendy J, Montell C. Musashi, a neural RNA‐binding protein required for Drosophila adult external sensory organ development. Neuron 1994; 13: 67–81. [DOI] [PubMed] [Google Scholar]
- 27. Ishibashi M, Ang SL, Shiota K, Nakanishi S, Kageyama R, Guillemot F. Targeted disruption of mammalian hairy and Enhancer of split homolog‐1 (HES‐1) leads to up‐regulation of neural helix‐loop‐helix factors, premature neurogenesis, and severe neural tube defects. Genes Dev 1995; 9: 3136–48. [DOI] [PubMed] [Google Scholar]
- 28. Lin EH, Hassan M, Li Y et al . Elevated circulating endothelial progenitor marker CD133 messenger RNA levels predict colon cancer recurrence. Cancer 2007; 110: 534–42. [DOI] [PubMed] [Google Scholar]
- 29. Mehra N, Penning M, Maas J et al . Progenitor marker CD133 mRNA is elevated in peripheral blood of cancer patients with bone metastases. Clin Cancer Res 2006; 12: 4859–66. [DOI] [PubMed] [Google Scholar]
- 30. Kuhn S, Koch M, Nübel T et al . A complex of EpCAM, claudin‐7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res 2007; 5: 553–67. [DOI] [PubMed] [Google Scholar]
- 31. Barker N, Van Es JH, Kuipers J et al . Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449: 1003–7. [DOI] [PubMed] [Google Scholar]
- 32. Ingham PW, McMahon AP. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev 2001; 15: 3059–87. [DOI] [PubMed] [Google Scholar]
- 33. Koenig BB, Cook JS, Wolsing DH et al . Characterization and cloning of a receptor for BMP‐2 and BMP‐4 from NIH 3T3 cells. Mol Cell Biol 1994; 14: 5961–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ten Dijke P, Yamashita H, Sampath TK et al . Identification of type I receptors for osteogenic protein‐1 and bone morphogenetic protein‐4. J Biol Chem 1994; 269: 16985–8. [PubMed] [Google Scholar]
- 35. Bray S. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 2006; 7: 678–89. [DOI] [PubMed] [Google Scholar]
- 36. Katoh Y, Katoh M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network. Int J Mol Med 2006; 18: 1019–23. [PubMed] [Google Scholar]
- 37. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008; 27: 1749–58. [DOI] [PubMed] [Google Scholar]
- 38. Nishizawa T, Suzuki H, Masaoka T, Minegishi Y, Iwasahi E, Hibi T. Helicobacter pylori eradication restored sonic hedgehog expression in the stomach. Hepatogastroenterology 2007; 54: 697–700. [PubMed] [Google Scholar]
- 39. Pepinsky RB, Rayhorn P, Day ES et al . Mapping sonic hedgehog–receptor interactions by steric interference. J Biol Chem 2000; 275: 10 995–1001. [DOI] [PubMed] [Google Scholar]
- 40. Murone M, Rosenthal A, De Sauvage FJ. Sonic hedgehog signaling by the patched‐smoothened receptor complex. Curr Biol 1999; 9: 76–84. [DOI] [PubMed] [Google Scholar]
- 41. Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature 2005; 437: 1018–21. [DOI] [PubMed] [Google Scholar]
- 42. Bolos V, Grego‐Bessa J, De La Pompa JL. Notchsignaling in development and cancer. Endocr Rev 2007; 28: 339–63. [DOI] [PubMed] [Google Scholar]
- 43. Artavanis‐Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science 1999; 284: 770–6. [DOI] [PubMed] [Google Scholar]
- 44. Katoh M, Katoh M. Notch signaling in gastrointestinal tract. Int J Oncol 2007; 30: 247–51. [PubMed] [Google Scholar]
- 45. Bailey JM, Singh PK, Hollingsworth MA. Cancer metastasis facilitated by developmental pathways: sonic hedgehog, Notch, and bone morphogenic proteins. J Cell Biochem 2007; 102: 829–39. [DOI] [PubMed] [Google Scholar]
- 46. Hay ED. An overview of epithelio‐mesenchymal transformation. Acta Anat (Basel) 1995; 154: 8–20. [DOI] [PubMed] [Google Scholar]
- 47. Hugo H, Ackland ML, Blick T et al . Epithelial‐mesenchymal and mesenchymal‐epithelial transitions in carcinoma progression. J Cell Physiol 2007; 213: 374–83. [DOI] [PubMed] [Google Scholar]
- 48. Kahn LB, Uys CJ, Dale J, Rutherfoord S. Carcinoma of the breast with metaplasia to chondrosarcoma: a light and electron microscopic study. Histopathology 1978; 2: 93–106. [DOI] [PubMed] [Google Scholar]
- 49. Ishikawa S, Kaneko H, Sumida T, Sekiya M. Ultrastructure of mesodermalmixed tumor of the uterus. Acta Pathol Jpn 1979; 29: 801–9. [DOI] [PubMed] [Google Scholar]
- 50. Krug EL, Mjaatvedt CH, Markwald RR. Extracellular matrix from embryonic myocardium elicits an early morphogenetic event in cardiac endothelial differentiation. Dev Biol 1987; 120: 348–55. [DOI] [PubMed] [Google Scholar]
- 51. Feldmann G, Dhara S, Fendrich V et al . Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res 2007; 67: 2187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial‐mesenchymal transition and myocardial patterning. Development 2005; 132: 5601–11. [DOI] [PubMed] [Google Scholar]
- 53. Timmerman LA, Grego‐Bessa J, Raya A et al . Notch promotes epithelial‐mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004; 18: 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Spaderna S, Schmalhofer O, Hlubek F, Jung A, Kirchner T, Brabletz T. Epithelial‐mesenchymal and mesenchymal‐epithelial transitions during cancer progression. Verh Dtsch Ges Pathol 2007; 91: 21–8. [PubMed] [Google Scholar]
- 55. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up‐regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 2007; 67: 1979–87. [DOI] [PubMed] [Google Scholar]
- 56. Yang AD, Fan F, Camp ER et al . Chronic oxaliplatin resistance induces epithelial‐to‐mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 2006; 12: 4147–53. [DOI] [PubMed] [Google Scholar]
- 57. Yauch RL, Januario T, Eberhard DA et al . Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 2005; 11: 8686–98. [DOI] [PubMed] [Google Scholar]
- 58. Kajiyama H, Shibata K, Terauchi M et al . Chemoresistance to paclitaxel induces epithelial‐mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 2007; 31: 277–83. [PubMed] [Google Scholar]
- 59. Tan B, Piwnica‐Worms D, Ratner L. Multidrug resistance transporters and modulation. Curr Opin Oncol 2000; 12: 450–8. [DOI] [PubMed] [Google Scholar]
- 60. Hazai E, Bikádi Z. Homology modeling of breast cancer resistance protein (ABCG2). J Struct Biol 2008; 162: 63–74. [DOI] [PubMed] [Google Scholar]
- 61. Challen GA, Little MH. A side order of stem cells: the SP phenotype. Stem Cells 2006; 24: 3–12. [DOI] [PubMed] [Google Scholar]
- 62. Polgar O, Robey RW, Bates SE. ABCG2: structure, function and role in drug response. Expert Opin Drug Metab Toxicol 2008; 4: 1–15. [DOI] [PubMed] [Google Scholar]
- 63. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo . J Exp Med 1996; 183: 1797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Loebinger MR, Giangreco A, Groot KR et al . Squamous cell cancers contain a side population of stem‐like cells that are made chemosensitive by ABC transporter blockade. Br J Cancer 2008; 98: 380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem‐like cells in the C6 glioma cell line. Proc Natl Acad Sci USA 2004; 101: 781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Szotek PP, Pieretti‐Vanmarcke R, Masiakos PT et al . Ovarian cancer side population defines cells with stem cell‐like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci USA 2006; 103: 11 154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem‐like cancer cells. Cancer Res 2007; 67: 4827–33. [DOI] [PubMed] [Google Scholar]
- 68. Haraguchi N, Inoue H, Tanaka F et al . Cancer stem cells in human gastrointestinal cancers. Hum Cell 2006; 19: 24–9. [DOI] [PubMed] [Google Scholar]
- 69. Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003; 22: 7340–58. [DOI] [PubMed] [Google Scholar]
- 70. Patrawala L, Calhoun T, Schneider‐Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem‐like cancer cells, whereas ABCG2+ and ABCG2− cancer cells are similarly tumorigenic. Cancer Res 2005; 65: 6207–19. [DOI] [PubMed] [Google Scholar]
- 71. Belpomme D, Gauthier S, Pujade‐Lauraine E et al . Verapamil increases the survival of patients with anthracycline‐resistant metastatic breast carcinoma. Ann Oncol 2000; 11: 1471–6. [DOI] [PubMed] [Google Scholar]
- 72. Yin L, Castagnino P, Assoian RK. ABCG2 expression and side population abundance regulated by a transforming growth factor beta‐directed epithelial‐mesenchymal transition. Cancer Res 2008; 68: 800–7. [DOI] [PubMed] [Google Scholar]
- 73. Dasika GK, Lin SC, Zhao S, Sung P, Tomkinson A, Lee EY. DNA damage‐induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 1999; 18: 7883–99. [DOI] [PubMed] [Google Scholar]
- 74. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science 1996; 274: 1664–72. [DOI] [PubMed] [Google Scholar]
- 75. Wang JY. Cellular responses to DNA damage. Curr Opin Cell Biol 1998; 10: 240–7. [DOI] [PubMed] [Google Scholar]
- 76. Weinert T. DNA damage and checkpoint pathways. molecular anatomy and interactions with repair. Cell 1998; 94: 555–8. [DOI] [PubMed] [Google Scholar]
- 77. Hartwell LH, Kastan MB. Cell cycle control and cancer. Science 1994; 266: 1821–8. [DOI] [PubMed] [Google Scholar]
- 78. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386: 623–7. [DOI] [PubMed] [Google Scholar]
- 79. Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol Ther 2003; 2: 233–5. [DOI] [PubMed] [Google Scholar]
- 80. Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA, Lobrich M. A double‐strand break repair defect in ATM‐deficient cells contributes to radiosensitivity. Cancer Res 2004; 64: 500–8. [DOI] [PubMed] [Google Scholar]
- 81. Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15: 2177–96. [DOI] [PubMed] [Google Scholar]
- 82. Bartek J, Lukas J. Mammalian G1‐ and S‐phase checkpoints in response to DNA damage. Curr Opin Cell Biol 2001; 13: 738–47. [DOI] [PubMed] [Google Scholar]
- 83. McGowan CH. Checking in on Cds1 (Chk2): a checkpoint kinase and tumor suppressor. Bioessays 2002; 24: 502–11. [DOI] [PubMed] [Google Scholar]
- 84. Rhind N, Russell P. Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J Cell Sci 2000; 113: 3889–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ieta K, Tanaka F, Haraguchi N et al . Biological and genetic characteristics of tumor‐initiating cells in colon cancer. Ann Surg Oncol 2008; 15: 638–48. [DOI] [PubMed] [Google Scholar]
- 86. Blazek ER, Foutch JL, Maki G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133− cells, and the CD133+ sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys 2007; 67: 1–5. [DOI] [PubMed] [Google Scholar]
- 87. Czarnowska E, Gajerska‐Dzieciatkowska M, Kuœmierski K et al . Expression of SDF‐1‐CXCR4 axis and an anti‐remodelling effectiveness of foetal‐liver stem cell transplantation in the infarcted rat heart. J Physiol Pharmacol 2007; 58: 729–44. [PubMed] [Google Scholar]
- 88. Liu G, Yuan X, Zeng Z et al . Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006; 5: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sancar A, Lindsey‐Boltz LA, Unsal‐Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73: 39–85. [DOI] [PubMed] [Google Scholar]
- 90. Phillips TM, McBride WH, Pajonk F. The response of CD24(–/low)/CD44+ breast cancer‐initiating cells to radiation. J Natl Cancer Inst 2006; 98: 1777–85. [DOI] [PubMed] [Google Scholar]
- 91. Shi W, Harris AL. Notch signaling in breast cancer and tumor angiogenesis: cross‐talk and therapeutic potentials. J Mammary Gland Biol Neoplasia 2006; 11: 41–52. [DOI] [PubMed] [Google Scholar]
- 92. Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med 2006; 6: 905–18. [DOI] [PubMed] [Google Scholar]
- 93. Efstratiadis A, Szabolcs M, Klinakis A. Notch, Myc and breast cancer. Cell Cycle 2007; 6: 418–29. [DOI] [PubMed] [Google Scholar]
- 94. Hu C, Dievart A, Lupien M, Calvo E, Tremblay G, Jolicoeur P. Overexpression of activated murine Notch1 and Notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. Am J Pathol 2006; 168: 973–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yamaguchi N, Oyama T, Ito E et al . NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2‐negative human breast cancer cells. Cancer Res 2008; 68: 1881–8. [DOI] [PubMed] [Google Scholar]
- 96. Wershil BK, Furuta GT. Gastrointestinal mucosal immunity. J Allergy Clin Immunol 2008; 121: S380–3; quiz S415. [DOI] [PubMed] [Google Scholar]
- 97. Sherr CJ. Cancer cell cycles. Science 1996; 274: 1672–7. [DOI] [PubMed] [Google Scholar]
- 98. Rubin JB, Kung AL, Klein RS et al . A small‐molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci USA 2003; 100: 13 513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Liang Z, Wu T, Lou H et al . Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res 2004; 64: 4302–8. [DOI] [PubMed] [Google Scholar]
- 100. Tavor S, Petit I, Porozov S et al . CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res 2004; 64: 2817–24. [DOI] [PubMed] [Google Scholar]
- 101. Larson RA, Sievers EL, Stadtmauer EA et al . Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33‐positive acute myeloid leukemia in first recurrence. Cancer 2005; 104: 1442–52. [DOI] [PubMed] [Google Scholar]
- 102. Ponsaing LG, Hansen MB. Therapeutic procedures for submucosal tumors in the gastrointestinal tract. World J Gastroenterol 2007; 13: 3316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sell S. Leukemia: stem cells, maturation arrest, and differentiation therapy. Stem Cell Rev 2005; 1: 197–205. [DOI] [PubMed] [Google Scholar]
- 104. Ince TA, Richardson AL, Bell GW et al . Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007; 12: 160–70. [DOI] [PubMed] [Google Scholar]
- 105. Mani SA, Guo W, Liao MJ. et al . The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell; 133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]