

Abstract
Wnt5a is a member of the Wnt family of proteins that signals through the non‐canonical Wnt/Ca2+pathway to suppress cyclin D1. Deregulation of this pathway has been found in animal models suggesting that it acts as tumour suppressor in acute myeloid leukemia (AML). Although DNA methylation is the main mechanism of regulation of the canonical Wnt pathway in AML, the role of WNT5A abnormalities has never been evaluated in this clinical setting. The methylation status of WNT5A promoter–exon 1 was analyzed by methylation‐specific PCR and sequencing in eleven AML‐derived cell lines and 252 AML patients. We observed WNT5A hypermethylation in seven cell lines and in 43% (107/252) of AML patients. WNT5A methylation was associated with decreased WNT5A expression (P < 0.001) that was restored after exposure to 5‐Aza‐2’‐deoxycytidine. Moreover, WNT5A hypermethylation correlated with upregulation of CYCLIN D1 expression (P < 0.001). Relapse (15%vs 37%, P < 0.001) and mortality (61%vs 79%, P = 0.004) rates were lower for patients in the non‐methylated group. Disease‐free survival and overall survival at 6 and 7 years, respectively, were 60% and 27% for unmethylated patients and 20% and 0% for hypermethylated patients (P = 0.0001 and P = 0.04, respectively). Interestingly, significant differences were also observed when the analysis was carried out according to cytogenetic risk groups. We demonstrate that WNT5A, a putative tumor suppressor gene in AML, is silenced by methylation in this disease and that this epigenetic event is associated with upregulation of CYCLIN D1 expression and confers poor prognosis in patients with AML. (Cancer Sci 2009)
Wnt proteins form a large family of secreted glycoproteins that activate the highly conserved Wnt pathway through their receptor Frizzled. Wnt–Frizzled signaling controls a wide variety of processes, including proliferation, differentiation, migration, and cell–cell adhesion, playing a crucial role not only during development but also in hematopoietic stem cell maintenance and oncogenesis.( 1 , 2 , 3 , 4 )
Wnt signaling occurs through the canonical and non‐canonical pathways. The canonical pathway has a central mediator, β‐catenin. In the absence of Wnt ligand, β‐catenin is phosphorylated and ubiquitinated by the activity of a multiprotein destruction complex that contains axin, adenomatous poliposis coli, glycogen‐synthase kinase 3β, and casein kinase 1, leading to proteasome‐mediated degradation. On the other hand, binding of Wnt to the Frizzled receptor leads to inhibition of the destruction complex through activation of the protein Dishevelled. Then, β‐catenin is stabilized and translocated into the nucleus. This allows binding of β‐catenin to transcription factors such as lymphoid enhancing factor‐1 and T‐cell factor, and leads to transcriptional activation of multiple target genes like CYCLIN D1 and C‐MYC.( 1 , 2 , 3 , 4 )
The non‐canonical signaling pathway, although not completely understood, is independent of β‐catenin and includes two defined pathways: the planar cell polarity and Wnt/Ca2+ pathways.( 5 ) Non‐canonical signaling is often initiated by Wnt5a, Wnt4, and Wnt11 through Frizzled and Dishevelled and might involve G proteins.( 6 )
Deregulation of Wnt signaling has become a hallmark of various types of tumours, including leukemias.( 1 , 2 , 3 , 4 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 ) Several lines of evidence have shown that aberrant canonical Wnt signaling contributes to the pathogenesis of acute myeloid leukemia (AML): (i) the Wnt pathway is constitutively activated in AML with normal karyotype or low‐risk cytogenetics( 17 , 18 ) and also in cases with FLT 3 internal tandem duplication;( 19 ) (ii) β‐catenin is upregulated in AML, representing an independent prognostic factor associated with relapse and shorter survival;( 20 ) (iii) comparative expression analysis between human hematopoietic stem cells and leukemic stem cells from AML patients identified 3005 differentially expressed genes, most of them implicated in Wnt signaling;( 21 ) and (iv) recent studies have shown that impaired regulation of Wnt antagonists such as secreted Frizzled‐related protein (sFRP) genes by promoter hypermethylation contributes to Wnt pathway activation in AML.( 22 , 23 )
Wnt5a is a member of the Wnt family of proteins that signals through the non‐canonical Wnt/Ca2+ pathway to suppress cyclin D1 expression. WNT5A hemizygous mice develop myeloid leukemias and display loss of Wnt5a function in tumor tissues. Furthermore, analyses of human AML reveal loss of Wnt5a expression in the majority of patient samples, including those patients who retain WNT5A coding sequences.( 6 ) Taken together, these data suggest that WNT5A could function as a tumor suppressor gene in myeloid leukemogenesis and also that downregulation of this gene in AML might be dependent on epigenetic mechanisms similar to what we and others have previously demonstrated in acute lymphoblastic leukemia and other malignancies.( 24 , 25 ) In the present study, we demonstrate that epigenetic regulation of WNT5A by promoter methylation in AML is associated with the outcome of this group of patients.
Materials and Methods
Cell lines and patients. Eleven AML‐derived cell lines (MUTZ‐3, F‐39P, TF‐1, HL‐60, NOMO‐1, KG‐1, MV4‐II, MOLM‐13, KASUMI‐1, EOL‐1, and HEL) were purchased from the Deutche Sammlung von Microorganismen und Zellkulturen (Braunschweig, Germany). Cells were grown at 37°C under 5% CO2 in humidified air in RPMI medium (Gibco‐BRL, Burlington, ON, Canada) supplemented with 20% fetal bovine serum (Biochrome, NY, USA), 1% penicillin/streptomycin, and 1% HEPES (Gibco‐BRL).
The study population included 252 consecutive patients (137 men, 115 women) who were diagnosed with de novo non‐M3 AML between September 1998 and September 2008 at the Hospital Universitario La Fe of Valencia and Hospital Reina Sofia of Cordoba, Spain. Approval was obtained from the Institutional Review Board. Informed consent was provided according to the Declaration of Helsinki. The diagnosis was made according to the morphological and cytochemical criteria of the French–American–British (FAB) classification.( 26 ) The mean age at diagnosis was 57 years (range 8–92 years). The median follow‐up of the series was 46 months. The main clinical characteristics of the patients are listed in Table 1. Fitted patients were enrolled into intensive chemotherapy trials in which induction therapy comprised standard combinations of anthracycline plus cytarabine. Patients achieving complete remission (CR) proceeded to consolidation therapy and eligible patients were selected for autologous or allogeneic stem cell transplantation. Seventy‐seven patients received stem cell transplantation (38 autologous, 39 allogeneic). Sixty‐two patients from the whole series relapsed, and 71 remain alive.
Table 1.
Main clinical characteristics of the patients
| Characteristics | Non‐methylated (%) (n = 145) | Methylated (%) (n = 107) | P‐value |
|---|---|---|---|
| Age (years) | NS | ||
| ≤60 | 70 (48) | 62 (58) | |
| >60 | 75 (52) | 45 (42) | |
| Sex | NS | ||
| Male | 80 (55) | 57 (53) | |
| Female | 65 (45) | 50 (47) | |
| WBC (×109/L) | NS | ||
| ≤10 | 66 (46) | 56 (52) | |
| >10 | 79 (54) | 51 (48) | |
| FAB classification | NS | ||
| M0 | 10 (7) | 6 (6) | |
| M1 | 35 (24) | 27 (25) | |
| M2 | 38 (26) | 28 (26) | |
| M4 | 26 (18) | 10 (9) | |
| M5 | 22 (15) | 15 (14) | |
| M6 | 9 (6) | 16 (15) | |
| M7 | 1 (1) | 1 (1) | |
| Unclassified | 4 (3) | 4 (4) | |
| Cytogenetic risk group | NS | ||
| Low | 5 (3) | 13 (12) | |
| Intermediate | 78 (54) | 63 (59) | |
| High | 29 (20) | 24 (22) | |
| Molecular alteration | NS | ||
| FLT3‐ITD | 20 (14) | 18 (17) | |
| NPM1 | 25 (17) | 12 (11) |
FLT3, Internal tandem duplication; NPM, nucleophosmin; NS, not significant; WBC, white blood cells.
Cytogenetic and molecular analysis. Karyotype analysis was carried out using unstimulated short‐term cultures according to the recommendations of the International System for Human Cytogenetics.( 27 ) Whenever possible, at least 20 metaphases were evaluated. Cytogenetic risk groups were defined as follows: high risk, –5/del(5q), –7/del(7q), abn 3q, complex aberrations (≥3 independent aberrations), t(9;22) and t(6;9); low risk, t(8;21) and inv(16); intermediate risk, all other karyotypic aberrations or a normal karyotype. FLT3 internal tandem duplications and nucleophosmin gene mutations (NPM1) were studied in cDNA samples following the methods described by Nakao et al. ( 28 ) and Schnittger et al. ( 29 ), respectively.
Methylation‐specific PCR. Analysis of WNT5A (GenBank: NC_000003) has revealed that WNT5A promoter and exon 1 possess a long 1246‐bp CpG island located between nt48 and nt1294, showing >60% C + G content and an observed‐over‐expected CpG frequency of >0.6. The methylation status of the CpG island in the WNT5A gene promoter was anaylsed by genomic DNA bisulfite treatment followed by methylation‐specific PCR (MSP) as reported by Herman et al. ( 30 ) Three different regions of this CpG island were analyzed for methylation (Fig. S1). The primers for the methylated (M) and unmethylated (U) promoter regions are reported in Figure S1. ‘Hot start’ PCR was carried out for 30 cycles consisting of denaturation at 95°C for 1 min, annealing at 60°C for 1 min, and extension at 72°C for 1 min, followed by a final 7‐min extension for all primer sets. The products were separated by electrophoresis on a 2% agarose gel. Bone marrow mononuclear cell DNA from healthy donors was used as a negative control for methylation‐specific assays. Human male genomic DNA universally methylated for all genes (Intergen Company, Purchase, NY, USA) was used as a positive control for methylated alleles. Water blanks were included with each assay. Results were always confirmed by repeat MSP assays after an independently performed bisulfite treatment.
Expression analyses. Expression of WNT5A and the WNT5A target gene CYCLIN D1 was analyzed by the RT‐PCR technique. Total RNA was extracted from marrow samples with Ultraspec (Biotecx, Houston, TX, USA) following the manufacturer’s instructions. Reverse transcription was carried out on 1 μg total RNA, after heating at 70°C for 5 min, with random hexamers as the reaction primer. The reaction was carried out at 42°C for 45 min in the presence of 12 U Avian Myeloblastosis virus reverse transcriptase. Quantitative real‐time PCR (qrt‐PCR) for gene expression was carried out with LightCycler technology, using 1 μL of cDNA in a 20‐μL reaction, 0.5 μmol/L of each primer and 2 μL of 10 × LightCycler FastStar DNA Master SYBR Green I (Roche Molecular Biochemicals, Barcelona, Spain). The final Mg2+ concentration in the reaction mixture was adjusted to 3.5 mmol/L. Primers were: for WNT5A expression, forward 5′‐CCACATGCAGTACATCGGAG‐3′ and reverse 5′‐TGCCGGAAGTGTATGCG‐3′; and for CYCLIN D1 expression, forward 5′‐CCCTCGGTGTCCTACTTCAAATGT‐3′ and reverse 5′‐TGATCTGTTTGTTCTCCTCCGCCT‐3′. The following program conditions were applied for qrt‐PCR running: denaturation program, one cycle at 95°C for 8 min; amplification program, 45 cycles at 95°C for 5 s, 60°C for 10 s, and 72°C for 15 s; melting program, one cycle at 95°C for 0 s, 40°C for 60 s, and 90°C for 0 s; and cooling program, one cycle at 40°C for 60 s. The temperature transition rate was 20°C/s, except in the melting program, which was 0.4°C/s between 40 and 90°C. The Abelson gene (ABL1) was used as a reference gene, and it was amplified in the same run and following the same procedure described above (forward 5′‐CCCAACCTTTTCGTTGCACTGT‐3′; reverse 5′‐CGGCTCTCGGAGGAGACGTAGA‐3′). In order to reduce the variation between different assays and samples, a procedure based on the relative quantification of target genes versus their controls/calibrators in relation to the reference gene was used. Calculations were automatically carried out by LightCycler software (RealQuant, version 1.0; Roche Molecular Biochemicals). The normalized ratios (N) for each gene were obtained from the next equation and expressed as a percentage of the control/calibrator:
The efficiencies (E) of each gene were calculated from the slopes of crossover points (Cp) versus the DNA concentration plot, according to the formula E = 10(−1/slope). ΔCp corresponded to the difference between control/calibrator Cp and sample Cp, either for the target or for the reference sequences. The selected controls/calibrators were normal marrow mononuclear cells from healthy donors. They were considered as 100% expression.
Wnt5a CpG island analysis by sequencing after sodium bisulfite modification. The methylation status of WNT5A CpG islands was analysed by bisulfite genomic sequencing in HL‐60, KASUMI‐1, and HEL AML‐derived cell lines and bone marrow mononuclear cells DNA from healthy donors. Genomic DNA (1 μg) was treated and modified using the CpGenomic DNA Modification Kit (Intergen Company). Human male genomic DNA universally methylated for all genes (Intergen Company) was used as a positive or methylated control. After bisulfite modification, region 2 from Wnt5a CpG island was amplified by PCR using 3 μL of modified DNA and Wnt5a‐SB1 (5′‐GGGGTTGATTTTTGTAGTTTAGATG‐3′) and Wnt5a‐SB2 (5′‐TTAAAACTTTCCAACCCCAAATATA‐3′) primers under the following conditions: 94°C for 10 min, 35 cycles at 94°C for 1 min, 57°C for 1 min, and 72°C for 1 min, and a final elongation cycle at 72°C for 10 min. PCR were carried out in a total volume of 25 μL, with 1 U high‐fidelity Platinum Taq DNA polymerase (Invitrogen Life Technologies, Paisley, UK), 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, and 50 pmol of each primer. The PCR products were separated on a 2% agarose gel, stained with ethidium bromide, and visualized under UV light. Amplification products, containing 21 CpG dinucleotides, were subcloned into the pCR 4‐TOPO plasmid using TOPO TA Cloning Kit for Sequencing (Invitrogen Life Technologies) and transformed into Escherichia coli according to the manufacturer’s recommendations. Colonies with recombinant plasmids containing the described PCR products were screened by digestion with EcoRI (Amersham Biosciences, Buckinghamshire, UK). Candidate plasmid clones were sequenced using T7 and T3 universal forward and reverse primers.
Treatment with the demethylating agent 5‐Aza‐2′‐deoxycytidine. AML‐derived KAG‐1 and HEL cell lines were grown at a density of 1 × 106 cells/mL in 25‐cm2 flasks with 10 mL of RPMI‐1640 medium supplemented with 20% fetal bovine serum and maintained at 37°C in a humid atmosphere containing 5% CO2. Cell lines were treated with 4 μmol/L of 5‐Aza‐2′‐deoxycytidine (DAC; Sigma‐Aldrich, Steinheim, Germany) for 4 days. After treatment, cells were washed with PBS, pelleted by centrifugation at 1500g for 5 min, and used for genomic DNA and RNA isolation. DNA was extracted using the QIAmp DNA Mini Kit (Qiagen, Hilden, Germany) and total RNA using Rneasy Mini Kit (Qiagen). Total RNA (1 μg) was used for cDNA synthesis using SuperScript II RNase H‐RT (Invitrogen Life Technologies) with random hexamers.
Statistical analysis. For statistical purposes, AML patients were classified into two different groups according to the methylation status of the Wnt5a promoter (methylated and non‐methylated groups). All descriptive statistics and tests (Mann–Whitney non‐parametric U‐test, χ2 and Fisher’s exact test) were calculated using the statistical package SPSS (Chicago, IL, USA) 11.0. A P < 0.05 was considered statistically significant. Unadjusted time‐to‐event analyses were carried out using the Kaplan–Meier method and log‐rank tests for comparisons. Overall survival (OS) was measured from the time of diagnosis to the time of last follow‐up or death from any cause. Disease‐free survival (DFS) was measured from the date of CR. In the analysis of DFS, relapse and death were considered uncensored events, whichever occurred first. All P‐values reported are two‐sided. Multivariate analysis was carried out using the Cox proportional hazards model. The follow up of the patients was updated on January 2009 and all follow‐up data were censored at that point.
Results
Expression of the WNT5A gene in AML‐derived cell lines is regulated by promoter hypermethylation. Quantitative expression of WNT5A transcripts was assessed by means of qrt‐PCR using cDNA from healthy donors as a control (it was considered as 100% of Nwnt5a ratio). Low levels of WNT5A expression were observed in seven AML cell lines (Fig. 1A). Among all the possible mechanisms of transcription regulation, we first decided to study the possible hypermethylation of the WNT5A promoter as we have previously demonstrated that WNT5A is a target for epigenetic regulation in leukemia.( 24 ) By MSP, region 2 of the WNT5A promoter was revealed to be methylated in AML‐derived cell lines with low WNT5A expression (Fig. 1B), which was clearly in contrast with the lack of WNT5A promoter methylation in this region observed in healthy MNC or AML cell lines with normal WNT5A expression (Fig. 1B). Methylation of regions 1 and 3 was observed in some cell lines but was always associated with region 2 methylation and never as an isolated event (Fig. 1B). Moreover, in order to confirm the results of the MSP, we screened bone marrow DNA from healthy donors and three AML‐derived cell lines (HL‐60, KASUMI‐1, and HEL) using bisulfite genomic sequencing. As described in ‘Materials and Methods’, the amplification products of the WNT5A promoter, which are composed of 21 CpG dinucleotides, were subcloned and sequenced. Genomic sequencing after bisulfite modification revealed that DNA from healthy donors and the HEL cell line was unmethylated. In contrast, HL‐60 and KASUMI‐1 cell lines were heavily methylated in almost all analysed CpG dinucleotides and clones (Fig. 1C). Exposure of AML cell lines to the demethylating agent DAC restored WNT5A expression in KASUMI‐1 cell lines that showed hypermethylation of the WNT5A promoter but it had not effect in the HEL cell line that showed an unmethylated promoter (Fig. 1D). These results indicate that WNT5A is downregulated in AML at least in part by hypermethylation and that region 2 of the WNT5A promoter is critical for this purpose.
Figure 1.
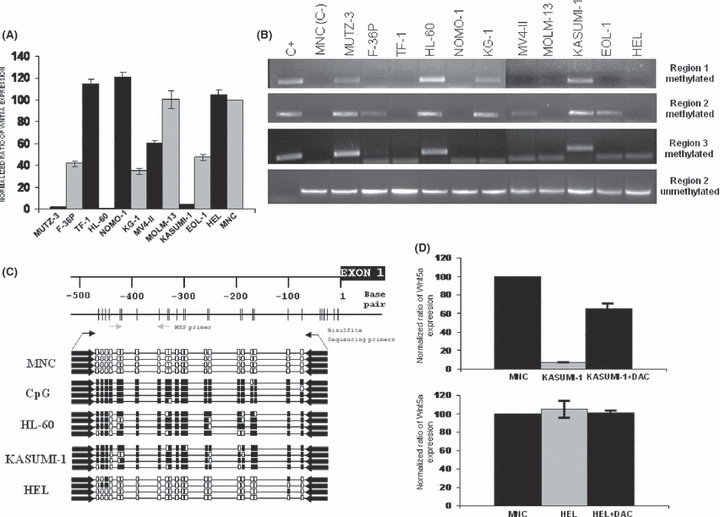
Expression and methylation analysis of the WNT5A gene in acute myeloid leukemia (AML)‐derived cell lines. (A) Quantitative real‐time PCR expression of WNT5A in AML‐derived cell lines demonstrating downregulation of gene expression in seven of the 11 cell lines studied. Gene expression was normalized using the expression in bone marrow mononuclear cells (BM‐MNC) from 30 healthy controls (normalized ratio = 100%). The figure represents the mean of three different studies in triplicate. (B) Methylation‐specific PCR (MSP) analysis of WNT5A in the same AML‐derived cell lines. Region 2 of WNT5A was methylated in all of the cell lines that had downregulated WNT5A expression. C+, positive methylated control; C–, BM‐MNC cells from healthy donors. (C) Schematic description of the WNT5A CpG island region 2. Each vertical bar represents a CpG dinucleotide. The gray arrows show the location of the MSP primers and the black arrows the location of bisulfite sequencing primers. The figure also represents the analysis of WNT5A CpG island methylation status by bisulfite sequencing in AML‐derived cell lines. Each box indicates a CpG dinucleotide (white box, unmethylated; black box, methylated) and each line represents the analysis of 21 CpG dinucleotides of a single clone of the WNT5A analysed region. (D) Expression analysis of the Wnt5a gene in the methylated KASUMI‐1 cell line and in the unmethylated HEL cell line, before and after treatment with the demethylating agent 5‐Aza‐2′‐deoxycytidine (DAC) demostrating upregulation of the gene after treatment in AML‐derived cell line that was methylated. Gene expression was normalized with expression in BM‐MNC from healthy controls (normalized ratio = 100%). The figure represents the mean of three different studies in triplicate.
WNT5A hypermethylation in AML patients is associated with decreased expression of WNT5A and upregulation of CYCLIN D1. To establish a more clinically relevant model of the role of the epigenetic regulation of WNT5A, we analyzed the methylation status of the three regions of the WNT5A CpG island by MSP in a group of 252 AML patients (Table 1). The methylation frequencies were as follows: 43% for region 2; 15% for region 3; and 11% for region 1. Unmethylated WNT5A was found in 145 of 252 patients (non‐methylated group, 57%) whereas 107 (43%) of 252 AML had methylation of the WNT5A CpG island (methylated group). In line with the studies in cell lines, region 2 methylation was always present in methylated patients. Expression of WNT5A was downregulated in patients with methylation of WNT5A in comparison with non‐methylated patients (41 ± 40%vs 250 ± 249%; P < 0.001; Fig. 2A).
Figure 2.
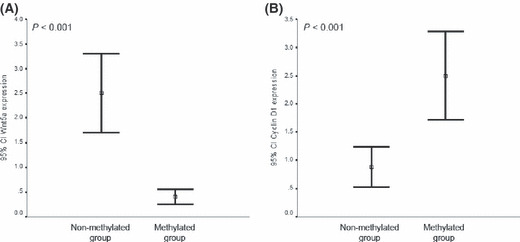
Expression of WNT5A and CYCLIN D1 genes in samples from acute myeloid leukemia (AML) patients. (A) Expression analysis of the WNT5A gene in AML patients. Methylated patients showed downregulation of WNT5A expression. Bars represent the mean expression (95% CI) in AML patients in comparison with the expression in Bone marrow mononuclear cells (BM‐MNC) from 30 healthy controls (normalized ratio = 100%). (B) Hypermethylation of the WNT5A gene is associated with upregulation of CYCLIN D1 in AML patients. Bars represent the mean expression (95% CI) in AML patients in comparison with the expression in BM‐MNC from 30 healthy controls (normalized ratio = 100%).
Recent studies have suggested that besides activating the canonical Wnt signaling, WNT5A may signal through the non‐canonical Wnt/Ca2+ pathway to suppress CYCLIN D1 expression.( 6 ) Consistent with this hypothesis, transcript levels of CYCLIN D1 (mean normalized ratio: 250 ± 210%vs 88 ± 114%, P < 0.001) were significantly higher among WNT5A methylated patients compared with non‐methylated patients (Fig. 2B).
WNT5A methylation, clinical presentation, and outcome. As shown in Table 1, aberrant WNT5A methylation was not associated with age, sex, white blood cell count, FAB classification, cytogenetic risk groups, or FLT3 or NPM1 mutations. Two hundred and one patients (80%) were eligible for intensive therapy. One hundred and thirty‐five patients (54%) achieved CR (124 after one course of induction therapy and 11 after a second cycle). No effect of the WNT5A methylation status on the response to induction therapy was observed (Table 2). However, patients in the non‐methylated group had a lower relapse rate than patients in the methylated group (15%vs 37%, P < 0.001; Table 2). The mortality rate was also lower for the non‐methylated group compared with the methylated group (61%vs 79%, P = 0.004; Table 2).
Table 2.
Clinical outcome of de novo myeloid acute leukemia patients
| Characteristics | Non‐methylated n (%) | Methylated n (%) | P‐value |
|---|---|---|---|
| Induction response | NS | ||
| Complete remission | 72 (50) | 63 (59) | |
| Failure | |||
| Resistance | 21 (14) | 18 (17) | |
| Death | 19 (13) | 8 (7) | |
| Not treated | 33 (23) | 18 (17) | |
| Post‐induction therapy | NS | ||
| Chemotherapy only | 66 (46) | 56 (52) | |
| Autologous SCT | 22 (15) | 16 (15) | |
| Allogenic SCT | 22 (15) | 17 (16) | |
| Relapse | 22 (15) | 40 (37) | <0.001 |
| Death | 89 (61) | 85 (79) | 0.004 |
NS, not significant; SCT, stem cell transplantation.
We analyzed the DFS among patients who achieved CR according to WNT5A methylation. Estimated DFS rates at 7 years were 60% and 20% for normal and methylated patients, respectively (P < 0.0001) (Fig. 3A). Among patients included in the cytogenetic low‐risk group, 5‐year DFS was 75% for unmethylated patients in contrast to 18% for methylated patients (P = 0.02; Fig. 3B). Among intermediate‐risk group patients, 6‐year DFS was 18% for methylated cases and 65% for normal cases (P < 0.0001; Fig. 3C). When the analysis was restricted to patients with high‐risk cytogenetics, the methylated group had a significantly reduced DFS (15%vs 69%, P = 0.005) at 1 year than the non‐methylated group (Fig. 3D).
Figure 3.
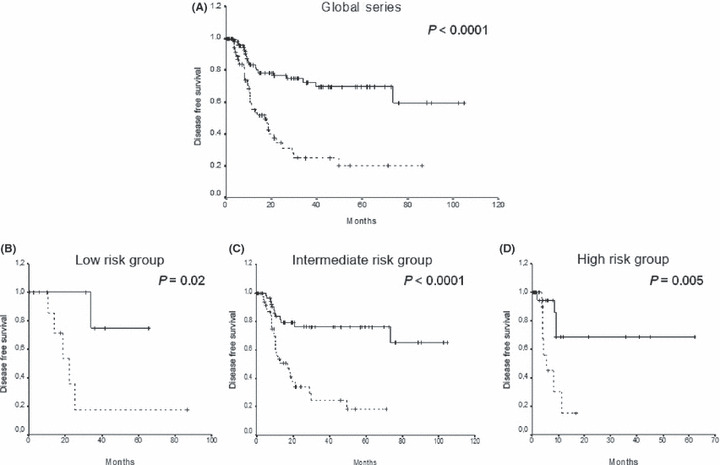
Kaplan–Meier disease‐free survival (DFS) function for acute myeloid leukemia (AML) patients. DFS curves for (A) all of the patients included in this study, and for patients classified into cytogenetic (B) low‐risk, (C) intermediate‐risk, and (D) high‐risk groups according to the WNT5A methylation status. Solid lines, WNT5A unmethylated patients. Dashed lines, WNT5A methylated patients.
The actual OS calculated for all leukemic patients was 27% and 0% at 9 years for cases with normal and hypermethylated WNT5A gene, respectively (P = 0.04; Fig. 4A). OS was also significantly different between normal and methylated patients in a separate analysis of the cytogenetic low‐risk group (100%vs 28%, P = 0.01; Fig. 4B) and intermediate‐risk group (34%vs 0%, P = 0.007; Fig. 4C) but not in the high‐risk group (20%vs 0%, P = not significant; Fig. 4D).
Figure 4.
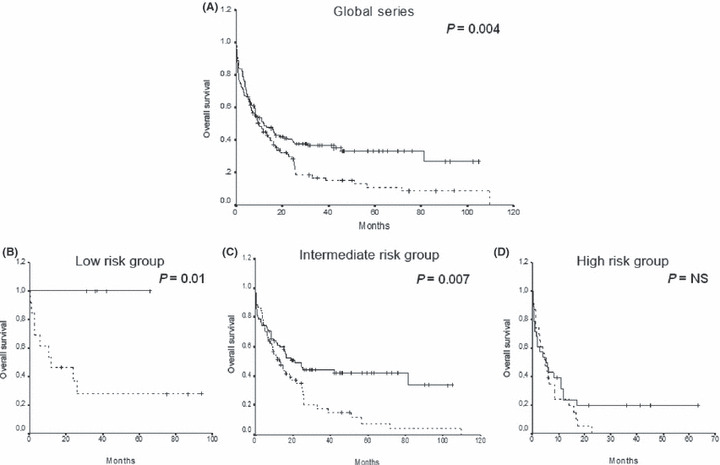
Kaplan–Meier overall survival (OS) function for acute myeloid leukemia (AML) patients. OS curves for (A) all of the patients included in this study, and for patients classified into cytogenetic (B) low‐risk, (C) intermediate‐risk, and (D) high‐risk groups according to the WNT5A methylation status. Solid lines, WNT5A unmethylated patients. Dashed lines, WNT5A methylated patients.
Multivariate modelling of the global series took into account all prognostic variables including age, sex, FAB subtype, leukocytosis, cytogenetic risk group, and FLT3 mutations, and the methylation profile showed that age over 60 years, cytogenetics, and WNT5A methylation status were independent prognostic factors for OS and DFS (Table S1).
Discussion
In the present study, we show for the first time that the WNT5A promoter is subject to methylation silencing in AML cell lines. We analyzed its methylation by MSP and detected methylation in all cell lines with downregulated WNT5A expression. Detailed methylation analysis with bisulfite genomic sequencing confirmed the MSP analysis and revealed a high density of methylated CpG sites in these cell lines. After exposure to DAC, WNT5A expression was dramatically increased in silenced cell lines, indicating that epigenetic mechanisms seemed to deregulate the expression of WNT5A in AML. Importantly, WNT5A methylation was also observed in primary tumor samples from a large series of AML patients. Methylated WNT5A was observed in 43% of our patients at diagnosis, supporting the role of WNT5A methylation in the early phases of myeloid leukemogenesis.
WNT5A is located at 3p14, a commonly deleted tumor suppressor locus in multiple tumors. Wnt5a has been classified as a non‐canonical and non‐transforming Wnt protein,( 31 ) with its role in tumorigenesis still ambiguous. There is evidence indicating that increased WNT5A expression is important for cancer progression, and that WNT5A was initially proposed as a proto‐oncogene.( 32 ) WNT5A has been shown to be a potent enhancer of cell motility and invasiveness of melanoma,( 33 ) upregulated in cancers of the lung, breast, stomach, and prostate.( 34 ) On the other hand, in other tumor models, including brain, breast, thyroid, and uroepithelial cancers, WNT5A has been shown to inhibit tumor cell proliferation,( 6 , 35 , 36 ) with its expression being a good prognostic marker for patients with breast and colon cancer.( 37 , 38 ) These results not only suggest that dysregulation of WNT5A expression is involved in tumor pathogenesis but also that the function of WNT5A either as a suppressor or as a promoter of malignant progression is cell type specific.
In hematopoietic tissues, we and others have shown that WNT5A is frequently silenced by methylation in lymphoid malignancies in a tumor‐specific manner, including natural killer/T‐cell lymphomas, acute lymphoblastic leukemia, and Burkitt lymphomas but not in normal lymph nodes and peripheral blood mononuclear cells.( 24 , 39 ) Further functional studies showed that ectopic expression of WNT5A resulted in inhibition of tumor cell growth and clonogenicity in lymphoid cell lines, consistent with previous studies showing that WNT5A inhibits B‐cell proliferation and WNT5A heterozygous mice develop B‐cell lymphomas,( 6 ) suggesting that WNT5A could serve as a tumor suppressor in lymphoid neoplams. However, the role of non‐canonical Wnt signaling in myeloid leukemias is unknown and still controversial. WNT5A hemizygous mice develop myeloid leukemias and deletion of the WNT5A gene, loss of WNT5A expression, and increased cyclin D1 expression is observed in some AML patient samples.( 6 ) Moreover, it has been recently demonstrated that mRNA expression of some of the Wnt antagonists is upregulated in response to apoptotic stimuli in myeloid leukemia cell lines. The correlation between the increased expression of non‐canonical Wnt genes and apoptotic stimulation in the HL‐60 AML‐derived cell line is a novel observation suggesting the possible involvement of the non‐canonical Wnt pathway in executing programmed cell death.( 40 ) These data and our present finding that methylation of WNT5A was present in 43% of AML patients with downregulated WNT5A expression further supports the hypothesis that WNT5A also functions as tumor suppressor gene in myeloid leukemogenesis.
In AML, hypermethylation of CpG islands in the promoter region is an epigenetic pathway that appears to be common to several genes involved in cell‐cycle control, apoptosis and signaling.( 41 ) Among the genes hypermethylated in AML, those that antagonize canonical Wnt signaling seem to play an important role. In that sense, we have recently demonstrated that expression of the Wnt inhibitors sFRP1, sFRP2, sFRP4, sFRP5, DKK ‐1, and DKK‐3 is downregulated due to abnormal promoter methylation in AML samples and this event correlates with constitutive activation of the canonical Wnt signaling pathway and upregulation of the CYCLIN D1 target gene in AML.( 23 ) Our present work completes the epigenetic picture of the Wnt pathways and shows that silencing of the WNT5A gene by inappropriate methylation is associated with high levels of CYCLIN D1 expression and has clinical consequences conferring a dismal prognosis in AML. However, one can speculate that the upregulation of CYCLIN D1 observed in our AML patients depends only on the activation of canonical Wnt signaling being the methylation of WNT5A an innocent bystander of the more extensive methylation of the canonical Wnt‐antagonists.( 23 ) Further studies are ongoing in our laboratory in order to rule out this point. In the present study we show that methylation of WNT5A provides important prognostic information in AML. The presence in individual tumors of WNT5A epigenetic events is a factor of poor prognosis in all AML cytogenetic risk groups. Patients with WNT5A methylation had a poorer DFS and OS than unmethylated patients. Multivariate analysis confirmed that methylation profile was associated with a shorter DFS and OS.
AML is a heterogeneous disease with different clinical behaviours, of which cytogenetics is the most important prognostic factor. In the present paper, we demonstrate that methylation status is able to redefine the prognosis of selected AML groups with well‐established cytogenetic features. Lack of WNT5A methylation improved the general poor outcome of patients presenting high‐risk cytogenetic abnormalities, whereas presence of methylation worsened the general good outcome of low‐risk cytogenetic patients. However, the most important finding of our report appears in the so‐called intermediate‐risk group, which comprised a heterogeneous group of patients with apparently normal karyotypes or patients with a variety of other aberrations whose prognostic outcome was uncertain. In this group of patients, better identification of prognostic indicators has been of particular interest in the past few years. FLT3 mutations have appeared as an important unfavorable prognostic factor in this subgroup, allowing the classification of this vast group of AML patients with normal karyotype into two categories with distinct responses to treatment.( 42 ) Our data show that the methylation profile of WNT5A is a new poor prognostic factor in the subgroup of patients with intermediate‐risk cytogenetics. Therefore, WNT5A methylation profiling in AML could have important clinical implications complementing standard cytogenetic and molecular studies for guiding the selection of therapy and providing a basis for developing novel therapies.
In conclusion, we have demonstrated that WNT5A is silenced by methylation in AML and that this epigenetic event is associated with high CYCLIN D1 expression and confers poor prognosis in this group of patients regardless of the cytogenetic features.
Authorship and Disclosure Statement
JR‐G, XA and FP contributed to the conception and design of the study, and to the analysis and interpretation of data; AV, JC, MAS, AT and CH provided the study materials and revised the article; JR‐G, XA, VM, ES‐E, PR‐O and AV‐Z carried out the majority of the experiments and drafted the article.
The authors report no potential conflicts of interest.
Supporting information
Fig. S1. Sequence of the WNT5A promoter CpG island after bisulfite modification showing the three regions studied and the primers for methylation‐specific PCR analyses.
Table S1. Multivariate analysis for overall survival (OS) and disease‐free survival (DFS).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Acknowledgments
This work was supported by grants from Fondo de Investigacion Sanitaria (FIS, Spain) 07/0602, 07/0608, 06/0003, 06/0285, CP07/00215; Beca Ortiz de Landazuri 2006, Departamento de Salud‐Gobierno de Navarra; ISCIII‐RETIC RD06/0020, Junta de Andalucia 0004/2007; and by funds from ‘UTE project CIMA’ and Asociacion Medicina e Investigacion. Vanesa Martin was supported by a Rio Hortega fellowship from Instituto Carlos III (Spain). We thank M.D. Odero for providing the AML‐derived cell lines.
References
- 1. Kestler HA, Kühl M. From individual Wnt pathways towards a Wnt signalling network. Philos Trans R Soc Lond B Biol Sci 2008; 363: 1333–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008; 8: 387–98. [DOI] [PubMed] [Google Scholar]
- 3. Clevers H. Wnt/beta‐catenin signalling in development and disease. Cell 2006; 127: 469–80. [DOI] [PubMed] [Google Scholar]
- 4. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–50. [DOI] [PubMed] [Google Scholar]
- 5. Kühl M. The WNT/calcium pathway: biochemical mediators, tools and future requirements. Front Biosci 2004; 9: 967–74. [DOI] [PubMed] [Google Scholar]
- 6. Liang H, Chen Q, Coles AH et al. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003; 4: 349–60. [DOI] [PubMed] [Google Scholar]
- 7. Suzuki H, Watkins DN, Jair KW et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 2004; 36: 417–22. [DOI] [PubMed] [Google Scholar]
- 8. Mazieres J, He B, You L et al. Wnt inhibitory factor‐1 is silenced by promoter hypermethylation in human lung cancer. Cancer Res 2004; 64: 4717–20. [DOI] [PubMed] [Google Scholar]
- 9. Yau TO, Chan CY, Chan KL et al. HDPR1, a novel inhibitor of the WNT/beta‐catenin signaling, is frequently downregulated in hepatocellular carcinoma: involvement of methylation‐mediated gene silencing. Oncogene 2005; 24: 1607–14. [DOI] [PubMed] [Google Scholar]
- 10. Liu TH, Raval A, Chen SS, Matkovic JJ, Byrd JC, Plass C. CpG island methylation and expression of the secreted frizzled‐related protein gene family in chronic lymphocytic leukemia. Cancer Res 2006; 66: 653–8. [DOI] [PubMed] [Google Scholar]
- 11. Nojima M, Suzuki H, Toyota M et al. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 2007; 26: 4699–713. [DOI] [PubMed] [Google Scholar]
- 12. Jiang X, Tan J, Li J et al. DACT3 is an epigenetic regulator of Wnt/beta‐catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell 2008; 13: 529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee EJ, Jo M, Rho SB et al. Dkk3, downregulated in cervical cancer, functions as a negative regulator of beta‐catenin. Int J Cancer 2009; 124: 287–97. [DOI] [PubMed] [Google Scholar]
- 14. Roman‐Gomez J, Jimenez‐Velasco A, Agirrex et al. Transcriptional silencing of the Dickkopfs‐3 (Dkk‐3) gene by CpG hypermethylation in acute lymphoblastic leukaemia. Br J Cancer 2004; 91: 707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Román‐Gómez J, Cordeu L, Agirre X et al. Epigenetic regulation of Wnt‐signaling pathway in acute lymphoblastic leukemia. Blood 2007; 109: 3462–9. [DOI] [PubMed] [Google Scholar]
- 16. Martin V, Agirre X, Jiménez‐Velasco A et al. Methylation status of Wnt signaling pathway genes affects the clinical outcome of Philadelphia‐positive acute lymphoblastic leukemia. Cancer Sci 2008; 99: 1865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simon M, Grandage VL, Linch DC, Khwaja A. Constitutive activation of the Wnt/beta‐catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005; 24: 2410–20. [DOI] [PubMed] [Google Scholar]
- 18. Mikesch JH, Steffen B, Berdel WE, Serve H, Müller‐Tidow C. The emerging role of Wnt signaling in the pathogenesis of acute myeloid leukemia. Leukemia 2007; 21: 1638–47. [DOI] [PubMed] [Google Scholar]
- 19. Tickenbrock L, Schwäble J, Wiedehage M et al. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood 2005; 105: 3699–706. [DOI] [PubMed] [Google Scholar]
- 20. Ysebaert L, Chicanne G, Demur C et al. Expression of beta‐catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia 2006; 20: 1211–6. [DOI] [PubMed] [Google Scholar]
- 21. Majeti R, Becker MW, Tian Q et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci USA 2009; 106: 3396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jost E, Schmid J, Wilop S et al. Epigenetic inactivation of secreted Frizzled‐related proteins in acute myeloid leukaemia. Br J Haematol 2008; 142: 745–53. [DOI] [PubMed] [Google Scholar]
- 23. Valencia A, Roman‐Gomez J, Cervera J et al. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia 2009; 23: 1658–66. [DOI] [PubMed] [Google Scholar]
- 24. Roman‐Gomez J, Jimenez‐Velasco A, Cordeu L et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur J Cancer 2007; 43: 2736–46. [DOI] [PubMed] [Google Scholar]
- 25. Ying J, Li H, Yu J et al. WNT5A exhibits tumor‐suppressive activity through antagonizing the Wnt/beta‐catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res 2008; 14: 55–61. [DOI] [PubMed] [Google Scholar]
- 26. Benett JM, Catovsky D, Daniel MT et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French–American–British Cooperative group. Br J Haematol 1976; 33: 451–8. [DOI] [PubMed] [Google Scholar]
- 27. ISCN . Guidelines for cancer cytogenetics. In: Shaffer LG, Tommerup N, eds. Supplement to: An International System for Human Cytogenetic Nomenclature. Karger: Basel, 2005; 1–157. [Google Scholar]
- 28. Nakao M, Yokota S, Iwai T et al. Internal tandem duplications of the FLT3 gene found in acute myeloid leukemia. Leukemia 1996; 10: 1911–8. [PubMed] [Google Scholar]
- 29. Schnittger S, Schoch C, Kern W et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 2005; 106: 3733–9. [DOI] [PubMed] [Google Scholar]
- 30. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wong GT, Gavin BJ, McMahon AP. Differential transformation of mammary epithelial cells by Wnt genes. Mol Cell Biol 1994; 14: 6278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clark CC, Cohen I, Eichstetter I et al. Molecular cloning of the human proto‐oncogene Wnt‐5A and mapping of the gene (WNT5A) to chromosome 3p14‐21. Genomics 1993; 18: 249–60. [DOI] [PubMed] [Google Scholar]
- 33. Weeraratna AT, Jiang Y, Hostetter G et al. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002; 1: 279–88. [DOI] [PubMed] [Google Scholar]
- 34. Iozzo RV, Eichstetter I, Danielson KG. Aberrant expression of the growth factor Wnt‐5A in human malignancy. Cancer Res 1995; 55: 3495–9. [PubMed] [Google Scholar]
- 35. Kremenevskaja N, Von WR, Rao AS et al. Wnt‐5a has tumor suppressor activity in thyroid carcinoma. Oncogene 2005; 24: 2144–54. [DOI] [PubMed] [Google Scholar]
- 36. Olson DJ, Gibo DM, Saggers G, Debinski W, Kumar R. Reversion of uroepithelial cell tumorigenesis by the ectopic expression of human wnt‐5a. Cell Growth Differ 1997; 8: 417–23. [PubMed] [Google Scholar]
- 37. Dejmek J, Dejmek A, Safholm A, Sjolander A, Andersson T. Wnt‐5a protein expression in primary dukes B colon cancers identifies a subgroup of patients with good prognosis. Cancer Res 2005; 65: 9142–6. [DOI] [PubMed] [Google Scholar]
- 38. Jonsson M, Dejmek J, Bendahl PO, Andersson T. Loss of Wnt‐5a protein is associated with early relapse in invasive ductal breast carcinomas. Cancer Res 2002; 62: 409–16. [PubMed] [Google Scholar]
- 39. Ying J, Li H, Chen Y‐W, Srivastava G, Gao Z, Tao Q. WNT5A is epigenetically silenced in hematological malignancies and inhibits leukemia cell growth as a tumor suppressor. Blood 2007; 110: 4130–1. [DOI] [PubMed] [Google Scholar]
- 40. Sercan HO, Pehlivan M, Simsek O, Ates H, Sercan Z. Induction of apoptosis increases expression of non‐canonical WNT genes in myeloid leukemia cell lines. Oncol Rep 2007; 18: 1563–9. [PubMed] [Google Scholar]
- 41. Desmond JC, Raynaud S, Tung E, Hofmann WK, Haferlach T, Koeffler HP. Discovery of epigenetically silenced genes in acute myeloid leukemias. Leukemia 2007; 21: 1026–34. [DOI] [PubMed] [Google Scholar]
- 42. Moreno I, Martín G, Bolufer P et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukaemia. Haematologica 2003; 88: 19–24. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Sequence of the WNT5A promoter CpG island after bisulfite modification showing the three regions studied and the primers for methylation‐specific PCR analyses.
Table S1. Multivariate analysis for overall survival (OS) and disease‐free survival (DFS).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item