

Abstract
α1,6‐Fucosyltransferase (Fut8), an enzyme that catalyzes the introduction of α1,6 core fucose to the innermost N‐acetylglucosamine residue of the N‐glycan, has been implicated in the development, immune system, and tumorigenesis. We found that α1,6‐fucosyltransferase and E‐cadherin expression levels are significantly elevated in primary colorectal cancer samples. Interestingly, low molecular weight population of E‐cadherin appeared as well as normal sized E‐cadherin in cancer samples. To investigate the correlation between α1,6‐fucosyltransferase and E‐cadherin expression, we introduced α1,6‐fucosyltransferase in WiDr human colon carcinoma cells. It was revealed that the low molecular weight population of E‐cadherin was significantly increased in α1,6‐fucosyltransferase‐transfected WiDr cells in dense culture, which resulted in an enhancement in cell–cell adhesion. The transfection of mutated α1,6‐fucosyltransferase with no enzymatic activity had no effect on E‐cadherin expression, indicating that core fucosylation is involved in the phenomena. In α1,6‐fucosyltransferase knock down mouse pancreatic acinar cell carcinoma TGP49 cells, the expression of E‐cadherin and E‐cadherin dependent cell–cell adhesion was decreased. The introduction of α1,6‐fucosyltransferase into kidney epithelial cells from α1,6‐fucosyltransferase–/– mice restored the expression of E‐cadherin and E‐cadherin‐dependent cell–cell adhesion. Based on the results of lectin blotting, peptide N‐glycosidase F treatment, and pulse‐chase studies, it was demonstrated that the low molecular weight population of E‐cadherin contains peptide N‐glycosidase F insensitive sugar chains, and the turnover rate of E‐cadherin was reduced in α1,6‐Fucosyltransferase transfectants. Thus, it was suggested that core fucosylation regulates the processing of oligosaccharides and turnover of E‐cadherin. These results suggest a possible role of core fucosylation in the regulation of cell–cell adhesion in cancer. (Cancer Sci 2009; 100: 888–896)
Abbreviations:
- AAL
Aleuria aurantia lectin
- BSA
bovine serum albumin
CHO, Chinese hamster ovary
- ConA
Concanavalia ensiformis
- DMEM
Dulbecco's modified Eagle's medium
- DSA
Datura stramonium lectin
- EDTA
ethylenediamine tetraacetic acid
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- FcγRIIIA
Fcγ receptor IIIA
- Fut8
α1,6‐fucosyltransferase
- GAPDH
glyceraldehydes‐3‐phosphate dehydrogenase
- GDP‐Fucose
guanosine diphosphate‐fucose
- GlcNAc
N‐acetyl glucosamine
- GnT‐III
N‐acetylglucosaminyltransferase III
- LRP‐1
lipoprotein receptor‐related protein‐1
- MDCK
Madin‐Darby canine kidney
- MES‐NaOH
2‐morpholinoethane sulfonic acid, memohydrate
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PNGase F
peptide N‐glycosidase F
- SDS‐PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TGF‐β1
transforming growth factor‐β1
It is generally accepted that glycosylation affects many properties of glycoproteins, including their conformation, flexibility, and hydrophilicity. As a result, it regulates protein sorting, stability, and protein–protein interactions.( 1 , 2 , 3 , 4 , 5 ) N‐Glycans have a common core structure, and their branching patterns are determined by glycosyltransferases.( 6 , 7 ) Fut8 is an enzyme that catalyzes the introduction of α1,6 core fucose on the asparagine‐branched N‐acetylglucosamine residue of the chitobiose unit of complex‐type N‐glycans.( 8 , 9 ) Fut8 has been investigated especially in terms of oncogenesis, since the α1,6‐fucosylation of α‐fetoprotein is a well‐known marker of hepatocellular carcinoma.( 10 ) In previous studies, our group reported that Fut8 expression is markedly enhanced in several types of cancer cell lines( 11 ) rat hepatoma tissues( 12 ) and in ovarian serous adenocarcinoma cells.( 13 )
E‐cadherin is a 120 kDa type I membrane protein, which belongs to the class of calcium‐dependent cell adhesion molecules.( 14 ) It mediates cell–cell adhesion through the assembly of multiprotein complexes linked to the actin cytoskeleton.( 15 ) Several models have been proposed to date for the cadherin homophilic interactions. Examples include the “linear zipper model”, which involves Trp‐mediated cis dimers and trans interactions between the outermost domains,( 16 ) a Trp‐dimer model, which involves the formation of a Trp‐mediated trans‐homophilic bond,( 17 ) a model which involves cis‐dimerization at the Ca2+‐binding site,( 18 ) and a model that invokes extensive overlap between ectodomains in the adhesive binding interface.( 19 , 20 ) The extracellular domain of human E‐cadherin consists of five repeats of about 110 amino acid residues, referred to as EC1 through 5, and contains four potential N‐glycosylation sites, two each in EC4 and EC5. It is synthesized in the form of a precursor polypeptide that is glycosylated and the precursor is then processed to the mature polypeptide.( 21 , 22 , 23 ) It has previously been reported that cells expressing unprocessed E‐cadherin by mutating recognition site(s) for processing protease showed no E‐cadherin‐dependent mediated adhesion.( 24 )
Our previous studies demonstrated that the introduction of GnT‐III and the addition of bisecting GlcNAc residues, products of GnT‐III, to E‐cadherin down‐regulated tyrosine phosphorylation of β‐catenin, enhanced cell–cell adhesion mediated by E‐cadherin, and suppressed lung metastasis in mouse melanoma cells.( 25 , 26 ) Consistent with these results, Guo et al. also reported that the overexpression of GnT‐V, which competes with GnT‐III for biantennary substrates, decreased cadherin‐mediated cell–cell adhesion.( 27 ) On the other hand, the overexpression of Fut8 in hepatoma cells suppressed intrahepatic metastasis after splenic injection into athymic mice.( 28 ) Liwosz et al. reported that the status of N‐glycosylation of E‐cadherin is altered in a cell density‐dependent manner, and the loss of complex type of N‐glycan reduces the molecular weight of E‐cadherin and enhanced its preferential association with the actin cytoskeleton, leading to the stabilization of E‐cadherin scaffolds.( 29 ) These studies suggest that N‐glycans play a role in modulating E‐cadherin status. In the present study, we found that Fut8 and E‐cadherin protein levels are significantly increased in colorectal cancer samples. E‐cadherin in Fut8 transfected WiDr cells, Fut8 knocked down cells, and Fut8 deficient cells from Fut8−/– mice were examined and our results demonstrate that the activity of Fut8 is involved in the appearance of a low molecular weight population of E‐cadherin and regulates the total amount of E‐cadherin. We propose the possible involvement of core fucosylation in changing the N‐glycosylation patterns of E‐cadherin, the subsequent stabilization of cell–cell contacts, and the regulation of metastatic potential.
Materials and Methods
Human tissues samples. All experiments were approved by ethical committees both in Osaka University and Osaka National Hospital. Tissues from six cases of primary colorectal cancer were surgically resected (Table 1). Written informed consent was obtained from each patient before surgery. The excised samples were obtained within 1 h after the operation from tumor tissues and corresponding non‐tumor tissues 5–10 cm remote from the tumor. All of the excised tissues were placed immediately in liquid nitrogen and stored at –80°C until additional analysis.
Table 1.
Clinical features of patients with colorectal cancer
| Sample number | Age | Sex | Location † | Dukes’ stages |
|---|---|---|---|---|
| 1 | 71 | Female | A | C |
| 2 | 41 | Male | R | B |
| 3 | 77 | Male | R | B |
| 4 | 87 | Male | T | B |
| 5 | 69 | Male | S | C |
| 6 | 71 | Male | A | D |
A, ascending colon; R, rectum; S, sigmoid colon; T, transverse colon.
Cell lines, culture, and transfection. Human colon carcinoma WiDr cells were obtained from the American Type Culture Collection (Rockville, MD, USA) and were maintained in DMEM supplemented with 10% FBS. A Fut8 expression vector was constructed by inserting the open reading frame of human Fut8 cDNA into a mammalian expression vector pCXN2 which was regulated by the β‐actin promoter. Mutant Fut8, which had no enzymatic activity, was produced by mutating arginine 365 to alanine.( 30 ) WiDr cells were transfected with pCXN2/Fut8 or pCXN2/R365A Fut8 or pCXN2 by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Selection was performed by 2‐week incubation in medium containing G418, and G418‐resistant colonies were isolated and recloned by serial dilution to ensure clonality. Fut8 knocked down mouse pancreatic aciuar cell catcinoma TGP49 cells were prepared as described previously.( 31 ) Fut8 deficient kidney epithelial cells were prepared from Fut8 −/– mice as described previously.( 32 ) The cells were serum starved for 8 h before harvest to achieve the cell cycle synchronization.
Activity assay of Fut8. The enzymatic activity of Fut8 was measured by high‐performance liquid chromatography using a fluorescence‐labeled sugar chain as the substrate, as previously described.( 33 ) A standard mixture included 80 mM MES‐NaOH (pH 7.0), 0.5% Triton X‐100, 2 µM 4‐(2‐pyridylamino)butylamine‐labeled sugar chain, and 50 µM GDP‐Fucose. After incubation at 37°C for 2 h, the reaction was terminated by incubating at 100°C for 1 min. The samples were then centrifuged at 15 000 g for 10 min and applied to high‐performance liquid chromatography on a TSK‐gel, ODS‐80TM column (4.6 × 150 mm) (Tosho, Tokyo, Japan). Elution was performed at 55°C with 20 mM sodium acetate buffer, pH 4.0, 0.1% butanol, in an isocratic manner. Fluorescence of the column elutes was detected with a fluorescence spectrometer (model RF 535; Shimadzu Corp., Kyoto, Japan), the excitation and emission wavelengths being 320 and 400 nm, respectively.
Protein extraction, immunoprecipitation, and western blotting. Frozen tissue samples were homogenized in 5 vol. of lysis buffer (20 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 5 mM ethylenediaminetetraacetic acid, 1%[w/v] Nonidet P‐40, 10%[w/v] glycerol, 5 mM sodium pyrophosphate, 10 mM NaF, 1 mM sodium orthovanadate, 10 mM β‐glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, 2 µg/mL aprotinin, 5 µg/mL leupeptin, and 1 mM dithiothreitol) using Polytron homogenizer (Kinematica, Littau‐Luzern, Switzerland). After centrifugation (15 000 g) for 20 min at 4°C the supernatant were collected. Cell cultures were rinsed twice with ice‐cold PBS and harvested in lysis buffer. Protein concentrations were determined using a Protein Assay CBB kit (Nacalai Tesque, Kyoto, Japan). For the immunoprecipitation of E‐cadherin, whole cell lysates (500 µg) were incubated with 4 µg of mouse anti‐E‐cadherin antibody (610182; BD Bioscience, San Jose, CA, USA) for 2 h at 4°C, and then with 20 µL of Protein G Sepharose 4 Fast Flow (GE Healthcare Biosciences, Buckinghamshire, UK) for 4 h at 4°C. For western blot analysis, protein samples or immunoprecipitates were subjected to SDS‐PAGE, and transferred to nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). Mouse anti‐E‐cadherin antibody (610182; BD Bioscience) or mouse anti‐Fut8 antibody( 34 ) was used as primary antibodies. Immunoreactive bands were visualized using an enhanced chemiluminescence kit (GE Healthcare Biosciences).
Cell surface biotinylation and immunoprecipitation of E‐cadherin. Cell surface biotinylation was performed as described previously.( 35 ) Briefly, cells were incubated with sulfosuccinimidobiotin (s‐NHS‐biotin; Pierce, Rockford, IL, USA) (1 mg/mL) for 10 min on ice, and the reaction was quenched with 50 mM NH4Cl. The cell lysate was immunoprecipitated with anti‐E‐cadherin antibody as described above. The biotinylated proteins were visualized using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) and an enhanced chemiluminescence kit.
Lectin blot analysis. Lectin blot analysis was performed as described previously.( 36 ) The immunoprecipitated E‐cadherin was electrophoresed on 8% SDS‐PAGE and transferred to nitrocellulose membranes. The membrane was blocked with 5% BSA (w/v) and then incubated with 2 µg/mL of biotinylated AAL, DSA, or ConA lectin (Seikagaku Corp., Tokyo, Japan) for 30 min at room temperature. After washing, lectin reactive proteins were detected using a Vectastain ABC kit and an enhanced chemiluminescence kit.
Reverse transcription–polymerase chain reaction (RT‐PCR) and quantitative real‐time PCR. Total RNA was prepared from WiDr cells. cDNAs were synthesized using an SYBY RT‐PCR kit (Perfect Real Time; Takara‐Bio Inc., Otsu, Japan) and Reverse Transcription Reagent (Takara‐Bio Inc.) according to the manufacturer's instructions. A random hexamer was used for cDNA synthesis. Real‐time PCR was performed using the SYBR RT‐PCR kit and was analyzed on Smart Cycler II system (Cepheid, Sunnyvale, CA, USA). Human E‐cadherin was amplified using the primers, sense (5′‐GGATTGCAAATTCCTGCCATTC‐3′) and antisense (5′‐AACGTTGTCCCGGGTGTCA‐3′). GAPDH was amplified as a control using the primers, sense (5′‐ATTGCCCTCAACGACCACTT‐3′) and antisense (5′‐AGGTCCACCACCCTGTTGCT‐3′). The levels of gene expression were determined using a Delta‐Delta Ct method.( 37 )
Immunofluorescence microscopy. Cells were plated on poly L‐lysine‐coated glass bottom dish, fixed by incubation with PBS containing 4% paraformaldehyde for 10 min at room temperature and permeabilized in PBS containing 0.1% Triton X‐100 for 1 min. After washing with PBS three times for 15 min each, the cells were incubated in ECCD‐2 (monoclonal antibody to mouse E‐cadherin M108; Takara‐Bio Inc.) (1:100 dilution) for 1 h at room temperature. Primary antibody binding was detected with a fluorescein isothiocyanate‐labeled goat antibody to mouse IgG. Glass bottom dishes were mounted under Permafluor aqueous mounting medium and the stained cells were viewed with a laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
Cell aggregation assay. Cells were washed twice with PBS and dissociated by incubation with PBS containing 2 mM EDTA for 30 min at 37°C. Single cell suspensions were prepared, washed, and resuspended in DMEM containing 1% (w/v) BSA. 1 × 106 cells were incubated on a rotation apparatus for 3 h at 37°C. In some experiments, 2 mM EDTA or 100 µg/mL HECD‐1 (M106 monoclonal antibody to human E‐cadherin; Takara‐Bio Inc.) was added. At the end of the incubation, cells were diluted into single wells of a 24‐well plate to prevent further aggregation. After allowing cells to settle for 40 min at 37°C, an equal volume of 7.4% formaldehyde in PBS was added to each well and the plate was incubated for 10 min at room temperature. Photos were taken at random under a phase contrast microscope to count single cells or cell aggregates (four or more cells).
Peptide N‐glycosidase F (PNGase F) treatment. Whole cell lysates were boiled in 0.1 M 2‐mercaptoethanol and 0.1% SDS for 10 min. After boiling, the 50 µg of proteins were incubated for 16 h at 37°C with 100 mM Tris‐HCl (pH 8.6), 1% NP‐40, and 40 mU/mL PNGase F (Takara Bio Inc.). Then the samples were subjected to 6% SDS‐PAGE as described above.
Metabolic labeling and pulse chase study. Eighty‐percent confluent monolayers of Fut8 and mock transfectants in 6‐well dishes were preincubated for 2 h at 37°C with methionine, cysteine‐free DMEM (Sigma) containing dialyzed 10% FBS. For pulse chase studies, L‐[35S]methionine and L‐[35S]cysteine (Pro‐mix; GE Healthcare Biosciences) were added at a concentration of 200 µCi/mL each to the culture media and incubated for 20 min at 37°C for protein labeling. After rinsing three times with PBS, the cells were incubated at 0, 8, 24, and 32 h in DMEM with 10% FBS. Another experiment was performed under conditions of a long‐pulse and a long‐chase. For the long‐pulse, L‐[35S]methionine and L‐[35S]cysteine were added at a concentration of 50 µCi/mL each to the culture media, followed by incubation for 24 h at 37°C for protein labeling, and after rinsing, the cells were incubated at 0, 24, and 48 h in DMEM with 10% FBS. E‐cadherin was immunoprecipitated and subjected to 8% SDS‐PAGE. After electrophoresis, the gel was autoradiographed using imaging plates and a BAS‐2500 system (FujiFilm, Tokyo, Japan).
Results
E‐cadherin and Fut8 expression were increased in primary colorectal cancers. Abnormal cell–cell adhesion that is observed in many types of cancer could result from the changes in E‐cadherin expression. We examined Fut8 and E‐cadherin expression levels in primary colorectal cancer, and found that they are significantly increased in five out of six examined samples (Fig. 1). Low molecular weight population of E‐cadherin appeared as well as normal sized E‐cadherin only in the cancer samples. The relative expression levels of Fut8 and E‐cadherin in five samples are 3.5–29 and 3.3–11, respectively.
Figure 1.
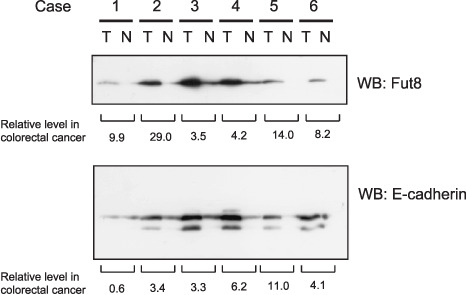
α1,6‐Fucosyltransferase (Fut8) and E‐cadherin expression levels were increased in primary colorectal cancer. Total protein lysate was prepared from matched samples of tumor (T) and adjacent non‐tumor tissue (N). 100 µg of protein from each pair were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), transferred to nitrocellulose membranes, and Fut8 and E‐cadherin were detected using an anti‐Fut8 antibody and an anti E‐cadherin antibody. WB, Western blotting.
Establishment of WiDr clones stably expressing Fut8. To investigate the correlation between Fut8 and E‐cadherin expression, we introduced Fut8 in WiDr human colon carcinoma cells in which Fut8 expression levels are low. WiDr cells were transfected with pCXN2/FUT8 or pCXN2, and G418‐resistant clones were selected as described under “Materials and Methods”. The selected Fut8 transfected clones showed elevated enzymatic activities, as 560, 470 and 190 nmol/h/mg protein, respectively. The following experiments were performed with three clones and similar results were observed for all data.
Analysis of E‐cadherin in the Fut8 transfected WiDr cells. Western blotting showed that the expression level of E‐cadherin in Fut8 transfectants was increased, especially in high‐density cultures (~11 × 104 cells/cm2) when compared to low density cultures (~3 × 104 cells/cm2) (Fig. 2a). In high‐density cultures, a low molecular weight population of E‐cadherin appeared in the Fut8 transfectants, in addition to the band with the same molecular weight as that in mock transfectants. We also established mutated Fut8 (R365A Fut8) which had no enzymatic activity, and examined its transfectants. Western blotting showed that the lower band was not expressed, even in high‐density cultures, suggesting that the Fut8 activity was involved in the appearance of the low molecular weight form of E‐cadherin (Fig. 2a). It was confirmed that both of the bands in Fut8 transfectants are expressed at the cell surface of Fut8 transfectants (Fig. 2b). A lectin blot analysis indicated that both bands in the Fut8 transfectants reacted with AAL, which binds preferentially to fucose linked α1,6 to GlcNAc although it binds to fucose linked α1,3 to N‐acetyllactosamine as well (Fig. 2c). The results suggested that the both forms were core‐fucosylated. We examined the reactivity toward other lectins such as DSA or ConA, which react with terminal galactose linked β1,6 to GlcNAc residues or mannose, respectively; however, there were no differences between mock and Fut8 transfectants (data not shown). The mRNA levels of E‐cadherin were evaluated by RT‐PCR and quantitative real‐time PCR. The results indicated that there was no significant difference between the Fut8 and mock transfectants (data not shown), suggesting that post‐translational modification is involved in the increase in E‐cadherin expression in the Fut8 transfectants.
Figure 2.
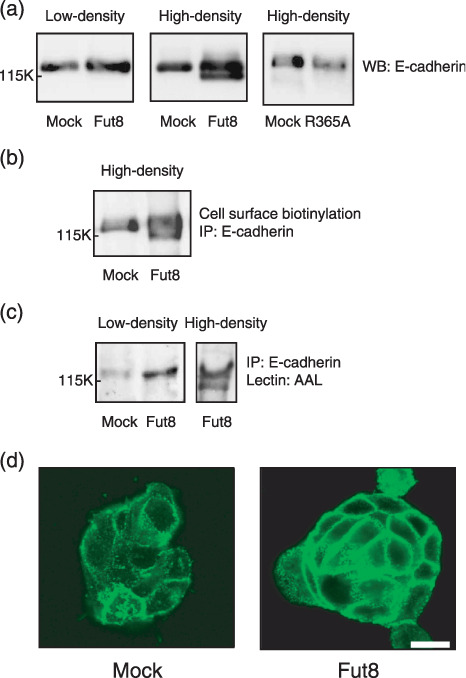
Effect of α1,6‐fucosyltransferase (Fut8) transfection on the characteristics and the expression of E‐cadherin in colon carcinoma WiDr cells. (a) Western blotting analysis of E‐cadherin in mock, Fut8, and R365A mutated Fut8 transfectants. A whole cell lysate (20 µg) was prepared from low (~3 × 104 cells/cm2) or high‐density (~11 × 104 cells/cm2) cultures and subjected to 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), transferred to a nitrocellulose membrane, and E‐cadherin was detected using an anti‐E‐cadherin antibody. WB, Western blotting. (b) Cell surface expression of E‐cadherin in the mock and Fut8 transfectants. E‐cadherin was immunoprecipitated from whole cell lysate of surface biotinylated mock and Fut8 transfectants, and subjected to 8% SDS‐PAGE, transferred to nitrocellulose membranes and the biotinylated E‐cadherin was visualized using a Vectastain ABC kit and an enhanced chemiluminescence kit. (c) Lectin blot analysis of E‐cadherin in the mock and Fut8 transfectants. E‐cadherin was immunoprecipitated from 400 µg of whole cell lysate, subjected to 8% SDS‐PAGE, and transferred to nitrocellulose membranes, which were probed by Aleuria aurantia lectin (AAL). IP, immunoprecipitation. (d) Distribution of E‐cadherin in mock and Fut8 transfectants. E‐cadherin was detected by a laser scanning confocal microscopy using an anti‐E‐cadherin antibody, ECCD‐2 (scale bar, 10 µm).
Accumulation of E‐cadherin at the cell–cell border in the Fut8 transfected WiDr cells. Morphologically, Fut8 transfected WiDr cells appeared as clusters with tight cell–cell contacts, whereas mock transfectants had relatively loose contacts. E‐cadherin expression was examined immunohistochemically. The Fut8 transfectants showed more intense fluorescence with condensation at the cell–cell contacts compared to mock transfectants, indicating that the expression of E‐cadherin was elevated in Fut8 transfectants (Fig. 2d).
Increased cell aggregation in the Fut8 transfected WiDr cells. To examine whether increased E‐cadherin in Fut8 transfectants functions, we performed a cell aggregation assay. As shown in Fig. 3, the cell aggregation rate of the Fut8 transfectants was significantly higher than that of the mock transfectants under high‐density condition (~11 × 104 cells/cm2), whereas there were no significant differences in aggregation status under low‐density condition (~3 × 104 cells/cm2). This increase in aggregation rate was inhibited in the presence of a calcium chelator, EDTA, or by an anti‐E‐cadherin monoclonal antibody, indicating that it was dependent on E‐cadherin.
Figure 3.
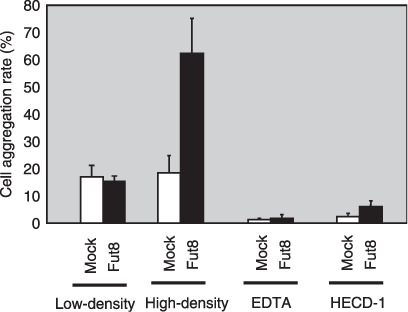
Effect of α1,6‐fucosyltransferase (Fut8) transfection on E‐cadherin‐dependent cell–cell adhesion in WiDr cells. Cell–cell aggregation was assayed with or without EDTA or the E‐cadherin inhibiting antibody, HECD‐1, as described in ‘Materials and Methods’. Data represent the mean (±SD) of six experiments.
Reduction of E‐cadherin and cell adhesion in the Fut8 knocked down TGP49 cells. We previously established Fut8 knocked down mouse pancreatic cancer cells TGP49.( 31 ) The clones showed a low expression of Fut8 and enzymatic activity was not detectable. Lectin blotting confirmed that the two bands of E‐cadherin in wild‐type cells reacted with AAL lectin, whereas the band in the Fut8 knocked down cells did not (Fig. 4a). Morphologically, Fut8 knocked down cells show more loose cell–cell contacts, and the expression levels of E‐cadherin at the cell–cell contacts were decreased compared to wild‐type cells (Fig. 4b). Consistently, E‐cadherin dependent cell aggregation decreased significantly in Fut8 knocked down cells (Fig. 4c).
Figure 4.
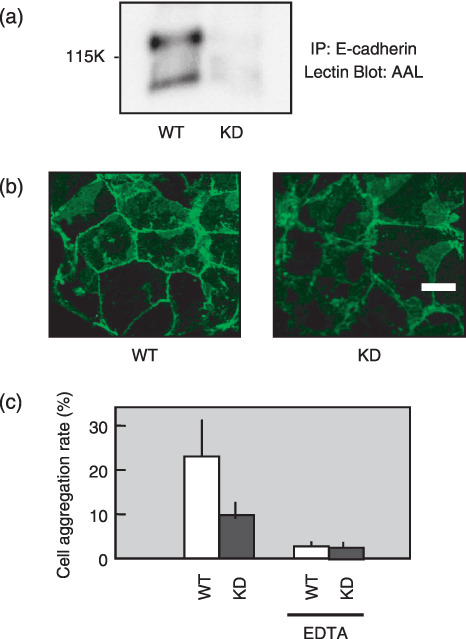
Changes of E‐cadherin expression and E‐cadherin‐dependent cell–cell adhesion in α1,6‐fucosyltransferase (Fut8) knock down cells. (a) Lectin blot analysis of E‐cadherin in Fut8 knocked down TGP49 cells and wild‐type TGP49 cells. E‐cadherin was immunoprecipitated from 400 µg of whole cell lysate, subjected to 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), and transferred to nitrocellulose membranes, which were probed by Aleuria aurantia lectin (AAL). IP, immunoprecipitation. (b) Distribution of E‐cadherin in Fut8 knocked down TGP49 cells and wild‐type TGP49 cells. E‐cadherin was detected by a laser scanning confocal microscopy using an anti‐E‐cadherin antibody, ECCD‐2. WT, wild‐type TGP49 cells; KD, Fut8 knocked down TGP49 cells (scale bar, 10 µm). (c) Cell–cell aggregation was assayed with or without EDTA. Data represent the mean (±SD) of six experiments.
Increase in E‐cadherin and cell adhesion by restoring Fut8 in Fut8−/– cells. Fut8 deficient kidney epithelial cells were prepared from Fut8 −/– mice as described previously.( 32 ) The cell surface expression of E‐cadherin (Fig. 5a) increased significantly in Fut8 restored cells compared to Fut8 −/– cells. Lectin blotting confirmed that the both of the two E‐cadherin bands in the Fut8 restored cells reacted with AAL lectin (Fig. 5b), although the lower band is relatively difficult to be detected. It was considered that the composition of glycosylation of kidney epithelial cells might be different from WiDr colon carcinoma cells. Morphologically, Fut8 −/– cells show loose cell–cell contacts and Fut8 restoring rescued them. An immunohistochemical study showed that the expression level of E‐cadherin at the cell–cell contacts in Fut8 −/– cells was also rescued by Fut8 transfection (Fig. 5c). E‐cadherin dependent cell aggregation (Fig. 5d) increased significantly in Fut8 restored cells compared to Fut8 −/– cells.
Figure 5.
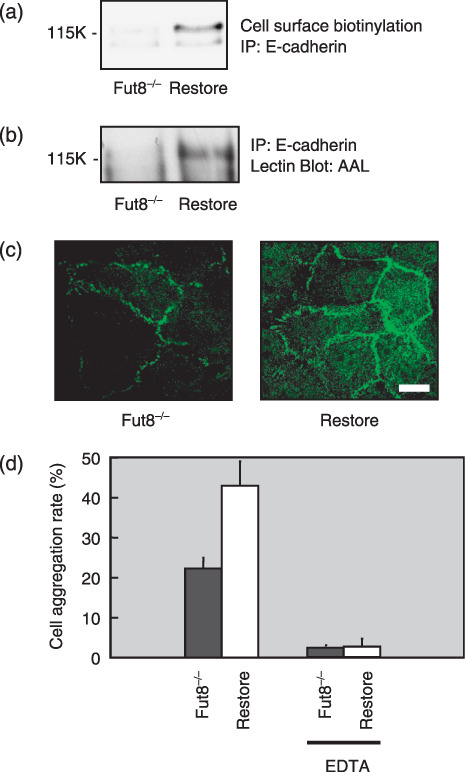
Changes of E‐cadherin expression and E‐cadherin‐dependent cell–cell adhesion in α1,6‐fucosyltransferase (Fut8)−/– cells. (a) Cell surface expression of E‐cadherin in the Fut8−/– mouse kidney epithelial cells and Fut8 restored cells. E‐cadherin was immunoprecipitated from whole cell lysate of surface biotinylated Fut8−/– cells and Fut8 restored cells, and subjected to 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), transferred to nitrocellulose membranes and the biotinylated E‐cadherin were visualized using a Vectastain ABC kit and an enhanced chemiluminescence kit. (b) Lectin blot analysis of E‐cadherin in the Fut8−/– cells and Fut8 restored cells. E‐cadherin was immunoprecipitated from 400 µg of whole cell lysates, subjected to 8% SDS‐PAGE and transferred to nitrocellulose membranes, which were probed by Aleuria aurantia lectin (AAL). (c) Distribution of E‐cadherin on Fut8−/– cells and Fut8 restored cells. E‐cadherin was detected by a laser scanning confocal microscopy using an anti‐E‐cadherin antibody, ECCD‐2. Fut8−/–, Fut8−/– mouse kidney epithelial cells; Restore, Fut8 restored Fut8−/– cells (scale bar, 10 µm). (d) Cell–cell aggregation was assayed with or without EDTA. Data represent the mean (±SD) of six experiments. IP, immunoprecipitation.
Peptide N‐glycosidase F (PNGase F) treatment of N‐glycan of E‐cadherin in the Fut8 transfected WiDr cells. To examine the N‐glycosylation status, Fut8 and mock transfectants were treated with PNGase F, which cleaves the N‐glycan between the innermost N‐acetylglucosamine and asparagines residues. As shown in Fig. 6(a), the upper band of E‐cadherin in the Fut8 transfectant appeared to be N‐glycosylated to a similar extent as the case of the mock transfectants. On the other hand, no decrease in molecular weight was observed in the PNGase F‐treated lower band in the Fut8 transfectants, although the band was reactive with AAL lectin (Fig. 2c), and notably, the molecular weight of the lower band appeared to be even smaller than that of the PNGase‐F digest of the upper band in the Fut8 transfectants. By molecular mass calculation, it was found that the molecular mass of the upper band is around 125 kDa, PNGase F digested upper band is around 110 kDa, and the lower band in Fut8 transfectant is around 105 kDa. Together with the lectin blotting results, these results indicate that the E‐cadherin corresponding to the lower band contains a form of N‐glycan that is resistant to PNGase F digestion, such as α1‐3 fucosylated N‐glycan.
Figure 6.
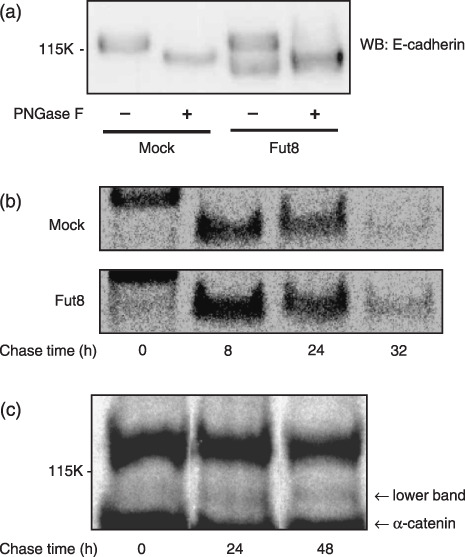
Analysis of E‐cadherin in α1,6‐fucosyltransferase (Fut8) transfected WiDr cells. (a) Whole cell lysates from mock and Fut8 transfectants were treated with peptide N‐glycosidase F (PNGase F) as described in ‘Materials and Methods’ and subjected to 6% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to nitrocellulose membrane, probed by an anti‐E‐cadherin antibody. WB, Western blotting. (b) Mock and Fut8 transfectants were radiolabeled with L‐[35S]methionine and L‐[35S]cysteine for 20 min. At 0, 8, 24, and 32 h after pulse labeling, E‐cadherin was immunoprecipitated from cell lysates using anti‐E‐cadherin antibody. After separation of the immunoprecipitated E‐cadherin by 6% SDS‐PAGE, the gel was dried and autoradiographed for 2 days using imaging plates and a BAS‐2500 system. The results were reproducible in three independent experiments. (c) Long‐pulse and long‐chase study. Fut8 transfectants were radiolabeled with L‐[35S]methionine and L‐[35S]cysteine for 24 h. At 0, 24, and 48 h after pulse labeling, E‐cadherin was immunoprecipitated from cell lysates using anti‐E‐cadherin antibody. After separation of the immunoprecipitated E‐cadherin by 6% SDS‐PAGE, the gel was dried and autoradiographed for 2 days using imaging plates and a BAS‐2500 system. The results were reproducible in three independent experiments.
Turnover of E‐cadherin in the Fut8 transfected WiDr cells. To elucidate the mechanisms by which the low molecular weight population of E‐cadherin is produced and the total expression levels of E‐cadherin are increased in Fut8 transfectants, the turnover of E‐cadherin was examined in pulse‐chase studies. As shown in Fig. 6(b), the turnover rate of E‐cadherin in Fut8 transfectants seemed slightly reduced and the band remained longer time. In this experiment, the upper band and the lower band in the Fut8 transfectants could not be clearly distinguished. When a long‐pulse and long‐chase experiment was performed, the low molecular weight form of E‐cadherin was detected after 24 h chase but not at 0 h in the Fut8 transfectants (Fig. 6c), whereas the normal form of E‐cadherin appeared 20 min after the pulse (data not shown).
Discussion
Changes in glycosylation status have been implicated in pathological status, especially cancer.( 38 ) We have been studying the functional regulation of signaling molecules by N‐glycosylation.( 36 , 39 , 40 , 41 , 42 , 43 ) E‐cadherin plays a central role in cancer metastasis, and it has been reported that its function is also affected by the modification of N‐glycans.( 25 , 26 , 44 ) In this study, we found that Fut8 and E‐cadherin expression levels are significantly increased in primary colorectal cancer samples. We established the Fut8 transfectants and examined the E‐cadherin. Since Fut8 is widely expressed in human tissues and human cancer cell lines, WiDr cells in which Fut8 expression levels are significantly low, and has malignant potential, were selected as the host cells. We have found that Fut8 activity is involved in the appearance of a low molecular weight population of E‐cadherin in high‐density culture, and regulates the total amount of cell surface E‐cadherin (Fig. 2a,b). E‐cadherin expression at cell–cell borders and E‐cadherin‐dependent cell aggregation were significantly enhanced in the Fut8 transfectants (2, 3). Studies with Fut8 knocked down cells and Fut8 −/– cells supported the conclusion that Fut8 activity was closely related to the appearance of low molecular weight population of E‐cadherin and total expression levels of E‐cadherin (4, 5). Since real‐time PCR showed that the levels of E‐cadherin mRNA were not changed in the Fut8 transfectants, the expression levels of E‐cadherin were most likely up‐regulated via a post‐translational process. The studies with PNGase F revealed that the glycosylation status was different in the lower band in high‐density culture of Fut8 transfectants (Fig. 6a). A study of Liwosz et al. indicated that the N‐glycosylation of E‐cadherin with complex N‐glycans is reduced in dense cultures, and that this change facilitates its association with the actin cytoskeleton, leading to the stabilization of E‐cadherin scaffolds.( 29 ) They observed that unstable adherens junctions in sparse cells contained E‐cadherin primarily modified with complex N‐glycans, whereas increased amounts of Triton‐insoluble E‐cadherin in dense cultures correlated with its modification with high mannose/hybrid oligosaccharides, which are small in size. Since CHO cells and MDCK cells, used as host cells in the report, have quite high Fut8 activity, it can be assumed that E‐cadherin was core fucosylated. We propose that Fut8 activity is involved in the glycosylation changes of E‐cadherin in dense culture and subsequent alterations in cell–cell adhesion. Appearance of low molecular weight population of E‐cadherin and increase of Fut8 expression in colorectal cancer samples might be independent phenomenon, and even if they are related, the cause and effect are not determined. However, the present data support the hypothesis that Fut8 increased in colorectal cancer affects the status of E‐cadherin.
The present study indicates that the low molecular weight population of E‐cadherin in the Fut8 transfectants was PNGase‐F insensitive; however, lectin blotting showed that it was glycosylated and core‐fucosylated (2, 6). The results of long‐pulse and long‐chase studies suggested that the low molecular weight E‐cadherin is produced from the normal form (Fig. 6c). Cell–cell adhesions mediated by E‐cadherin could activate signaling pathways such as receptor tyrosine kinase signaling or Wnt signaling, and it might be possible that cell–cell adhesion‐derived signaling affects the processing of N‐glycan with core fucose. Changes in glycosylation patterns according to cell density were also observed in our previous work( 45 ) and we consider that cell‐cell adhesion‐derived signaling is involved in processing of glycoproteins. Thus, it is suggested that the N‐glycans processing and turnover rate of E‐cadherin were altered, which caused the accumulation of low molecular population of E‐cadherin and total E‐cadherin, in Fut8 transfectants in dense culture. It is also possible that increase in E‐cadherin‐dependent cell aggregation in Fut8 transfectants is due to enhancement of E‐cadherin cell adhesive activity, and we must consider the both possibilities at present.
As seen in Fig. 6a, the PNGase‐F treated upper band of E‐cadherin seems still larger than the low molecular weight population. It is possible that this occurred as the result of deamidation in the upper band, since SDS‐PAGE often reflects amino acid modifications.( 46 )
We previously reported that E‐cadherin turnover is significantly delayed in melanoma cells transfected with GnT‐III, which is involved in the regulation of branch formation in N‐glycans.( 25 ) We also showed that the EGFR or Src‐mediated tyrosine phosphorylation of β‐catenin was decreased in GnT‐III transfectants.( 26 ) Guo et al. reported that the EGF‐induced tyrosine phosphorylation of β‐catenin and P120ctn increased in GnT‐V transfectants, which led to a reduction in cell–cell adhesion.( 27 ) The fact that GnT‐III activity was not changed by Fut8 transfection suggests that Fut8 activity regulates the expression of E‐cadherin independent of GnT‐III, and the accumulation of both normal form and the low molecular weight E‐cadherin enhances cell–cell adhesion.
It has been revealed that core fucosylation catalyzed by Fut8 is involved in various biological phenomena. Fut8 transgenic mice caused steatosis in the liver and kidney due to a decreased lysosomal acid lipase activity.( 47 ) Core fucose deficient IgG1 showed an improved binding to FcγRIIIA, and consequently, antibody‐dependent cellular cytotoxicity activity was up‐regulated.( 48 , 49 ) We developed Fut8 −/– mice and reported that the mice showed semi lethality, growth retardation, and emphysema‐like changes, and the experiments indicated that dysfunction of TGF‐β1 receptor( 32 ), impairment in the low‐density LRP‐1( 41 ) and functional changes of α3β1 integrin( 50 ) are involved in the phenomena. The regulation of the cell surface expression levels of E‐cadherin, reported herein, could be involved in the pathology observed in Fut8 transgenic mice and Fut8 knock out mice. Metastasis analysis using those animals would indicate the role of Fut8 in cell–cell adhesion in vivo.
Acknowledgments
We thank Dr Tadashi Suzuki for helpful comments and discussions. This work was supported by grants from the 21st Century COE Program from Japan Society for the Promotion of Science; Core Research for Evolutional Science and Technology from Japan Science and Technology Agency; and Special Coordination Funds for Promoting Science and Technology and Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Hakomori S. Tumor malignancy defined by aberrant glycosylation and sphingo (glyco) lipid metabolism. Cancer Res 1996; 56: 5309–18. [PubMed] [Google Scholar]
- 2. Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science 2001; 291: 2370–6. [DOI] [PubMed] [Google Scholar]
- 3. Taniguchi N, Ekuni A, Ko JH et al . A glycomic approach to the identification and characterization of glycoprotein function in cells transfected with glycosyltransferase genes. Proteomics 2001; 1: 239–47. [DOI] [PubMed] [Google Scholar]
- 4. Lau KS, Partridge EA, Grigorian A et al . Complex N‐glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007; 129: 123–34. [DOI] [PubMed] [Google Scholar]
- 5. Taniguchi N, Miyoshi E, Gu J, Honke K, Matsumoto A. Decoding sugar functions by identifying target glycoproteins. Curr Opin Struct Biol 2006; 16: 561–6. [DOI] [PubMed] [Google Scholar]
- 6. Taniguchi N, Miyoshi E, Ko JH, Ikeda Y, Ihara Y. Implication of N‐acetylglucosaminyltransferases III and V in cancer: gene regulation and signaling mechanism. Biochim Biophys Acta 1999; 1455: 287–300. [DOI] [PubMed] [Google Scholar]
- 7. Miyoshi E, Noda K, Yamaguchi Y et al . The alpha1–6‐fucosyltransferase gene and its biological significance. Biochim Biophys Acta 1999; 1473: 9–20. [DOI] [PubMed] [Google Scholar]
- 8. Uozumi N, Yanagidani S, Miyoshi E et al . Purification and cDNA cloning of porcine brain GDP‐L‐Fuc: N‐acetyl‐beta‐D‐glucosaminide alpha1–>6fucosyltransferase. J Biol Chem 1996; 271: 27810–7. [DOI] [PubMed] [Google Scholar]
- 9. Yanagidani S, Uozumi N, Ihara Y et al . Purification and cDNA cloning of GDP‐L‐Fuc: N‐acetyl‐beta‐D‐glucosaminide: alpha1‐6 fucosyltransferase (alpha1–6 FucT) from human gastric cancer MKN45 cells. J Biochem (Tokyo) 1997; 121: 626–32. [DOI] [PubMed] [Google Scholar]
- 10. Shimizu K, Katoh H, Yamashita F et al . Comparison of carbohydrate structures of serum alpha‐fetoprotein by sequential glycosidase digestion and lectin affinity electrophoresis. Clin Chim Acta 1996; 254: 23–40. [DOI] [PubMed] [Google Scholar]
- 11. Miyoshi E, Uozumi N, Noda K et al . Expression of alpha1‐6 fucosyltransferase in rat tissues and human cancer cell lines. Int J Cancer 1997; 72: 1117–21. [DOI] [PubMed] [Google Scholar]
- 12. Noda K, Miyoshi E, Uozumi N et al . High expression of alpha‐1‐6 fucosyltransferase during rat hepatocarcinogenesis. Int J Cancer 1998; 75: 444–50. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi T, Ikeda Y, Miyoshi E et al . alpha1,6fucosyltransferase is highly and specifically expressed in human ovarian serous adenocarcinomas. Int J Cancer 2000; 88: 914–19. [DOI] [PubMed] [Google Scholar]
- 14. Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science 1991; 251: 1451–5. [DOI] [PubMed] [Google Scholar]
- 15. Gumbiner BM. Regulation of cadherin‐mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol 2005; 6: 622–34. [DOI] [PubMed] [Google Scholar]
- 16. Shapiro L, Fannon AM, Kwong PD et al . Structural basis of cell‐cell adhesion by cadherins. Nature 1995; 374: 327–37. [DOI] [PubMed] [Google Scholar]
- 17. Boggon TJ, Murray J, Chappuis‐Flament S et al . C‐cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 2002; 296: 1308–13. [DOI] [PubMed] [Google Scholar]
- 18. Pertz O, Bozic D, Koch AW et al . A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E–cadherin homoassociation. Embo J 1999; 18: 1738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chappuis‐Flament S, Wong E, Hicks LD, Kay CM, Gumbiner BM. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. J Cell Biol 2001; 154: 231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu B, Chappuis‐Flament S, Wong E et al . Functional analysis of the structural basis of homophilic cadherin adhesion. Biophys J 2003; 84: 4033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peyrieras N, Hyafil F, Louvard D, Ploegh HL, Jacob F. Uvomorulin: a nonintegral membrane protein of early mouse embryo. Proc Natl Acad Sci USA 1983; 80: 6274–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vestweber D, Kemler R. Some structural and functional aspects of the cell adhesion molecule uvomorulin. Cell Differ 1984; 15: 269–73. [DOI] [PubMed] [Google Scholar]
- 23. Shore EM, Nelson WJ. Biosynthesis of the cell adhesion molecule uvomorulin (E‐cadherin) in Madin‐Darby canine kidney epithelial cells. J Biol Chem 1991; 266: 19672–80. [PubMed] [Google Scholar]
- 24. Ozawa M, Kemler R. Correct proteolytic cleavage is required for the cell adhesive function of uvomorulin. J Cell Biol 1990; 111: 1645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshimura M, Ihara Y, Matsuzawa Y, Taniguchi N. Aberrant glycosylation of E‐cadherin enhances cell–cell binding to suppress metastasis. J Biol Chem 1996; 271: 13811–5. [DOI] [PubMed] [Google Scholar]
- 26. Kitada T, Miyoshi E, Noda K et al . The addition of bisecting N‐acetylglucosamine residues to E‐cadherin down‐regulates the tyrosine phosphorylation of beta‐catenin. J Biol Chem 2001; 276: 475–80. [DOI] [PubMed] [Google Scholar]
- 27. Guo HB, Lee I, Kamar M, Pierce M. N‐acetylglucosaminyltransferase V expression levels regulate cadherin‐associated homotypic cell‐cell adhesion and intracellular signaling pathways. J Biol Chem 2003; 278: 52412–24. [DOI] [PubMed] [Google Scholar]
- 28. Miyoshi E, Noda K, Ko JH et al . Overexpression of alpha1‐6 fucosyltransferase in hepatoma cells suppresses intrahepatic metastasis after splenic injection in athymic mice. Cancer Res 1999; 59: 2237–43. [PubMed] [Google Scholar]
- 29. Liwosz A, Lei T, Kukuruzinska MA. N‐glycosylation affects the molecular organization and stability of E‐cadherin junctions. J Biol Chem 2006; 281: 23138–49. [DOI] [PubMed] [Google Scholar]
- 30. Takahashi T, Ikeda Y, Tateishi A et al . A sequence motif involved in the donor substrate binding by alpha1,6‐fucosyltransferase: the role of the conserved arginine residues. Glycobiology 2000; 10: 503–10. [DOI] [PubMed] [Google Scholar]
- 31. Li W, Nakagawa T, Koyama N et al . Down‐regulation of trypsinogen expression is associated with growth retardation in alpha1,6‐fucosyltransferase‐deficient mice: attenuation of proteinase‐activated receptor 2 activity. Glycobiology 2006; 16: 1007–19. [DOI] [PubMed] [Google Scholar]
- 32. Wang X, Inoue S, Gu J et al . Dysregulation of TGF‐beta1 receptor activation leads to abnormal lung development and emphysema‐like phenotype in core fucose‐deficient mice. Proc Natl Acad Sci U S A 2005; 102: 15791–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Uozumi N, Teshima T, Yamamoto T et al . A fluorescent assay method for GDP‐L‐Fuc: N‐acetyl‐beta‐D‐glucosaminide alpha1‐6fucosyltransferase activity, involving high performance liquid chromatography. J Biochem (Tokyo) 1996; 120: 385–92. [DOI] [PubMed] [Google Scholar]
- 34. Ito Y, Miyauchi A, Yoshida H et al . Expression of alpha1,6‐fucosyltransferase (FUT8) in papillary carcinoma of the thyroid: its linkage to biological aggressiveness and anaplastic transformation. Cancer Lett 2003; 200: 167–72. [DOI] [PubMed] [Google Scholar]
- 35. Hobert ME, Kil SJ, Medof ME, Carlin CR. The cytoplasmic juxtamembrane domain of the epidermal growth factor receptor contains a novel autonomous basolateral sorting determinant. J Biol Chem 1997; 272: 32901–9. [DOI] [PubMed] [Google Scholar]
- 36. Sato Y, Takahashi M, Shibukawa Y et al . Overexpression of N‐acetylglucosaminyltransferase III enhances the epidermal growth factor‐induced phosphorylation of ERK in HeLaS3 cells by up‐regulation of the internalization rate of the receptors. J Biol Chem 2001; 276: 11956–62. [DOI] [PubMed] [Google Scholar]
- 37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2 (‐Delta Delta C(T) method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- 38. Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A 2002; 99: 10231–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shibukawa Y, Takahashi M, Laffont I, Honke K, Taniguchi N. Down‐regulation of hydrogen peroxide‐induced PKC delta activation in N‐acetylglucosaminyltransferase III‐transfected HeLaS3 cells. J Biol Chem 2003; 278: 3197–203. [DOI] [PubMed] [Google Scholar]
- 40. Takahashi M, Tsuda T, Ikeda Y, Honke K, Taniguchi N. Role of N‐glycans in growth factor signaling. Glycoconj J 2004; 20: 207–12. [DOI] [PubMed] [Google Scholar]
- 41. Lee SH, Takahashi M, Honke K et al . Loss of core fucosylation of low‐density lipoprotein receptor‐related protein‐1 impairs its function, leading to the upregulation of serum levels of insulin‐like growth factor‐binding protein 3 in Fut8−/– mice. J Biochem (Tokyo) 2006; 139: 391–8. [DOI] [PubMed] [Google Scholar]
- 42. Yokoe S, Takahashi M, Asahi M et al . The Asn418‐linked N‐glycan of ErbB3 plays a crucial role in preventing spontaneous heterodimerization and tumor promotion. Cancer Res 2007; 67: 1935–42. [DOI] [PubMed] [Google Scholar]
- 43. Li W, Takahashi M, Shibukawa Y et al . Introduction of bisecting GlcNAc in N‐glycans of adenylyl cyclase III enhances its activity. Glycobiology 2007; 17: 655–62. [DOI] [PubMed] [Google Scholar]
- 44. Przybylo M, Hoja‐Lukowicz D, Litynska A, Laidler P. Different glycosylation of cadherins from human bladder non‐malignant and cancer cell lines. Cancer Cell Int 2002; 2: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iijima J, Zhao Y, Isaji T et al . Cell–cell interaction‐dependent regulation of N‐acetylglucosaminyltransferase III and the bisected N‐glycans in GE11 epithelial cells. Involvement of E‐cadherin‐mediated cell adhesion. J Biol Chem 2006; 281: 13038–46. [DOI] [PubMed] [Google Scholar]
- 46. Fujiwara N, Miyamoto Y, Ogasahara K et al . Different immunoreactivity against monoclonal antibodies between wild‐type and mutant copper/zinc superoxide dismutase linked to amyotrophic lateral sclerosis. J Biol Chem 2005; 280: 5061–70. [DOI] [PubMed] [Google Scholar]
- 47. Wang W, Li W, Ikeda Y et al . Ectopic expression of alpha1,6 fucosyltransferase in mice causes steatosis in the liver and kidney accompanied by a modification of lysosomal acid lipase. Glycobiology 2001; 11: 165–74. [DOI] [PubMed] [Google Scholar]
- 48. Shields RL, Lai J, Keck R et al . Lack of fucose on human IgG1 N‐linked oligosaccharide improves binding to human Fcgamma RIII and antibody‐dependent cellular toxicity. J Biol Chem 2002; 277: 26733–40. [DOI] [PubMed] [Google Scholar]
- 49. Shinkawa T, Nakamura K, Yamane N et al . The absence of fucose but not the presence of galactose or bisecting N‐acetylglucosamine of human IgG1 complex‐type oligosaccharides shows the critical role of enhancing antibody‐dependent cellular cytotoxicity. J Biol Chem 2003; 278: 3466–73. [DOI] [PubMed] [Google Scholar]
- 50. Zhao Y, Itoh S, Wang X et al . Deletion of core fucosylation on alpha3beta1 integrin down‐regulates its functions. J Biol Chem 2006; 281: 38343–50. [DOI] [PubMed] [Google Scholar]