

Abstract
Amplification of chromosomal DNA is thought to be one of the mechanisms that activates cancer‐related genes in tumors. In a previous genome‐wide screening of DNA copy number aberrations in a panel of non‐small cell lung cancer (NSCLC) cell lines using an in‐house bacterial artificial chromosome‐based array, we identified a novel amplification at 14q11.2 in HUT29 cells derived from human lung adenocarcinoma. To identify the most likely target for the 14q11.2 amplification, we determined the extent of the amplicon by fluorescence in situ hybridization and then analyzed NSCLC cell lines for the expression levels of 28 genes present within the 1‐Mb amplified region. Significant overexpression in the HUT29 cell line with amplification, relatively frequent overexpression in additional NSCLC cell lines compared with an immortalized normal lung epithelial cell line, and reported information about the function of each candidate gene prompted us to characterize the BCL2‐like2 (BCL2L2) gene, a prosurvival member of the BCL2 family, as the most likely target for the 14q11.2 amplicon. Immunohistochemical analysis of 61 primary cases of lung adenocarcinoma demonstrated that BCL2L2 overexpression was significantly associated with tumor stage and differentiation status, and tended to be associated with a poorer prognosis. Downregulation of BCL2L2 expression using small interfering RNA dramatically inhibited the growth of HUT29 cells, but showed no effect on anticancer reagent‐induced cell death of the same cell line. These findings demonstrate that overexpressed BCL2L2, through amplification or other mechanisms, promotes the growth of NSCLC, especially the adenocarcinoma subtype, and might be a therapeutic target. (Cancer Sci 2007; 98: 1070–1077)
Lung cancer is the most common malignancy, the leading cause of cancer death worldwide (including Japan) and its incidence is increasing.( 1 , 2 ) Non‐small cell lung cancers (NSCLC), which include squamous cell carcinoma (SCC), adenocarcinoma and large cell carcinoma, make up almost 80% of all lung cancer cases. Despite advances in cytotoxic drugs, radiotherapy and clinical management, the treatment approaches for NSCLC have remained generic and largely ineffective, and the prognosis is generally unfavorable for patients with NSCLC: the 5‐year survival rate for all stages of NSCLC has remained fixed at 15% for the last 15 years.( 1 , 2 ) The recent success of molecularly targeted therapies for a limited subset of cancer genotypes has solidified the view that a more detailed knowledge of the spectrum of genetic lesions in NSCLC will, in turn, lead to meaningful therapeutic progress.( 2 )
Significant genomic copy number alterations, such as amplifications and homozygous deletions, are believed to be useful landmarks for identifying oncogenes and tumor‐suppressor genes, respectively, which are critical to tumorigenesis.( 3 , 4 , 5 ) Gene amplification, a phenomenon characteristic of numerous human cancers, appears to be a key mechanism whereby a cancer cell activates molecules that confer a selective advantage,( 6 ) although mechanisms other than amplification may also contribute to the overexpression and functional activation of these genes. We have identified many oncogenes and other cancer‐related genes from amplified chromosomal regions using conventional comparative genomic hybridization (CGH) and in‐house bacterial artificial chromosome (BAC)‐based array‐CGH in various tumors, including lung cancer.( 7 , 8 , 9 , 10 , 11 ) Therefore, the characterization of significant amplifications and the genes affected by them represents an excellent way to identify novel genes involved in growth control and carcinogenesis of NSCLC.
To search through the entire NSCLC cell genome for significant changes in copy number with precise and rapid identification of tumor‐suppressor genes as well as oncogenes, we applied array‐CGH using an in‐house BAC array for a relatively large panel of NSCLC cell lines.( 12 , 13 ) In the course of this program, we identified the amplicon at 14q11.2 containing BCL2L2 as one of the regions with a significant copy number increase in one NSCLC cell line derived from adenocarcinoma.( 12 , 13 ) Overexpression of BCL2L2 occurred through significant amplification of the gene, and was observed relatively frequently in other cell lines compared with the control counterpart derived from normal pulmonary epithelial cells. Immunohistochemical analysis in lung adenocarcinomas and functional analysis using small interfering RNA (siRNA) revealed that frequent overexpression of BCL2L2 may be involved in the pathogenesis of NSCLC, especially adenocarcinoma, at least partly through unknown mechanisms other than its antiapoptotic function.
Materials and Methods
Cell lines and primary tumors. Of the 27 NSCLC cell lines used,( 8 , 12 ) 11 were derived from SCC (EBC‐1, HS‐24, LK‐2, PC10, VMRC‐LCP, LC1‐sq, SK‐MES‐1, ACC‐LC‐73, HUT‐15, Sq‐1 and KNS‐62), 10 from adenocarcinomas (11‐18, A549, VMRC‐LCD, SK‐LC‐3, ABC‐1, RERF‐LC‐MS, RERF‐LC‐OK, PC‐14, HUT29 and RERF‐LC‐KJ), and six from large‐cell carcinomas (86‐2, LU65, PC‐13, NCI‐H460, ACC‐LC‐33 and LU99A). All cell lines were maintained in appropriate media, RPMI‐1640 or Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum, 100 units/mL penicillin and 100 µg/mL streptomycin. Two immortalized lung epithelial cell lines, HPL1D derived from human peripheral lung epithelial cells and BEAS2B derived from human bronchial epithelial cells,( 14 , 15 ) were used as controls.
Primary tumor samples were obtained during surgery from 61 NSCLC patients undergoing tumor resection at the National Defense Medical College (Saitama, Japan) between January 1991 and December 2000. All cases were adenocarcinomas. The tissues had been embedded in paraffin after 24 h of formalin fixation, and relevant clinical and survival data were available for all 61 patients. Written consent was always obtained in the formal style and after approval by the local ethics committees. None of the patients in either group had received chemotherapy or radiation before surgery. Disease stage was defined in accordance with the sixth revision of the Union Internationale Contre le Cancer international tumor‐node‐metastasis (TNM) staging system for lung cancer.( 16 , 17 , 18 ) Patients with cT4 disease, cN3 disease or distant organ metastases were not included in this study. The median follow‐up period for the surviving patients was 54.5 months (ranging from 12 to 83.8 months).
Fluorescence in situ hybridization. Metaphase chromosomes were prepared from normal male lymphocytes and from NSCLC cell lines. Fluorescence in situ hybridization (FISH) analyses were carried out as described elsewhere( 7 , 8 ) using BAC located around the region of interest as probes. The copy number and molecular organization of the region of interest were assessed according to the hybridization patterns of both metaphase and interphase chromosomes.( 7 )
Reverse transcription–polymerase chain reaction. Single‐stranded cDNA was generated from total RNA( 12 , 13 ) and amplified with primers specific for each gene (Supplementary Table S1). Reverse transcription–polymerase chain reaction (RT‐PCR) products were electrophoresed on a 3% agarose gel, and band quantification was done using LAS‐3000 with MultiGauge software (Fujifilm, Tokyo, Japan). The glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) gene was amplified at the same time as an internal control. The mRNA expression of each gene in each sample was normalized on the basis of the respective GAPDH content and recorded as a relative expression level.
Western blotting. Monoclonal antibodies specific to BCL2L2 and anti‐β‐actin were purchased from Novocastra (Newcastle, UK) and Sigma (St Louis, MO, USA), respectively. Anti‐stress‐activated protein kinase (SAPK)/c‐Jun NH2‐terminal kinase (JNK) and antiphospho‐SAPK/JNK (Thr183/Tyr185) antibodies were from Cell Signaling Technology (Beverly, MA, USA). Cells were lysed in RIPA buffer (10 mM Tris‐HCl, 150 mM NaCl, 1 mM ethylenediaminetetracetic acid, 1% sodium deoxycholate, 0.1% sodium dodecylsulfate, and 1% Triton X‐100, pH 7.4) with a protease inhibitor cocktail (Roche Diagnostics, Tokyo, Japan). After the protein concentration of each lysate was estimated by BCA assay (Pierce Chemicals, Rockford, IL, USA), 10 µg of protein was separated on sodium dodecylsulfate–polyacrylamide gels and transferred to Hybond‐P membranes (GE Healthcare Biosciences, Tokyo, Japan). Proteins were detected after incubation with appropriate primary antibodies and peroxidase‐conjugated secondary antibodies, followed by development with WestDura chemiluminescent reagent (Pierce Chemicals).
Immunohistochemistry (IHC). Expression of BCL2L2 protein in formalin‐fixed, paraffin‐embedded tissue sections from primary lung adenocarcinoma was detected by indirect IHC, as described elsewhere.( 7 ) The sections were mounted on silane‐coated glass slides, deparaffinized, and rehydrated through a graded ethanol series. Antigens were retrieved by autoclave pretreatment in Target Retrieval Solution High pH (DAKO, Carpinteria, CA, USA) for 15 min at 95°C. Endogenous peroxidase was blocked using 5% hydrogen peroxide. After blocking in 2% normal swine serum, sections were treated overnight at 4°C with 0.75 mM anti‐BCL2L2 antibody (Novocastra), then reacted for 1 h at room temperature with a dextran polymer reagent combined with secondary antibodies and peroxidase (Envision Plus; DAKO). Antigen–antibody reactions were visualized with 0.2% 3,3′‐diaminobenzidine tetrahydrochloride and hydrogen peroxide, and the slides were counterstained with Mayer's hematoxylin.
Formalin‐fixed HUT29 and A549 cell lines were used as positive and negative controls, respectively. Overexpression of BCL2L2 was graded as either positive (tumor‐cell cytoplasm showing higher immunopositivity compared with normal alveolar type II cells, which were considered to be the source of lung adenocarcinomas),( 19 ) or negative (tumor‐cell cytoplasm showing immunonegativity compared with normal alveolar type II cells). Each section was examined at ×200 magnification.
Transfection with siRNA and cell growth assays. BCL2L2 (BCL‐W)‐specific siRNA (BCL2L2‐siRNA) was purchased from SantaCruz Biotechnology (Santa Cruz, CA, USA). PSMB5‐ and THTPA‐specific siRNA (PSMB5‐siRNA and THTPA‐siRNA, respectively) were from Dharmacon (Lafayette, CO, USA). A control siRNA for the luciferase gene (CGUACGCGGAAUACUUCGA, Luc‐siRNA) was synthesized by Sigma Aldrich (Tokyo, Japan). For each experiment, 1 × 105 cells in logarithmic growth phase (seeded in six‐well plates) were transfected with 50 nmol/L synthetic siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Knockdown efficiency was determined 48 h and/or 120 h after transfection by RT‐PCR and/or western blotting. For measurements of cell growth, transfected cells were seeded in 96‐well plates. For drug treatment experiments using cisplatin (cDDP) and CPT‐11 (CPT), cells were exposed to various concentrations of cDDP or CPT for 48 h after plating in 96‐well plates. The numbers of viable cells were assessed using a colorimetric water‐soluble tetrazolium salt (WST) assay.( 11 ) Experiments were repeated three times, and each series was carried out in triplicate.
Statistical analysis. The χ2 or Fisher's exact test was used to examine categorical data. The Kaplan–Meier survival curve was used for the analysis of overall survival, and differences between the groups were tested with the log‐rank test. Differences in cell viability between BCL2L2‐ and control Luc‐siRNA‐treated cells were tested by the non‐parametric Mann–Whitney U‐test with a determination of the associated probability. All tests were considered significant at the level of P < 0.05.
Results
Analysis of 14q11.2 amplification in NSCLC cell lines by FISH. We had already carried out genome‐wide screening for DNA copy number aberrations in a panel of 27 NSCLC cell lines using an in‐house BAC‐based array (MCG Cancer Array‐800 and MCG Whole Genome Array‐4500),( 5 ) and had reported several novel genes involved in lung carcinogenesis.( 12 , 13 ) In the present study, we focused on the amplification at 14q11.2 that had been identified in HUT29 cells by the previous array‐CGH analyzes.( 12 ) Genome profiling in the HUT29 cell line revealed remarkable amplification at 14q11.2. The highest log2 ratio (3.9) was detected within the 14q11.2 amplicon (BAC RP11‐68M15) with the exception of regions for known target oncogenes, such as MYCN at 2p24.3 (log2 ratio = 5.3, data not shown).( 12 , 13 )
In order to construct a precise amplicon map, we carried out a series of FISH analyses. Significant amplification with double minute chromosome (dmin) patterns was observed with BAC within the amplicon (Fig. 1A). The estimated extent of the amplification was approximately 1 Mb between RP11‐137H15 and RP11‐76P17 at 14q11.2, and according to information archived by human genome databases (http://www.ncbi.nlm.nih.gov/ and http://genome.ucsc.edu/, May 2006) the 14q11.2 amplicon detected in HUT29 cells contains at least 28 genes (Fig. 1B, Supplementary Table S2).
Figure 1.
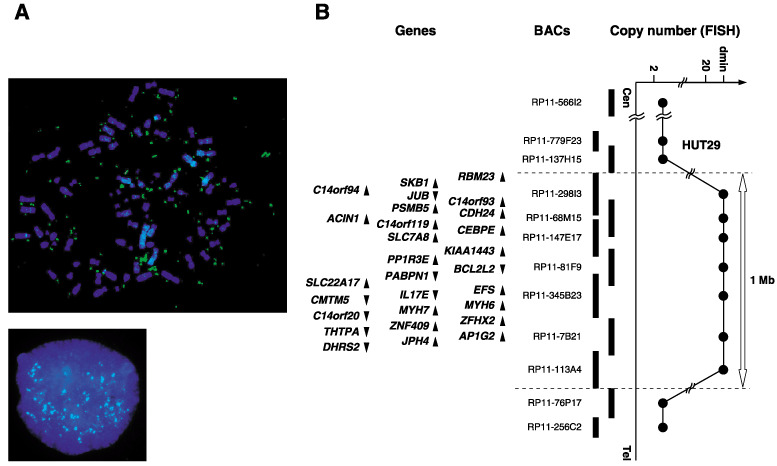
(A) Fluorescence in situ hybridization (FISH) images with RP11‐68M15 as a probe (green signals) hybridized to metaphase (upper) and interphase (lower) chromosomes from the non‐small cell lung cancer line HUT29 clearly showed amplification with a double minute chromosome (dmin) pattern. (B) Map of 14q11.2 covering the region of amplifications observed in the HUT29 cell line. Bacterial artificial chromosomes used for FISH are indicated as thick vertical bars. Within the amplicon, 28 genes exist and are indicated by arrowheads showing the direction of transcription (see Supplementary Table S2).
BCL2L2 overexpression in NSCLC cell lines. We next determined the expression levels of the candidate target genes located within the 14q11.2 amplicon by means of semiquantitative RT‐PCR in NSCLC cell lines and two immortalized lung epithelial cell lines. For screening genes overexpressed through amplification and showing higher levels of expression in cancer cells than in their normal counterparts, 28 genes were investigated using a smaller panel of NSCLC cell lines including HUT29, a cell line with 14q11.2 amplification derived from adenocarcinoma, the control cell line HPL1D derived from peripheral lung epithelial cells (which are considered to be the source of adenocarcinomas) and BEAS2B derived from human bronchial epithelial cells. Among 28 candidate genes, 10 genes were overexpressed (>1.6‐fold increase relative to the control HPL1D cell line) in HUT29 cells compared with other NSCLC cell lines and control cell lines, especially HPL1D (group A in Supplementary Fig. S1).
We next carried out a semiquantitative RT‐PCR analysis for those 10 genes in all 27 NSCLC cell lines to evaluate expression patterns in NSCLC cells compared with control cell lines. Four of the genes showed a relatively frequent overexpression pattern (>1.6‐fold increase relative to the control HPL1D cell line) in NSCLC cells (≥5 of 27 cell lines; Fig. 2). Although these genes are likely to be potential targets for amplification based on their expression pattern, their functions of most of them (especially the cancer‐associated functions) remain unknown, except BCL2L2 encoding one of the prosurvival factors in the BCL2 family and PSMBS5 according to the human genome database (http://www.ncbi.nlm.nih.gov/, Supplementary Table S2). Because (1) BCL2L2 is known to inhibit cell death( 20 , 21 ) and is expressed frequently in human tumor cells and primary adenocarcinomas such as gastric and colorectal cancers( 23 , 24 , 25 ) but (2) its significance in the carcinogenesis of NSCLC, especially adenocarcinoma, has never been reported, we further examined the biological and clinicopathological significance of BCL2L2 in lung carcinogenesis as a possible target for the 14q11.2 amplification.
Figure 2.
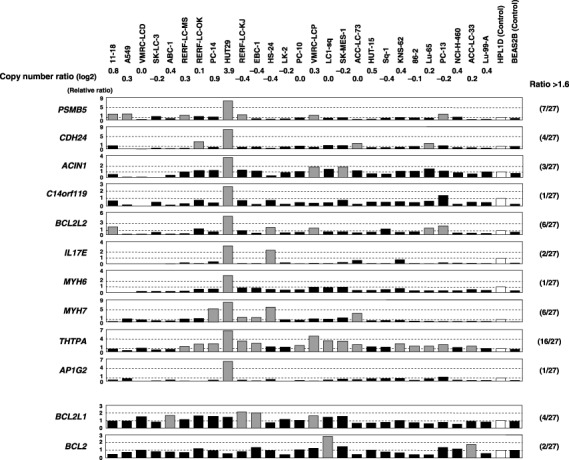
Expression levels of the 10 candidate target genes within the 14q11.2 amplicon (upper) and prosurvival members of the BCL2 family (lower) in 27 non‐small cell lung cancer (NSCLC) cell lines and immortalized lung epithelial cell lines detected by semiquantitative reverse transcription–polymerase chain reaction (RT‐PCR). RT‐PCR products were electrophoresed on 3% agarose gel, and band quantification was carried out. The mRNA expression of each gene in each sample was normalized on the basis of the corresponding glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) level. Results were then normalized to the expression level of the gene in the control cell line HPL1D as a normal counterpart, and were recorded as a relative expression level (relative ratio). Expression levels were considered ‘overexpression’ (gray columns) when the expression level of each gene in each sample relative to that in the control cell line HPL1D (open columns) was increased >1.6‐fold. The frequency of overexpression of each gene in the NSCLC cell lines is indicated in parentheses. Relatively frequent (>5 of 27 cell lines) overexpression in NSCLC cell lines was observed for four genes including BCL2L2. Copy number ratio (log2 ratio) at 14q11.2 (BAC RP11‐68M15) was determined in our previous array‐comparative genomic hybridization (CGH) analysis.( 12 )
In order to compare the expression patterns of antiapoptotic members of the BCL2 family,( 21 ) RT‐PCR was carried out for BCL2L2, BCL2L1 and BCL2. The expression of BCL2L2 was increased in several NSCLC cell lines compared with the normal cell‐derived cell line, whereas the expression levels of BCL2L1 and BCL2 in cancer cell lines were similar to or lower than those in the normal control cell line (Fig. 2).
Expression of BCL2L2 protein in primary NSCLC tumors. To verify BCL2L2 overexpression in primary NSCLC, we undertook immunohistochemical analyses of BCL2L2 protein in 61 primary tumors, all adenocarcinomas. The specificity of anti‐BCL2L2 was confirmed using cell lines overexpressing and almost lacking BCL2L2 as positive and negative controls, respectively (Fig. 3A,B). In neighboring non‐neoplastic cells, especially in alveolar type II cells, staining for BCL2L2 was very weak or was not observed (Fig. 3E). In cancerous tissues, however, positive immunostaining, which was defined as staining with BCL2L2 antibody stronger than alveolar type II cells, was observed in most tumors (40/61, 65.6%; Fig. 3C,D). BCL2L2 immunoreactivity was detected mainly in the cytoplasm of primary adenocarcinoma cells as well as in normal alveolar type II cells.( 23 )
Figure 3.
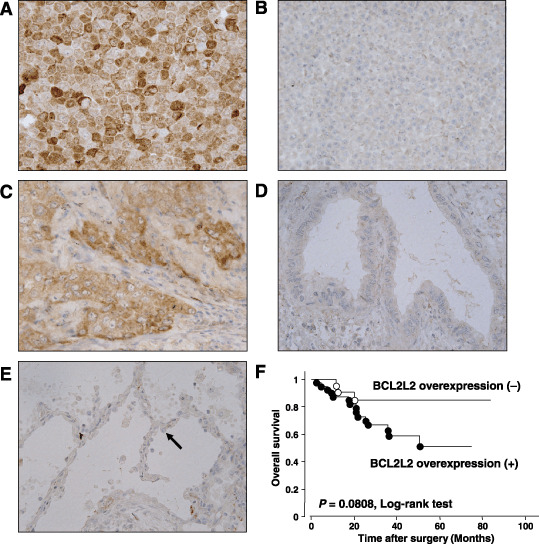
Immunohistochemical staining of BCL2L2 protein. (A) HUT29 cells used as a positive control. (B) A549 cells used as a negative control. (C) Representative case of primary lung adenocarcinoma showing BCL2L2 overexpression compared with normal type II alveolar cells (×400). (D) Representative case of primary lung adenocarcinoma without BCL2L2 overexpression compared with normal type II alveolar cells (×400). (E) Normal primary lung epithelial cells. Black arrowhead indicates the type II alveolar cell. (F) Kaplan–Meier analyses and log‐rank tests in 61 primary lung adenocarcinomas demonstrated that cases with BCL2L2 overexpression tended to be associated with a poorer overall survival compared with those without BCL2L2 overexpression, although the difference was not significant (P = 0.0808).
Association between clinicopathological characteristics and expression status of BCL2L2 protein in primary cases. To obtain some hint as to the significance of BCL2L2 expression in the pathogenesis of NSCLC, we investigated the relationship between the expression status of BCL2L2 protein determined by immunohistochemistry and the clinicopathological characteristics of all 61 primary tumors. The expression of BCL2L2 was significantly associated with tumor staging (P = 0.044, Fisher's exact test) and differentiation status (P = 0.039, χ2‐test), but was not associated with age, sex, histological subtype or the T and N categories of the TNM classification (Table 1). A univariate analysis of overall survival by the log‐rank test demonstrated that BCL2L2 positivity tended to be associated with a poorer prognosis, although the difference was not significant (P = 0.0808, log‐rank test; Fig. 3F).
Table 1.
Relationship between clinicopathological data and BCL2L2 overexpression
| Clinicopathological characteristics | n | BCL2L2 protein overexpression | P‐value* |
|---|---|---|---|
| Total | 61 | 40 (65.6%) | |
| Sex | |||
| Male | 30 | 22 (73.3%) | 0.21 |
| Female | 31 | 10 (58.1%) | |
| Age (years) | |||
| Median | 68 | ||
| ≥70 | 27 | 18 (66.7%) | 0.873 |
| <70 | 34 | 22 (64.7%) | |
| Histological subtype† | |||
| Bronchioloalveolar cell type | 39 | 24 (61.5%) | 0.135 |
| Papillary type | 10 | 9 (90.9%) | |
| Differentiation | |||
| Well | 24 | 12 (50.0%) | 0.039 |
| Moderate or poor | 37 | 28 (75.7%) | |
| Stage | |||
| I–II | 49 | 29 (59.2%) | 0.044 |
| IIIa | 12 | 11 (91.7%) | |
| TNM classification | |||
| T category | |||
| T1 | 35 | 23 (65.7%) | 0.979 |
| T2–T3 | 26 | 17 (65.4%) | |
| N category | |||
| N0 | 42 | 27 (64.3%) | 0.753 |
| N1–N2 | 19 | 13 (68.4%) | |
The χ2 or Fisher's exact test were used to examine categorical data. All tests were considered significant at the P < 0.05 level. Statistically significant values are in bold type. Histological subtypes in 12 cases were unknown and were excluded from the statistical analysis. BAC, bronchioloalveolar cell type; PAP, papillary type.
Suppression of cell growth after downregulation of BCL2L2 expression. To gain further insight into the potential role of BCL2L2 as an oncogene whose overexpression could be associated with lung carcinogenesis, we carried out a cell growth assay using siRNA specific to BCL2L2 to investigate whether knockdown of BCL2L2 expression would suppress proliferation of NSCLC cells showing amplification or overexpression of the gene. In HUT29 cells, the expression of BCL2L2 mRNA and protein was knocked down 48–120 h after the transient introduction of a BCL2L2‐specific siRNA (BCL2L2‐siRNA) compared with a control siRNA (Luc‐siRNA, Fig. 4A). The knockdown of endogenous BCL2L2 expression by BCL2L2‐siRNA decreased cell growth in HUT29 cells compared with the Luc‐siRNA transfected counterpart (P < 0.05 vs Luc‐siRNA‐treated control on both day 3 and day 5, Mann–Whitney U‐test; Fig. 4B). However, morphologically detected apoptotic change to nuclei was not clear in cells treated with BCL2L2‐siRNA (data not shown). The growth inhibitory effect of BCL2L2‐siRNA treatment was not observed in the A549 cell line, which shows low expression of BCL2L2 in mRNA and protein levels (2, 4), indicating that off‐target effects of BCL2L2‐siRNA on cell growth could be excluded. The phosphorylation status of SAPK/JNK was not affected by the knockdown of endogenous BCL2L2 in HUT29 cells (Supplementary Fig. S2), although it has been reported that highly expressed Bcl2L2 suppresses cell death by blocking SAPK/JNK activation in gastric cancer cell lines.( 25 ) We also tested the effect of knocking down BCL2L2 on cDDP or CPT‐induced cell death in HUT29 cells. No difference in sensitivity to the anticancer drugs was observed between cells treated with BCL2L2‐siRNA and those treated with Luc‐siRNA (Fig. 4C). Those results suggest that BCL2L2 may promote cell growth and survival in NSCLC cells through its unknown functions apart from antiapoptotic activity.
Figure 4.
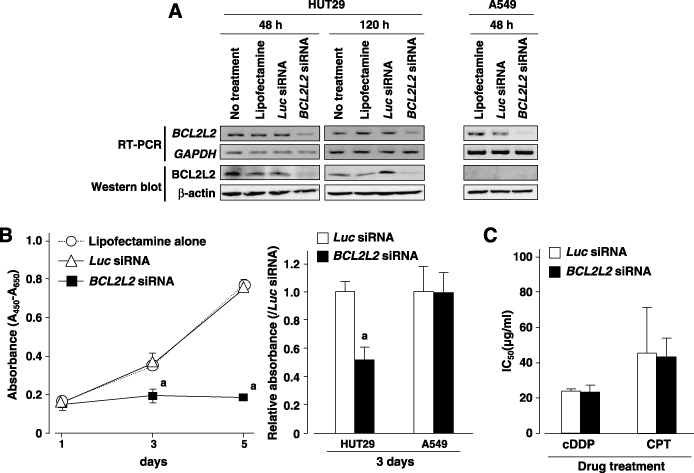
(A) Downregulation of BCL2L2 expression at the mRNA and protein levels was observed after transfection of BCL2L2 small interfering RNA (siRNA) compared with Luc‐siRNA or vehicle (Lipofectamine 2000 reagent) alone in HUT29 cells. The knockdown effect of siRNA was observed even 120 h after transfection in this cell line. In A549 cell line showing low expression of BCL2L2 (Fig. 2), downregulation of BCL2L2 mRNA was also observed 48 h after transfection. However, BCL2L2 protein was not detected by western blotting using 20 µg of protein, even in A549 cells treated with Lipofectamine 2000 alone or the control Luc‐siRNA, indicating that it is difficult to evaluate the effect of BCL2L2‐siRNA treatment on the BCL2L2 protein in this cell line. (B) The effect of downregulation of BCL2L2 expression on cell growth was observed in BCL2L2‐siRNA‐transfected HUT29 cells and control A549 cells. Cell viability was determined by water‐soluble tetrazolium salt (WTS) assay at the indicated times. Knockdown of endogenous BCL2L2 expression by BCL2L2‐siRNA in HUT29 cells significantly decreased cell growth compared with the Luc‐siRNA‐transfected counterpart (left and right). However, no inhibitory effect of BCL2L2‐siRNA treatment on cell growth was observed in A549 cells with low expression of BCL2L2 compared with the Luc‐siRNA treatment (right). Statistical analysis used the Mann–Whitney U‐test: a, P < 0.05 versus Luc‐siRNA treatment. (C) Effect of BCL2L2 knockdown on anticancer reagent‐induced cell death in HUT29 cells. siRNA‐transfected cells were treated with various concentrations of cisplatin (cDDP) or CPT‐11 (CPT) for 48 h. Cell viability was determined by WTS assay, and the inhibition concentration 50% (IC50) was calculated. No difference in viability was observed between BCL2L2‐ and Luc‐siRNA‐transfected cells after treatment with either anticancer reagent.
Because genes other than BCL2L2, which showed frequent overexpression in a panel of NSCLC cell lines (Fig. 2), may also be candidate amplification targets, we screened the effect of PSMB5 and THTPA knockdown on cell growth and viability in HUT29 cells using gene‐specific siRNA. We excluded the MYH7 gene from this cell viability screen using siRNA because this gene encodes cardiac muscle‐specific motor protein (http://www.ncbi.nlm.nih.gov/, Supplementary Table S2). Knockdown of PSMB5 and THTPA by siRNA did not affect growth and survival of HUT29 cells (Supplementary Fig. S3).
Discussion
In the study presented here, we characterized the amplicon at 14q11.2, which we detected in a NSCLC cell line derived from adenocarcinoma in our previous analysis using array‐CGH,( 12 ) and identified BCL2L2 as at least one of the targets for amplification. We further showed that (1) overexpression of BCL2L2 protein occurred in primary adenocarcinoma of the lung, especially advanced tumors with a poorer differentiation status, and (2) knockdown of this gene using siRNA inhibits cell growth, at least in part by inducing apoptosis, in cell lines with BCL2L2 amplification and overexpression. The overall results suggest that BCL2L2 contributes to the pathogenesis of NSCLC, particularly the progression of lung adenocarcinoma.
BCL2L2 is a prosurvival member of the BCL2 protein family and acts as an inhibitor of apoptosis,( 26 , 27 , 28 ) although only limited information is available about its physiological and pathophysiological roles, as well as its modes of action. Knockdown of BCL2L2 expression using gene‐specific siRNA dramatically inhibited growth of the HUT29 cell line, which shows amplification and overexpression of the gene, without any exogenous apoptotic stimuli. However, no protective effect of BCL2L2 on anticancer reagent‐induced apoptosis was observed in HUT29 cells, suggesting that overexpressed BCL2L2 may not function as a simple antiapoptotic molecule in these cancer cells. The BCL2L2‐dependent growth promotion observed in this cell line regardless of apoptotic stimuli may be explained by a recently reported widespread and important phenomenon, termed ‘oncogene addiction’.( 29 , 30 ) Although no potential molecular mechanism for BCL2L2 addiction is known, some cellular pathways that play an important role in cell growth, differentiation or survival in malignant cells may be exclusively occupied by BCL2L2, and disruption of this gene may consequently lead to dramatic growth inhibition. Therefore, BCL2L2 and BCL2L2‐dependent pathways could be attractive therapeutic targets for treatment purposes.( 31 )
One of the most striking findings in the study reported here is that primary lung adenocarcinomas frequently (65.6%) showed increased expression of BCL2L2 compared with their neighboring non‐neoplastic cells, especially in alveolar type II cells, which are considered to be the source of adenocarcinoma. In addition, expression of BCL2L2 was frequently observed in more advanced tumors (P = 0.044, Fisher's exact test) and poorly differentiated tumors (P = 0.039, χ2‐test), and cases with greater expression of the gene showed poorer overall survival, though with marginal significance (P = 0.0808, log‐rank test). Although overexpression of BCL2L2 was previously reported in various human cancer cell lines and primary tumors, such as gastric and colon cancers,( 22 , 23 , 24 , 25 ) and amplification of BCL2L2 was also found in small cell lung cancer cell lines by cDNA microarray‐based CGH (without information of its expression status),( 32 ) this is the first report supporting the notion that expression of this molecule is also deregulated in lung adenocarcinoma and may contribute to the pathogenesis of this disease. These findings support our contention that BCL2L2 is a good candidate for a therapeutic target in adenocarcinoma of the lung. In addition, further study using larger cohorts will be needed to clarify the clinical significance of BCL2L2 as a prognosticator.
In gastric cancer, it was reported that BCL2L2 overexpression is mainly observed in a particular histopathological type, the diffuse–mixed type, but, inconsistent with our findings in primary NSCLC, is not related to stage and differentiation status.( 25 ) It was also reported that overexpressed BCL2L2 promotes cancer cell invasion by inducing matrix metalloproteinase (MMP) 2 expression in gastric cancer cells.( 33 ) In our preliminary study using a Matrigel‐coated modified Boyden chamber, however, HUT29 cells did not show an invasive phenotype (Kawasaki T and Inazawa J, unpublished data), which discouraged us from evaluating the effect of BCL2L2 knockdown on cell invasion in NSCLC. Indeed, downregulation of BCL2L2 expression did not affect the level of MMP2 mRNA (Kawasaki T and Inazawa J, unpublished data). In gastric cancer cells, it was also reported that overexpressed BCL2L2 suppresses cell death by blocking SAPK/JNK activation. In the present study, however, the phosphorylation status of SAPK/JNK was not affected by knockdown of endogenous BCL2L2 in HUT29 cells. Those results suggest that the dependency of cancer cells on the overexpressed or activated BCL2L2 for various tumor‐promoting phenotypes, such as sustained survival, proliferation, invasion and metastasis, may be different among tissues or cell types. Due to this tissue‐ or cell lineage‐specific and context‐dependent ‘oncogene addiction’( 34 ) of BCL2L2, it will be necessary to determine the expression status of BCL2L2 and the biological and clinicopathological significance of overexpressed BCL2L2 in each type of tumor, including gastric, colon and lung cancers.
Among nine candidate target genes for 14q11.2 amplification other than BCL2L2, PSMB5, encoding a subunit of the 20S proteasome that has ATP‐dependent proteolytic activity, was reported to be overexpressed in breast cancer at the mRNA but not the protein level.( 35 ) It is therefore unclear whether this gene is a strong candidate target gene, even though the proteasome is known to have relevance to carcinogenesis.( 36 ) Genomic amplification in tumors does not usually involve only the core domain but may extend several hundred kilobases to several megabases flanking the selected genes. Genes coactivated via coamplification may be factors that influence the phenotype or clinical outcome of tumors. It is still unclear whether target genes within a given amplicon are merely physically linked because they share the same origin, or whether there is any functional correlation among coamplified genes in the same amplicon.( 37 , 38 ) As 10 of 28 genes within the 14q11.2 amplicon observed in HUT29 cells were overexpressed through amplification, further study will be needed to clarify the individual and cooperative functional roles of possible target genes in lung carcinogenesis. Almost no functional or clinicopathological significance has been reported, except for BCL2L2, and our preliminary cell viability screen after knockdown of gene function using gene‐specific siRNA demonstrated no significant growth regulatory effect of the PSMB5 and THTPA genes, which showed relatively frequent overexpression in NSCLC cell lines (Supplementary Fig. S3).
Supporting information
The following supplementary material is available for this article:
Fig. S1. Expression levels of the 28 candidate target genes within the 14q11.2 amplicon in four non‐small cell lung cancer (NSCLC) cell lines and immortalized lung epithelial cell lines HPL1D and BEAS2B determined by semiquantitative reverse transcription–polymerase chain reaction. See legend for Fig. 2 for interpretation. Among 28 candidate genes, 10 genes (group A) were overexpressed in HUT29 cells, which contain 14q11.2 amplification, compared with other NSCLC cell lines and control cell lines, especially HPL1D (open columns).
Fig. S2. Effect of BCL2L2‐specific small interfering RNA on activation of SAPK/JNK in HUT29 cells transfected for 48 h. Lysates were collected and subjected to western blotting with appropriate antibodies. Downregulation of BCL2L2 expression did not affect the expression of SAPK/JNK protein (SAPK/JNK) and its phosphorylation status (p‐SAPK/JNK).
Fig. S3. Effect of downregulation of PSMB5 (A) or THTPA (B) expression using small interfering RNA (siRNA) specific for each gene in HUT29 cells. Expression of PSMB5 (A) and THTPA (B) mRNA decreased after transfection of gene‐specific siRNA compared with the control Luc‐siRNA or vehicle (Lipofectamine 2000 reagent) alone (upper). However, no inhibitory effect of downregulation of PSMB5 (A) or THTPA (B) expression on cell growth was observed in gene‐specific siRNA‐transfected HUT29 cells (lower). Cell viability was determined by water‐soluble tetrazolium salt assay at the indicated times.
Table S1. Primer sequences used in this study
Table S2. Candidate target genes within the 14q11.2 amplicon
This material is available as part of the online article from: http://www.blackwell‐synergy.com/doi/abs/10.1111/j.1349‐7006.2007.00491.x (This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Acknowledgments
We are grateful to Professor Takashi Takahashi (Laboratory of Cancer Cell Biology, Research Institute for Disease Mechanism and Control, Nagoya University Graduate School of Medicine), Professor Akira Horii (Department of Molecular Pathology, Tohoku University Graduate School of Medicine), and Professor Takehiko Fujisawa (Department of Thoracic Surgery, Chiba University Graduate School of Medicine) for providing NSCLC cell lines and cell lines from human lung and bronchial epithelial cells. This work was supported by Grants‐in‐Aid for Scientific Research and 21st Century Center of Excellence Program for Molecular Destruction and Reconstitution of Tooth and Bone from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by Core Research for Evolutional Science and Technology of the Japan Science and Technology Corporation; and by the Haraguchi Memorial Foundation.
References
- 1. Watanabe Y, Ozasa K, Nagura J et al . Mortality in the JACC study till 1999. J Epidemiol 2005; 15 (Suppl. 1): S74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pao W, Miller VA. Epidermal growth factor receptor mutations, small‐molecule kinase inhibitors, and non‐small‐cell lung cancer: current knowledge and future directions. J Clin Oncol 2005; 23: 2556–68. [DOI] [PubMed] [Google Scholar]
- 3. Snijders AM, Nowak N, Segraves R et al . Assembly of microarrays for genome‐wide measurement of DNA copy number. Nat Genet 2001; 29: 263–4. [DOI] [PubMed] [Google Scholar]
- 4. Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet 2003; 12: 145–52. [DOI] [PubMed] [Google Scholar]
- 5. Inazawa J, Inoue J, Imoto I. Comparative genomic hybridization (CGH)‐arrays pave the way for identification of novel cancer‐related genes. Cancer Sci 2004; 95: 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwab M. Oncogene amplification in solid tumors. Semin Cancer Biol 1999; 9: 319–25. [DOI] [PubMed] [Google Scholar]
- 7. Imoto I, Yang ZQ, Pimkhaokham A et al . Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell. Cancer Res 2001; 61: 6629–34. [PubMed] [Google Scholar]
- 8. Yokoi S, Yasui K, Saito‐Ohara F et al . A novel target gene, SKP2, within the 5p13 amplicon that is frequently detected in small cell lung cancers. Am J Pathol 2002; 161: 207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takada H, Imoto I, Tsuda H et al . Screening of DNA copy‐number aberrations in gastric cancer cell lines by array‐based comparative genomic hybridization. Cancer Sci 2005; 96: 100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tanami H, Tsuda H, Okabe S et al . Involvement of cyclin D3 in liver metastasis of colorectal cancer, revealed by genome‐wide copy‐number analysis. Laboratory Invest 2005; 85: 1118–29. [DOI] [PubMed] [Google Scholar]
- 11. Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene 2007; 26: 1178–87. [DOI] [PubMed] [Google Scholar]
- 12. Izumi H, Inoue J, Yokoi S et al . Frequent silencing of DBC1 is by genetic or epigenetic mechanisms in non‐small cell lung cancers. Hum Mol Genet 2005; 14: 997–1007. [DOI] [PubMed] [Google Scholar]
- 13. Imoto I, Izumi H, Yokoi S et al . Frequent silencing of the candidate tumor suppressor PCDH20 by epigenetic mechanism in non‐small‐cell lung cancers. Cancer Res 2006; 66: 4617–26. [DOI] [PubMed] [Google Scholar]
- 14. Masuda A, Kondo M, Saito T et al . Establishment of human peripheral lung epithelial cell lines (HPL1) retaining differentiated characteristics and responsiveness to epidermal growth factor, hepatocyte growth factor, and transforming growth factor β1. Cancer Res 1997; 57: 4898–904. [PubMed] [Google Scholar]
- 15. Reddel RR, Ke Y, Gerwin BI et al . Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus‐12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res 1998; 48: 1904–9. [PubMed] [Google Scholar]
- 16. UICC . TNM. Classification of Malignant Tumours, 6th edn. Geneva: UICC, 2002. [Google Scholar]
- 17. Watanabe Y. TNM classification for lung cancer. Ann Thorac Cardiovasc Surg 2003; 9: 343–50. [PubMed] [Google Scholar]
- 18. Tsuchiya R. The Japanese database for the staging of lung cancer. Ann Ital Chir 1999; 70: 905. [PubMed] [Google Scholar]
- 19. Kitinya JN, Sueishi K, Tanaka K, Katsuda Y. Immunoreactivity of surfactant‐apoprotein in adenocarcinomas, large cell and small cell carcinomas of the lung. Acta Pathol Jpn 1986; 36: 1271–8. [DOI] [PubMed] [Google Scholar]
- 20. Kaufmann T, Schinzel A, Borner C. Bcl‐w (edding) with mitochondria. Trends Cell Biol 2004; 14: 8–12. [DOI] [PubMed] [Google Scholar]
- 21. Gibson L, Holmgreen SP, Huang DC et al . Bcl‐w, a novel member of the bcl‐2 family, promotes cell survival. Oncogene 1996; 13: 665–75. [PubMed] [Google Scholar]
- 22. O’Reilly LA, Print C, Hausmann G et al . Tissue expression and subcellular localization of the pro‐survival molecule Bcl‐w. Cell Death Differ 2001; 8: 486–94. [DOI] [PubMed] [Google Scholar]
- 23. Wilson JW, Nostro MC, Balzi M et al . Bcl‐w expression in colorectal adenocarcinoma. Br J Cancer 2000; 82: 178–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kitamura S, Kondo S, Shinomura Y. Met/HGF receptor modulates bcl‐w expression and inhibits apoptosis in human colorectal cancers. Br J Cancer 2000; 83: 668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee HW, Lee SS, Lee SJ, Um HD. Bcl‐w is expressed in a majority of infiltrative gastric adenocarcinomas and suppresses the cancer cell death by blocking stress‐activated protein kinase/c‐Jun NH2‐terminal kinase activation. Cancer Res 2003; 63: 1093–100. [PubMed] [Google Scholar]
- 26. Gibson L, Holmgreen SP, Huang DC et al . Bcl‐w, a novel member of the bcl‐2 family, promotes cell survival. Oncogene 1996; 13: 665–75. [PubMed] [Google Scholar]
- 27. Antonsson B. Bax and other pro‐apoptotic Bcl‐2 family killer proteins and their victim, the mitochondrion. Cell Tissue Res 2001; 306: 347–61. [DOI] [PubMed] [Google Scholar]
- 28. Print CG, Loveland KL, Gibson L et al . Apoptosis regulator bcl‐w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA 1998; 95: 12 424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weinstein IB. Cancer: addiction to oncogenes – The Achilles heal of cancer. Science 2002; 297: 63–4. [DOI] [PubMed] [Google Scholar]
- 30. Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction – a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 2006; 3: 448–57. [DOI] [PubMed] [Google Scholar]
- 31. Hubner A, Jaeschke A, Davis RJ. Oncogene addiction: role of signal attenuation. Dev Cell 2006; 11: 752–4. [DOI] [PubMed] [Google Scholar]
- 32. Kim YH, Girard L, Giacomini CP et al . Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 2006; 25: 130–8. [DOI] [PubMed] [Google Scholar]
- 33. Bae IH, Park MJ, Yoon SH et al . Bcl‐w promotes gastric cancer cell invasion by inducing matrix metalloproteinase‐2 expression via phosphoinositide 3‐kinase, Akt, and Sp1. Cancer Res 2006; 66: 4991–5. [DOI] [PubMed] [Google Scholar]
- 34. Garraway LA, Sellers WR. Lineage dependency and lineage‐ survival oncogenes in human cancer. Nat Rev Cancer 2006; 6: 593– 602. [DOI] [PubMed] [Google Scholar]
- 35. Deng S, Zhou H, Xiong R et al . Over‐expression of genes and proteins of ubiquitin specific peptidases (USPs) and proteasome subunits (PSs) in breast cancer tissue observed by the methods of RFDD‐PCR and proteomics. Breast Cancer Res Treat Sep 27 2006 [Epub ahead of print]. [DOI] [PubMed]
- 36. Voorhees PM, Dees EC, O’Neil B et al . The proteasome as a target for cancer therapy. Clin Cancer Res 2003; 9: 6316–25. [PubMed] [Google Scholar]
- 37. Zender L, Spector MS, Xue W et al . Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125: 1253–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang ZQ, Streicher KL, Ray ME, Abrams J, Ethier SP. Multiple interacting oncogenes on the 8p11‐p12 amplicon in human breast cancer. Cancer Res 2006; 66: 11 632–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following supplementary material is available for this article:
Fig. S1. Expression levels of the 28 candidate target genes within the 14q11.2 amplicon in four non‐small cell lung cancer (NSCLC) cell lines and immortalized lung epithelial cell lines HPL1D and BEAS2B determined by semiquantitative reverse transcription–polymerase chain reaction. See legend for Fig. 2 for interpretation. Among 28 candidate genes, 10 genes (group A) were overexpressed in HUT29 cells, which contain 14q11.2 amplification, compared with other NSCLC cell lines and control cell lines, especially HPL1D (open columns).
Fig. S2. Effect of BCL2L2‐specific small interfering RNA on activation of SAPK/JNK in HUT29 cells transfected for 48 h. Lysates were collected and subjected to western blotting with appropriate antibodies. Downregulation of BCL2L2 expression did not affect the expression of SAPK/JNK protein (SAPK/JNK) and its phosphorylation status (p‐SAPK/JNK).
Fig. S3. Effect of downregulation of PSMB5 (A) or THTPA (B) expression using small interfering RNA (siRNA) specific for each gene in HUT29 cells. Expression of PSMB5 (A) and THTPA (B) mRNA decreased after transfection of gene‐specific siRNA compared with the control Luc‐siRNA or vehicle (Lipofectamine 2000 reagent) alone (upper). However, no inhibitory effect of downregulation of PSMB5 (A) or THTPA (B) expression on cell growth was observed in gene‐specific siRNA‐transfected HUT29 cells (lower). Cell viability was determined by water‐soluble tetrazolium salt assay at the indicated times.
Table S1. Primer sequences used in this study
Table S2. Candidate target genes within the 14q11.2 amplicon
This material is available as part of the online article from: http://www.blackwell‐synergy.com/doi/abs/10.1111/j.1349‐7006.2007.00491.x (This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item