

Abstract
The DRH is an inbred rat strain established by selective mating of the 3′‐Me‐DAB resistant progeny of closed colony Donryu rats over 20 generations. Genetic analysis shows that two semidominant QTLs, Drh1 and Drh2, are responsible for strong resistance to chemical‐induced hepatocarcinogenesis in DRH strain rats. To evaluate the effect of the single Drh1 locus on various stages of liver carcinogenesis, we constructed a speed congenic strain DRH.F344‐Drh1 by transferring a susceptible Drh1 allele of F344 to DRH rats by marker‐assisted backcrossing. The DRH.F344‐Drh1 rats had a ∼43 cM segment of chromosome 1 bearing Drh1 but the Drh2 was of the DRH allele. After oral administration of 3′‐Me‐DAB for 8 weeks, DRH.F344‐Drh1 had as many enzyme altered foci as F344, whereas the quantitative parameters of fibrosis, enzyme altered foci, GST‐P expression and proliferation of liver cells in DRH.F344‐Drh1 rats were intermediate between F344 and DRH. In the liver of carcinogen‐fed DRH rats, there was intensive apoptosis as detected by TUNEL stain, but not in the liver of F344 and DRH.F344‐Drh1 rats. Injection of lead nitrate (100 µmol/kgB.W) induced a wave of liver cell proliferation, as seen by BrdU uptake within a few days in F344 and DRH.F344‐Drh1 rats, but not in DRH rats. Instead, there were numerous TUNEL‐positive nuclei in the DRH liver after lead nitrate injection. Apparently, the hepatocytes were removed by apoptosis during transition from G0 to G1. The major role of Drh1 is effective removal of the hepatocytes newly recruited to proliferate after chemical injury. Resistance to preneoplastic lesions in DRH rats may well be based on similar mechanism. (Cancer Sci 2005; 96: 164–169)
Chemical‐induced hepatocarcinogenesis in rats occurs in the multistep process, each of which is under regulation of a number of host genetic and epigenetic factors. 1 , 2 The administration of a hepatocarcinogen to rats leads to the development of enzyme‐altered foci (EAF) of liver cells intensely expressing phase II enzymes, such as glutathione S‐transferase placental form (GST‐P) or γ‐glutamyltranspeptidase (GGT‐P) 3 , 4 within 4–6 weeks. The subsequent step is the promotion of the initiated cells within the EAF foci, which are subjected to further mutational events. New clones with more malignant phenotypes emerge during the progression step to give rise to hepatocellular carcinomas (HCC). However, EAF may also undergo the remodeling pathway 5 , 6 either by reversion or apoptotic death. To dissect such polygenic traits of chemical hepato‐carcinogenesis it is indispensable to study appropriate animal models.
The inbred rat strain DRH offers a useful experimental model to elucidate the genetic susceptibility and resistance to chemical‐induced liver carcinomas. 7 , 8 , 9 This strain is established by inbreeding closed colony Donryu rats for more than 20 generations under continuous feeding of a diet containing 3′‐methyl‐4‐dimethyl‐aminoazobenzene (3′‐Me‐DAB) and by selecting for reduced HCC induction during inbreeding for more than 10 years. DRH rats are highly resistant not only to 3′‐Me‐DAB and its derivatives, but also to other structurally different hepatocarcinogens such as 2‐acetylaminofluorene, and N‐nitrosodimethylamine. (10) Moreover, DRH rats are resistant to mammary cancers induced by 7,12‐dimethybenz(a)anthracene, (10) which is metabolically activated by the mechanism different from aminoazo carcinogens. In our previous study, analyzing (F344 × DRH)F 2 rats fed 3′‐Me‐DAB, two resistant quantitative trait loci (QTLs), Drh1 and Drh2, were mapped on rat chromosome 1 (RNO1) and RNO4, 11 , 12 both loci yield resistance to EAF formation at preneoplastic stage (% variance of phenotype explained 18% and 16%, respectively), (11) and Drh2 suppresses their progression. (12) The complex genetic trait is, however, a hazard to understand the functional role of an individual locus. In the present study, to evaluate the contribution of Drh1 to resistance to preneoplastic lesions, we constructed a speed congenic strain DRH.F344‐Drh1, namely, DRH rats introgressed an F344 RNO1 segment bearing Drh1 by marker‐assisted backcrossing. Subsequently, we examined the quantitative parameters of earlier stage of 3′‐Me‐DAB induced hepatocarcinogenesis, such as liver fibrosis, EAF formation, GST‐P induction, proliferation of liver cells, and apoptosis in the age‐matched congenic and parental rats. In addition, proliferative activity induced after a single injection of lead nitrate was studied. The effective apoptotic removal of the hepatocytes newly recruited to proliferation after chemical injury is a likely mechanism of resistance by Drh1.
Materials and Methods
Construction of speed congenic strain rats. All animal experiments were approved by the Ethical Committee for Animal Experiments and carried out under the Guidelines for Animal Experiments of Kyoto University.
Inbred DRH and F344 rats were purchased from SEAC Yoshitomi Co. (Fukuoka, Japan). The animals were housed individually in suspended wire‐bottomed cages in a room with constant temperature and humidity with a 12‐h light‐dark cycle and allowed free access to food and water. The speed congenic strain was developed by a marker‐assisted selection method. (14) A (DRH × F344)F 1 male was mated with DRH female rats to produce the progenies and a male heterozygous at F344‐derived Drh1 segment and homozygous at DRH‐derived Drh2 segment was selected out of 16 N2 pups as described later. Therefore, the males selected in every generation were successively backcrossed to DRH females until N6 generations, at which time Drh1 heterozygous brother and sister siblings were intercrossed, and among their progeny, homozygous pairs were selected for subsequent inbreeding. The speed congenic strain was designated as DRH.F344‐Drh1 according to the nomenclature suggested by Mouse Genome Informatics (2004). In the present study, the DRH.F344‐Drh1 rats were at the fourth to fifth generation after resuming inbreeding. We also attempted to generate a reverse speed congenic strain F344.DRH‐Drh1, but they were accidentally lost during backcrossing. DRH and DRH.F344‐Drh1 rats have been deposited in the National Bio Resource Project (http://www.anim.med.kyoto‐u.ac.jp/nbr/home.htm) in Kyoto University.
Genotype analysis. A 1‐cm long tail was sampled from each rat at 4 weeks of age and genomic DNA was extracted. All primers for PCR‐based microsatellite analysis were purchased from Research Genetics, Inc. (Huntsville, AL, USA). Polymorphic microsatellite markers between DRH and F344 used in this study were for RNO1, D1Mgh2, D1Mit9, D1Wox6, D1Mgh8, D1Rat50, D1Rat47, D1Got155, D1Got157, D1Rat51, D1Got156, D1Rat54, D1Rat139, D1Rat114, D1Got193, D1Rat112, D1Rat70, D1Wox10, D1Rat73, D1Rat75, D1Mgh12, D1Wox25, D1Mit9, D1Wox11, and D1Wox21; for RNO2, D2Mgh14, D2Mit15, and D2Mit14; for RNO3, D3Mit9, D3Mit13, D3Mgh2, and D3Mgh16; for RNO4, D4Mgh2, D4Mgh6, D4Mgh13, D4Rat51, D4Rat34, D4Rat38, and D4Rat48; for RNO5, D5Mgh5, D5Mit12, and D5Mgh14; for RNO6, D6Mgh5, D6Mit12, and D6Mgh3; for RNO7, D7Mit4, D7Mit28, and D7Mit11; for RNO8, D8Mgh11, D8Mgh3, and D8Mit6; for RNO9, D9Rat135, D9Mit3, and D9Bro1; for RNO10, D10Mit5, D10Mgh6, and D10Mit1; for RNO11, D11Mgh2, and D11Mit2; for RNO12, D12Mgh2, and D12Mit5; for RNO13, D13Mgh2, and D13Mit4; for RNO14, D14Mit1; for RNO15, D15Mgh7; for RNO16, D16Mgh5, and D16Mit2; for RNO17, D17Mgh5, D17Mit2, and D17Mit5; for RNO18, D18Mit8, and D18Mgh7; for RNO19, D19Mgh8; for RNO20; D20Rat1, and D20Mit1. Genotype screening was done very densely for RNO1 and RNO4. For other chromosomes, average and maximal distances between markers were 30.2 cM and 55.2 cM. Theoretically, the percentage DRH/F344 segments was < 1%. (14)
For routine screening of Drh1, D1Mgh12, D1Wox10, and D1Mgh8 were used, and for Drh2, D4Rat34, D4Rat38, and D4Rat48. (11) The method for PCR and agarose electrophoresis of PCR products was described previously. (13)
Induction of preneoplastic lesion. 3′‐Me‐DAB was purchased from Tokyo Kasei Kogyo Co. (Tokyo, Japan). To induce preneoplastic lesions, 4‐week‐old male rats were allowed free access to water and rat pellets containing 0.06% 3′‐Me‐DAB. All rats were killed after 8 weeks or noted as otherwise under ether anesthesia, and a full postmortem examination was carried out.
Immunohistochemistry. The standard Catalyzed Signal Amplification System (DAKO, Carpinteria, CA, USA) was employed in immunohistochemistry as described by Miyagawa‐Hayashino et al. (15) Liver tissues were fixed in 4% paraformaldehyde and paraffin embedded by standard techniques. After deparaffinization, the sections were treated with 3% hydrogen peroxide and protein blocking solution. For GST‐P, 1 : 1000 diluted rabbit anti‐GST‐P antibody (MBL Co. Ltd, Nagoya, Japan) and biotin‐labeled antirabbit IgG as the second antibody were used. The foci of GST‐P‐positive cells > 0.2 mm diameter were counted as EAF. For CyclinD1 or PCNA, 1 : 100 diluted mouse monoclonal anti‐CyclinD1 (DAKO) or anti‐PCNA antibody (DAKO), and biotin‐labeled antimouse IgG were used. Subsequently the sections were treated with avidin‐biotin‐peroxidase complex (DAKO). The positive staining of peroxidase binding was visualized by the diaminobenzidine method. TUNEL (TdT‐mediated dUTP nick end labeling) stain was performed with Apoptosis in situ Detection Kit (WAKO, Osaka, Japan). De‐paraffinized liver sections were treated with proteinase K (DAKO), and hybridized with TdT solution and peroxidase‐conjugated antibody according to the kit protocol. For single stranded DNA (ssDNA), 1 : 200 diluted polyclonal rabbit antissDNA antibody (DAKO) and biotin‐labeled antirabbit IgG were used.
BrdU uptake. Temporary liver regeneration was studied by injecting 4‐week‐old male rats i.p. with lead nitrate (100 µmol/kg B.W.) (WAKO), (16) and subsequently with 30 mg/kg B.W. 5‐bromo‐2′‐deoxyuridine (BrdU) (WAKO) 2 h before killing. 17 , 18 Paraffin sections (3‐µm thickness) of liver tissues were placed on poly L‐lysine‐coated slides. The uptake of BrdU in liver cell nuclei was visualized with the 5‐Bromo‐2′‐deoxy‐uridine Labeling and Detection Kit II (Roche, USA), according to the manufacturer's instruction. After counterstaining with hematoxylin, 1000 nuclei were counted per 40× view fields to determine percentage of BrdU‐positive nuclei.
Evaluation of fibrosis. Liver fibrosis induced by 3′‐Me‐DAB was quantitatively evaluated by screening 12 randomly selected X40‐view fields of Azan‐stained sections with a color image processor. The percentage of blue‐stained area, representing fibrosis in the total liver area, was measured.
Image analysis. All histological sections were processed quantitatively using a color image processor AnalySIS 3.1 (Soft Imaging System GmbH, Muenster, Germany). For each sample, 12 independent high‐power fields were analyzed for each section for quantitative parameters. The highest and the lowest counts were discarded and 10 counts were used for statistical analysis.
Quantitative real‐time RT‐PCR. The protocol of real‐time RT‐PCR was identical to that in Yabe et al. (19) Briefly, total RNA was prepared from livers of rats by using ISOGEN (Wako) (11) and treated with DNase I (DNA‐free; Ambion Dignostics Inc., Austin, TX, USA). First‐strand cDNA was synthesized from DNase I‐treated total RNA with random hexamer primers by using the ABI cDNA Synthesis Kit (Applied Biosystems, Foster City, CA, USA). Synthesized cDNA was subsequently mixed with 2X SYBR Green PCR Master Mix (Applied Biosystems) and GST‐P forward primer: 5′‐GCT GGA AGG AGG AGG TGG T‐3′ and reverse primer: 5′‐CTC AAG ATG GCA TTA GAT TGG TAA AGG‐3′ and subjected to real‐time PCR quantification using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). All reactions were performed in triplicate. The relative amounts of mRNAs were calculated by using the comparative CT method. Rat β‐actin mRNA was used as a control for this assay.
Statistical analysis. All the numerical data were shown as mean standard deviation (SD), and statistical analysis was performed with either ANOVA or Student's t‐test using StatView software. When the P‐value was < 0.05, the difference was considered to be significant, and when the P‐value was < 0.01, highly significant.
Results
Establishment of speed congenic DRH.F344‐Drh1 rats. Speed congenic DRH.F344‐Drh1 rats were generated by selective transfer of the F344 RNO1 segment bearing Drh1 to DRH rats by marker assisted backcrossing. As shown in Fig. 1, the congenic rats had a ∼43 cM F344‐derived introgressed RNO1 segment between D1Mgh8 (86 cM from centromere) and D1Mgh12 (129 cM). On this segment, we found 11 polymorphic marker loci and all of them were homozygous for F344 allele. Marker loci outside this segment, D1Mgh2, D1Mit9, D1Wox25, and D1Wox6 were homozygous for DRH allele. Our previous study (11) showed that the QTL peak for Drh1 (an arrowhead in Fig. 1) is between D1Wox10 (113 cM) and D1Mgh12 (129 cM). In this study, we identified two new marker loci D1Rat73 (116 cM) and D1Rat75 (124.5 cM) that were closer to the Drh1 peak. For allocation of Drh1 on the ∼8.5 cM segment between D1Rat73 and D1Rat75 further study will be required. However, the RNO4 segment housing Drh2 was determined to be completely of DRH origin by genotyping of D4Rat34, D4Rat38, and D4Rat48, markers loci in the Drh2 segment (data not shown). In addition, 55 microsatellite markers randomly scattered in whole genome were genotyped, but all were homozygous for DRH allele.
Figure 1.
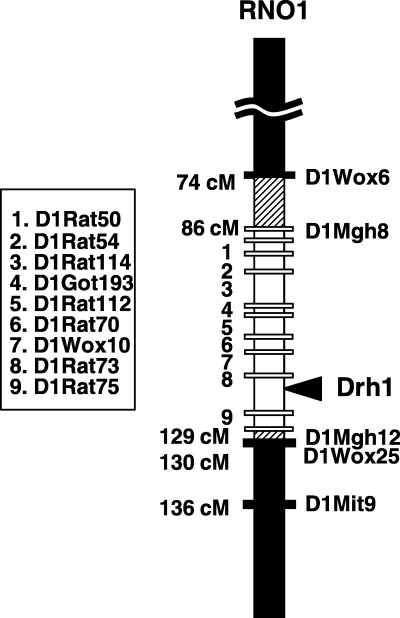
Drh1 segment of RNO1 in the DRH.F344‐Drh1 rat. Numbers on the left indicate the map location of microsatellite marker loci listed in the box. The closed column represents a DRH‐derived segment; The hatched column, undetermined; and the open column, an F344‐derived segment. The closed bars represent the loci with the DRH allele, and open bars, the loci with the F344 allele. The arrowhead shows the map location of the Drh1 QTL peak.
Effects of F344‐Drh1 on 3′‐Me‐DAB induced preneoplastic lesions. Induction of GST‐P and enzyme altered foci: Induction of GST‐P and EAF, the preneoplastic lesions of hepatocarcinogenesis, was examined after 8 weeks feeding of 3′‐Me‐DAB (Fig. 2a) and the results are summarized in Table 1. The number of EAF was equivalent in F344 and DRH.F344‐Drh1 (P = 0.23), but significantly higher than that in DRH (P < 0.0001). This suggests that Drh1 plays a major role in determining the number of EAF. A kinetic study showed that EAF formation was low in DRH even at 4 weeks of 3′‐Me‐DAB administration (Fig. 3). As shown in Table 1, the average sizes of EAF and the percentage liver area occupied by EAF were high in F344, intermediate in DRH.F344‐Drh1, and low in DRH (P < 0.0001). The level of GST‐P mRNA determined by real time RT‐PCR in the congenic rat liver was an intermediate of both parental strains (P < 0.001) (Table 1). Data for cyclin D1 and PCNA obtained by immunohistochemistry of livers were also intermediate of both parental strains (P < 0.0001) (Figs 2b,c) (Table 1). These findings suggest that Drh1 locus is an important determinant for the GST‐P expression and growth of EAF cells but that another locus in DRH, probably Drh2, also serves as the second determinant.
Figure 2.
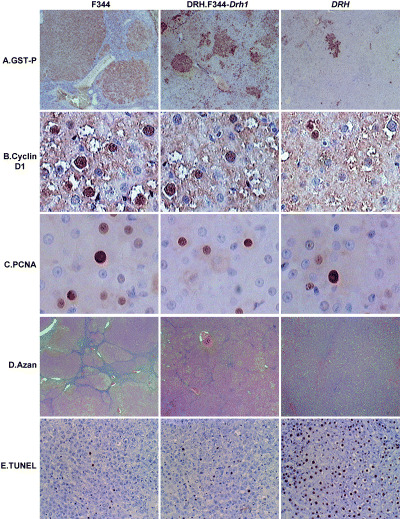
Histochemical analysis of the livers after 8 weeks of administration of 3′‐DAB. (a) GST‐P (×40), (b) Cyclin D1 (×200), (c) PCNA (×200), (d) Azan stain to show fibrosis (×20), (e) TUNEL stain (×40). See Table 1 for quantitative analysis data by image analyzer.
Table 1.
Effects of Drh1 on preneoplastic liver lesion induced by 3′‐Me‐DAB
| Phenotype parameters | F344 | DRH.F344‐Drh1 | DRH |
|---|---|---|---|
| No. of EAF per a 40X filed | 9.6 ± 2.7*, † | 8.6 ± 1.4 † | 2.5 ± 0.8 |
| Percent GST‐P positive area | 44.2 ± 5.2 | 12.6 ± 2.8 | 3.0 ± 1.6 |
| Mean area of EAF (mm2) | 0.08 ± 0.02 | 0.02 ± 0.006 | 0.007 ± 0.004 |
| GST‐P mRNA (fold) | 36.1 ± 10.5 | 12.2 ± 2.7 | 2.2 ± 0.51 |
| Percent CyclinD1 positive cells | 29.1 ± 7.3 | 17.1 ± 3.1 | 7.1 ± 2.5 |
| Percent PCNA positive cells | 23.1 ± 4.4 | 10.6 ± 4.2 | 3.7 ± 0.9 |
| Percent area of fibrosis | 17.9 ± 3.3 | 11.3 ± 2.9 | 5.7 ± 0.8 |
| Percent TUNEL positive cells | 1.3 ± 0.8 ‡ | 2.9 ± 1.5 ‡ | 57.9 ± 7.3 |
Mean ± SD, n = 6. Significantly different in ANOVA (< P, 0.05) except for
(P = 0.23) and
(P = 0.64).
Figure 3.
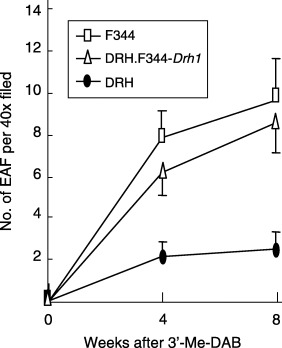
Time course of GST‐P EAF formation. Each point represents an average of six rats.
Fibrosis after 3′‐Me‐DAB feeding. The development of liver fibrosis is an indispensable consequence of damage by liver carcinogens. After 3′‐Me‐DAB administration for 8 weeks, extensive liver fibrosis developed in F344 but much less in DRH rats, except for mild steatosis (Fig. 2d). In the DRH.F344‐Drh1 rats, the development was intermediate. The extent of fibrosis was quantitatively evaluated with an image processor (Table 1) and the difference was statistically significant (P < 0.0001). The data indicate that DRH rats were highly resistant to fibrosis formation after 3′‐DAB administration, for which Drh1 locus was partially responsible.
Apoptosis in preneoplastic lesions. It is claimed that EAF can be modulated by apoptosis of liver cells in resistant rats. The administration of S‐adenosyl‐L‐methionine to Wistar rats decreases the number and size of diethylnitrosamine (DEN)‐induced EAF by inhibition of DNA synthesis, increased apoptotic cell deaths, remodeling, and loss of biochemical markers. (5) To see the possible effect of Drh1 on liver cell apoptosis, we stained preneoplastic liver sections with TUNEL (Fig. 2e). A remarkable higher TUNEL‐positive signal was detected in DRH liver than F344 and DRH.F344‐Drh1 liver (P < 0.0001) whereas the signals in the latter two were equally low (P = 0.64) (Table 1). The specificity of the TUNEL reaction was verified by immuno‐staining with anti‐ssDNA (data not shown). Therefore, induction of apoptosis may be predominantly determined by Drh1, but not by Drh2.
Effects of lead nitrate on hepatocytes proliferation and apoptosis. Cell proliferation is another response of liver after injury. We studied the effect of a single i.p. injection of lead nitrate into rats that induces a synchronized wave of hepatocyte proliferation. It was found that the percentage of BrdU‐positive liver cells was significantly lower in DRH than in F344 rats 2 days after treatment (P < 0.001). In DRH.F344‐Drh1, the percentage of BrdU positive liver cells was as high as in F344 rats (Fig. 4). TUNEL stain of the liver 2 days after lead nitrate injection revealed increased apoptosis in DRH (36.8 ± 5.7%), but much less in the DRH.F344‐Drh1 (3.4 ± 1.8%) and F344 rats (2.1 ± 1.9%). Drh1 may induce apoptosis to the hepatocytes newly recruited to proliferation after chemical injury.
Figure 4.
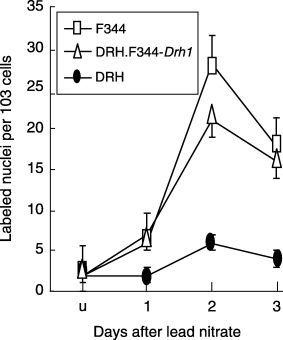
BrdU uptake after a single i.p. injection of lead nitrate. Each point represents an average of three rats.
Growth characteristics. One of the remarkable properties of DRH rats was their consistently larger body size than F344 (P < 0.001 at all age). DRH and DRH.F344‐Drh1 rats shared similar gain of body weight that was significantly higher than F344 (Fig. 5). Therefore, the Drh1 locus was not a determinant of growth and body weight.
Figure 5.
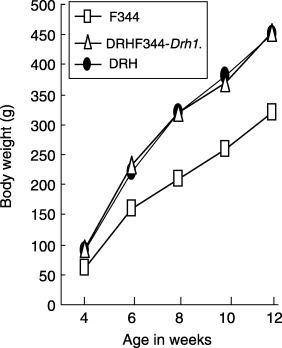
Growth of DRH, DRH.F344‐Drh1, and F344 rats. Each point represents an average of six rats.
Discussion
Genetic resistance of DRH rats to 3′‐Me‐DAB is provided by two QTLs on RNO1 and RNØ4. 11 , 12 Judged from quantitative parameters relevant to these QTL, both Drh1 and Drh2 strongly inhibited GST‐P induction and formation of EAF in an earlier stage of liver carcinogenesis. The effect of Drh1 is limited to the preneoplastic lesion, whereas that of Drh2 extends to their progression and the growth of liver cancers. (11) The present study aims to evaluate the function of the single locus Drh1. For this purpose, we constructed speed congenic DRH.F344‐Drh1 rats by marker‐assisted backcrossing (i.e. DRH rats carrying a ∼43 cM F344 RNO1 segment with Drh1). Drh1 is a QTL locus mapped from study of a relatively small number of (DRH × F344)F 2 rats. (11) The locus itself showed a very high LOD score, however, it may contain several genes clustering in this segment. Furthermore, the relatively long introgressed Drh1 segment may contain other loci affecting EAF formation. Selective backcrossing of Drh1F344/DRHDrh2DRH/DRH males to DRH females for six generations and subsequent four or more generations inbreeding of Drh1F344/F344Drh2DRH/DRH progenies made F344 allele at the loci outside the Drh1 segment < 1%. This does not warrant total absence of contribution of F344 derived loci except for Drh1, although none of 55 marker loci located outside Drh1 segment including those for Drh2 had F344 allele. Beyond these shortcomings, the data obtained from the speed congenic rats seem to provide important clues for genetic resistance to liver carcinogenesis.
From the observation of DRH.F344‐Drh1 rats given 3′‐Me‐DAB, the number of EAF is primarily determined by the Drh1 genotype, since F344 and DRH.F344‐Drh1 rats had a comparable number of EAF, but DRH had a much lower number. The higher TUNEL reaction in DRH liver at this stage may suggest that the reduced number of EAF may be due to the apoptotic death of GST‐P positive cells. However, this hypothesis requires careful consideration. It is not clear whether the initiated cells in DRH rats disappeared from established EAF by modulation either through apoptotic removal of enzyme‐altered cells or re‐differentiation, or by elimination of initiated cells immediately after exposure to the chemical. The observation that the number of EAF was fewer in DRH than F344 and congenic rats even at 4 weeks of 3′‐Me‐DAB administration seems to support the latter possibility. Chemical injury with lead nitrate is known to recruit the liver cells resting in G0 to enter cell cycle and induce phase II enzymes such as GST‐P. (20) The DRH rats, but not F344 and DRH.F344‐Drh1 rats, failed to start DNA synthesis and showed higher apoptosis after lead nitrate. It is shown that the resistance of DRH rats to hepatocarcinogens was not specific to their chemical structure. (10) Therefore, it is plausible that the reduced number of EAF is due to apoptotic elimination of the cells affected by a chemical to enter the cell cycle.
In contrast, other parameters for EAF in the DRH.F344‐Drh1 rats (i.e. GST‐P mRNA, average size of EAF, and growth markers for liver cells) were intermediate between F344 and DRH, indicating the resistance was also conferred by other loci segregating independently from Drh1. Consistent with our previous report, (21) the growth of GST‐P positive cells and the formation of EAF are therefore under the control of at least two genes. The extent of liver fibrosis in the congenic rats was also an intermediate of F344 and DRH. In contrast, Drh1 is not responsible for the heavier body weight in DRH rats.
Our previous study (11) revealed that Drh2 is also a determinant of the EAF number, although its contribution is less than Drh1. Considering the function of Drh1 in the early stage of EAF formation, it seems reasonable to assume that Drh1 is epistatic to Drh2 in respect of the number of EAF.
Genetic resistance to carcinogen‐induced hepatocarcinogenesis has been studied in several strains of rats. 22 , 23 , 24 , 25 It is not easy to compare these models directly, because the carcinogenic protocols, rat strains, and quantitative parameters to evaluate the steps of carcinogenesis are variable among different models. Feo et al. 22 , 26 , 27 , 28 , 29 reported that Brown Norway, Wistar, and Copenhagen rats show genetic resistance to liver cancers induced by combined treatment of DEN and acetoaminofluorene plus partial hepatectomy. Re‐differentiation or apoptotic death of EAF cells 5 , 6 , 29 is assumed to be a mechanism of resistance. Among numerous QTL mapped in these models, Hcs3 and Hcs5 in Copenhagen rats (29) and Hcs3 and Hcs2 in Brown Norway rats (22) are mapped on the distal segment of RNO1 and they determine the number and volume of EAF. Although their phenotypic effects are much less than those of Drh1 and the exact map location is difficult to assess, it is possible that one of them may be identical to Drh1. In Copenhagen rats, unlike DRH, susceptibility to the number of EAF is semidominant. However, this does not necessarily support or exclude their identity, as different alleles may be used in different combinations of rat strains. Further study would be required to elucidate their relationship and the molecular basis of the resistance.
Acknowledgments
We are grateful to Drs Sugiyama, Toyokuni, Tsuruyama, and Yamada for their helpful discussions and to Messrs. Toda and Koda for histological sections and histochemistry. This study was supported by Grants‐in‐Aid for Cancer Research from the Ministry of Education, Culture, Culture and Sports, and Ministry of Health, Welfare and Labor, Japan.
References
- 1. Mills JJ, Jirtle RL, Boyer IJ. Mechanisms of liver tumor promotion. In: Jirtle RL (eds). Liver regeneration and carcinogenesis. Academic Press, San Diego 1995; 199–226. [Google Scholar]
- 2. Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002; 31: 339–45. [DOI] [PubMed] [Google Scholar]
- 3. Moore MA, Nakagawa K, Satoh K, Ishikawa T, Sato K. Single GST‐P positive liver cells – putative initiated hepatocytes. Carcinogenesis 1987; 8: 483–6. [DOI] [PubMed] [Google Scholar]
- 4. Cameron RG. Identification of the putative first cellular step of chemical hepatocarcinogenesis. Cancer Lett 1989; 47: 163–7. [DOI] [PubMed] [Google Scholar]
- 5. Garcea R, Daino L, Pascale R, Simile MM, Puddu M, Frassetto S, Cozzolino P, Seddaiu MA, Gaspa L, Feo F. Inhibition of promotion and persistent nodule growth by S‐adenosyl‐L‐methionine in rat liver carcinogenesis. role of remodeling and apoptosis. Cancer Res 1989; 49: 1850–6. [PubMed] [Google Scholar]
- 6. Wood GA, Korkola JE, Archer MC. Tissue‐specific resistance to cancer development in the rat: phenotypes of tumor‐modifier genes. Carcinogenesis 2002; 23: 1–9. [DOI] [PubMed] [Google Scholar]
- 7. Yoshimoto F, Masuda S, Higashi T, Nishii T, Takamisawa I, Tateishi N, Sakamoto Y. Comparison of drug‐metabolizing activities in the livers of carcinogen‐sensitive parent and carcinogen‐resistant descendants. Cancer Res 1985; 45: 6155–9. [PubMed] [Google Scholar]
- 8. Higashi T, Fukui R, Sekiyama M, Yoshimoto F, Tateishi N, Sakamoto Y. Decreased induction of γ–glutamyltransferase activity by 3′‐methyl‐4‐dimethyl‐aminoazobenzene in the liver of rats given carcinogen‐containing diet for several generations. Jpn J Cancer Res 1986; 77: 139–44. [PubMed] [Google Scholar]
- 9. Yano M, Higashi T, Kano S, Tateishi N, Kido Y, Mori T, Higashi K, Sakamoto Y. Decreased activation of carcinogens in the liver of carcinogen‐resistant rats. Cancer Res 1989; 49: 5352–7. [PubMed] [Google Scholar]
- 10. Yan Y, Higashi K, Yamamura K, Fukamachi Y, Abe T, Gotoh S, Sugiura T, Hirano T, Higashi T, Ichiba M. Different responses other than the formation of DNA‐adducts between the livers of carcinogen‐resistant rats (DRH) and carcinogen‐sensitive rats (Donryu) to 3′methyl‐4‐dimethylaminoazobenzene administration. Jpn J Cancer Res 1998; 89: 806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zeng ZZ, Higashi S, Kitayama W, Denda A, Yan Y, Matsuo K, Konishi Y, Hiai H, Higashi K. Genetic resistance to Chemical Carcinogen‐induced preneoplastic hepatic lesions in DRH Strain Rats. Cancer Res 2000; 60: 2876–81. [PubMed] [Google Scholar]
- 12. Yan Y, Zeng ZZ, Higashi S, Denda A, Konishi Y, Onishi S, Ueno H, Higashi K, Hiai H. Resistance of DRH strain rats to chemical carcinogenesis of liver: genetic analysis of later progression stage. Carcinogenesis 2002; 23: 189–96. [DOI] [PubMed] [Google Scholar]
- 13. Yamada Y, Matsushiro H, Ogawa MS, Okamoto K, Nakakuki Y, Toyokuni S, Fukumoto M, Hiai H. Genetic predisposition to pre‐B lymphomas in SL/Kh strain mice. Cancer Res 1994; 54: 403–7. [PubMed] [Google Scholar]
- 14. Markel P, Shu P, Ebeling C, Carlson GA, Nagle DL, Smutko JS, Moore KJ. Theoretical and empirical issues for marker‐assisted breeding of congenic mouse strains. Nat Genet 1997; 17: 280–4. [DOI] [PubMed] [Google Scholar]
- 15. Miyagawa‐Hayashino A, Tsuruyama T, Haga H, Oike F, Kim I‐D, Egawa H, Hiai H, Tanaka K, Manabe T. Arteriopathy in chronic allograft rejection in liver transplantation. Liver Transpl 2004; 10: 513–7. [DOI] [PubMed] [Google Scholar]
- 16. Denda A, Kitayama W, Konishi Y, Yan Y, Fukamachi Y, Miura M, Gotoh S, Ikemura K, Abe T, Higashi T, Higashi K. Genetic properties for the suppression of development of putative preneoplastic glutathione S‐transferase placental form‐positive foci in the liver of carcinogen‐resistant DRH strain rats. Cancer Lett 1999; 140: 59–67. [DOI] [PubMed] [Google Scholar]
- 17. Ilio CD, Aceto A, Columbano A, Ledda‐Columbano GM, Federici G. Induction of rat glutathione transferase subunit 7 by lead nitrate. Cancer Lett 1989; 46: 167–71. [DOI] [PubMed] [Google Scholar]
- 18. Miyake N, Sawada M, Hiai H. Regeneration of Paneth cell after dithizone treatment. Acta Histochem Cytochem 1995; 28: 549–53. [Google Scholar]
- 19. Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver specific mRNA for Insig‐2 down‐regulated by insulin: Implications for fatty acid synthesis. Proc Natl Acad Sci USA 2003; 100: 3155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roomi MW, Columbano A, Ledda‐Columbano GM, Sarma DS. Lead nitrate induces certain biochemical properties characteristic of hepatocyte nodules. Carcinogenesis 1986; 7: 1643–6. [DOI] [PubMed] [Google Scholar]
- 21. Higashi K, Denda A, Higashi T, Hiai H. Genetic resistance to chemical hepatocarcinogenesis in the DRH rat strain. Com Med 2004; 54: 252–6. [PubMed] [Google Scholar]
- 22. De Miglio MR, Pascale RM, Simile MM, Muroni MR, Calvisi DF, Virdis P, Bosinco GM, Frau M, Seddaiu MA, Ladu S, Feo F. Chromosome mapping of multiple loci affecting the genetic predisposition to rat liver carcinogenesis. Cancer Res 2002; 62: 4459–63. [PubMed] [Google Scholar]
- 23. Wood GA, Sarma DS, Archer MC. Inheritance of resistance to promotion of preneoplastic liver lesions in Copenhagen rats. Exp Biol Med 2001; 226: 831–5. [DOI] [PubMed] [Google Scholar]
- 24. Wood GA, Sarma DS, Archer MC. Resistance to the promotion of glutathione S‐transferase 7–7‐positive liver lesions in Copenhagen rats. Carcinogenesis 1999; 20: 1169–75. [DOI] [PubMed] [Google Scholar]
- 25. Melhem MF, Kunz HW, Gill TJ III A major histocompatibility complex‐linked locus in the rat critically influence resistance to diethylnitrosamine carcinogenesis. Proc Natl Acad Sci USA 1993; 90: 1967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pascale RM, Simile MM, De Miglio MR, Muroni MR, Gaspa L, Dragani TA, Feo F. The BN rat strain carries dominant hepatocarcinogen resistance loci. Carcinogenesis 1996; 17: 1765–8. [DOI] [PubMed] [Google Scholar]
- 27. De Miglio MR, Canzian F, Pascale RM, Simile MM, Muroni MR, Calvisi D, Romeo G, Feo F. identification of genetic loci controlling hepatocarcinogenesis on Rat Chromosomes 7 and 10. Cancer Res 1999; 59: 4651–7. [PubMed] [Google Scholar]
- 28. De Miglio MR, Simile MM, Muroni MR, Pusceddu S, Calvisi D, Carru A, Seddaiu MA, Daino L, Deiana L, Pascale RM, Feo F. Correlation of c‐myc overexpression and amplification with progression of preneoplastic liver lesions to malignancy in the poorly susceptible Wistar rat strain. Mol Carcinog 1999; 25: 21–9. [PubMed] [Google Scholar]
- 29. De Miglio MR, Pascale RM, Simile MM, Muroni MR, Virdis P, Kwong KM, Wong LK, Bosinco GM, Pulina FR, Calvisi DF, Frau M, Wood GA, Archer MC, Feo F. Polygenic control of hepatocarcinogenesis in Copenhagen x, F344 rats. International J Cancer, 2004: 111, 9–16. [DOI] [PubMed] [Google Scholar]