

Abstract
Double‐stranded RNA (dsRNA) plays a major role in RNA interference (RNAi), a process in which segments of dsRNA are initially cleaved by the Dicer into shorter segments (21–23 nt) called small interfering RNA (siRNA). These siRNA then specifically target homologous mRNA molecules causing them to be degraded by cellular ribonucleases. RNAi downregulates endogenous gene expression in mammalian cells. Vascular endothelial growth factor (VEGF) is a key molecule in vasculogenesis as well as in angiogenesis. Tumor growth is an angiogenesis‐dependent process, and therapeutic strategies aimed at inhibiting angiogenesis are theoretically attractive. To investigate the feasibility of using siRNA for VEGF in the specific knockdown of VEGF mRNA, thereby inhibiting angiogenesis, we have performed experiments with a DNA vector based on a siRNA system that targets VEGF (siVEGF). It almost completely inhibited the expression of three different isoforms (VEGF120, VEGF164 and VEGF188) of VEGF mRNA and the secretion of VEGF protein in mouse squamous cell carcinoma NRS‐1 cells. The siVEGF released from cationized gelatin microspheres suppressed tumor growth in vivo. A marked reduction in vascularity accompanied the inhibition of a siVEGF‐transfected tumor. Fluorescent microscopic study showed that the complex of siVEGF with cationized gelatin microspheres was still present around the tumor 10 days after injection, while free siVEGF had vanished by that time. siVEGF gene therapy increased the fraction of vessels covered by pericytes and induced expression of angiopoietin‐1 by pericytes. These data suggest that cationized‐gelatin microspheres containing siVEGF can be used to normalize tumor vasculature and inhibit tumor growth in a NRS‐1 squamous cell carcinoma xenograft model. (Cancer Sci 2006; 97: 313 – 321)
Numerous growth factors have been identified that can positively regulate angiogenesis. Researchers were initially spurred to identify, characterize and purify vascular endothelial growth factor (VEGF) because of its ability to induce vascular leakage and permeability, as well as its ability to promote vascular endothelial cell proliferation.( 1 , 2 ) Since then, various members of the VEGF family have been identified based on their homology to VEGF.( 3 ) VEGF has been shown to play a pivotal role in tumor angiogenesis.( 4 ) There are at least five isoforms of both human and mouse VEGF. Each isoform is produced by a different splicing of the full‐length mRNA. The mouse VEGF variants encode proteins composed of 120, 164 and 188 amino acids.( 5 ) VEGF164, which possesses exon 7, has much greater heparin‐binding affinity than does VEGF120. VEGF188, which possesses both exon 6 and 7, has far greater charge and much more association with the extracellular matrix than do the other two isoforms.( 4 ) Various VEGF isoforms have different functions which together enable tumorogenic vascularization. Previous research suggests that VEGF expression and induction of this protein are related to radiosensitivity of oral squamous cell carcinoma.( 6 ) In addition, the expression of VEGF may be a predictor of lymph node metastasis of oral squamous cell carcinoma.( 7 ) These data indicate that inhibiting the expression or functioning of VEGF may be a useful component of cancer therapy.
RNA interference (RNAi) is the sequence‐specific, post‐transcriptional gene silencing method initiated by double‐stranded RNA (dsRNA) molecules, which are homologs of the gene being degraded.( 8 , 9 ) There are several recent reports of vector‐based strategies for delivering small interfering RNA (siRNA) into mammalian cells in order to expand the utility of RNAi.( 10 , 11 , 12 ) The siRNAs are then unwound by RISC (siRNA‐induced silencing complex) in the presence of adenosine triphosphate (ATP). The activated RISC binds to and degrades target mRNA guided by the single strand siRNA. In mammals, long dsRNA can activate protein kinase R (PKR) and RNaseL, which are key components of the interferon‐signaling pathway, thus causing unspecific effects, whereas 21–23 nt siRNAs are short enough to bypass them.( 13 ) The vector‐based RNAi technique, in which an endogenous U6 promoter generates siRNA in vivo, is a new and economical system to achieve gene silencing.( 14 ) Although vector‐based siRNA overcomes two disadvantages of synthetic dsRNA (transience and high cost), there are some problems to be solved, such as the transience and low level of gene transfection.
To increase the biological activity of the expressing plasmid DNA, it is necessary to increase its transfection efficiency to cells in vivo. Some synthetic materials have been designed to promote the successful transfection of plasmid DNA to mammalian cells in vivo. Because plasmid DNA is a large and negatively charged molecule, it is impossible to allow the plasmid DNA to internalize into cells even though the attachment onto the cell membrane of negative charges takes place. Gelatin has been extensively used in industrial, pharmaceutical and medical applications because of the ease with which chemical modification of a physicochemical nature takes place. For example, positively charged, cationized gelatin can readily be prepared by introducing amine residues to the carboxyl groups of gelatin. These advantages have stimulated researchers to produce a new gene delivery system using cationized gelatin hydrogel as a carrier. This system allows controlled biodegradation of the local delivery agent, and nucleases protect plasmid DNA from rapid degradation.( 15 , 16 , 17 )
In this study, we hypothesized that using cationized gelatin hydrogel as a carrier of VEGF vector‐based siRNA would increase cellular uptake and prolong the release of the siRNA injected into the tumor. Following the injection of VEGF siRNA expression vector complexed with cationized gelatin hydrogel to mouse tumors via intratumoral delivery, we measured tumor growth, the level of VEGF expression in tumors, and tumor angiogenesis in order to quantify the anti‐angiogenic effect.
Materials and Methods
Tumor cell line and cell cultures
The NRS‐1 tumors were cutaneous squamous cell carcinomas that spontaneously arose in the C3H/He mouse strain. NRS‐1 tumor cells were passed in RPMI 1640 medium supplemented with 10% heat‐inactivated fetal calf serum (FCS) at 37°C in an incubator with 5% CO2.
Expression plasmid constructs
For this study we chose a siRNA sequence that targets mouse VEGF (from mRNA sequence; GeneBank accession number NM‐009505). We purchased the siRNA expression vector pSilencer 1.0‐U6 from Ambion (Austin, TX, USA) and constructed siRNA‐expressing plasmids targeting mouse VEGF according to the manufacturer's instructions. Five pairs of cDNA oligonucleotides targeting mouse VEGF mRNA at different locations were synthesized. Each pair of cDNA oligonucleotides was annealed at 90°C for 3 min, cooled to 37°C and incubated for 1 h. The annealed dsDNA oligonucleotides were ligated between the Apa I and Eco RI sites on the pSilencer 1.0‐U6 siRNA expression vector. The target mouse VEGF sequences were: siVEGF‐1, gccagcacataggagagat; siVEGF‐2, gttcatggatgtctaccag; siVEGF‐3, gctactgccgtccgattga; siVEGF‐4, gacagaacaaagccagaaa; siVEGF‐5, ggagagcagaagtcccatg. The control siRNA was constructed by inserting a sequence targeting on the enhanced green fluorescent protein (EGFP) gene, and the targeted sequence was gacgtaaacggccacaagt.( 18 ) All the plasmid constructs were confirmed by DNA sequencing.
Transfection of siRNAs
NRS‐1 cells were seeded in six‐well culture plates at 2 × 105 cells/well in serum‐free RPMI 1640 medium and grown overnight to ∼50% confluence prior to transfection. All plasmids were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. We mixed 1 µg of each siRNA expression plasmid and Lipofectamine 2000 in Opti‐MEM serum‐free medium in a small sterile tube, and the mixture was left at room temperature for 20 min. The NRS‐1 culture medium was removed and the complex containing siRNA expression plasmid and Lipofectamine 2000 (1 mL) was added. After incubation for 6 h at 37°C, the cells were washed with phosphate‐buffered saline (PBS) and allowed to recover in RPMI1640/10% FCS medium. After incubating for 18 h, the cell culture supernatant was collected in order to measure VEGF levels. To monitor transfection efficiency, fluorescein‐labeled siRNA expression plasmid using Label IT siRNA Tracker Fluorescein Kit (Mirus, Madison, WI, USA) was used. Eighteen hours after transfection, adherent NRS‐1 cells were detached from wells by Trypsin‐EDTA solution and completely separated into single cells with gentle pipetting. The transfection efficiency of siRNA was evaluated by FACScan and CELLQuest software (Becton Dickinson). A total of at least 10 000 cells were counted for each sample. Gates were set according to forward and side scatters.
VEGF enzyme‐linked immunosorbent assay
Secretion of VEGF in the cell‐culture supernatant, serum and tumor contents of VEGF in the NRS‐1 xenografts were determined by a Quantikine mouse VEGF Immunoassay kit (R&D System, Minneapolis, MN, USA) according to the manufacturer's instructions.
Quantitative reverse transcription‐polymerase chain reaction
Total RNA was extracted using Isogen (Nippon gene, Toyama, Japan). Single‐stranded cDNAs were synthesized from total RNA using reverse transcriptase using the Super Script II (Invitrogen). Quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) was carried out with specific primers for VEGF120, VEGF164, VEGF188 and angiopoietin‐1 (Ang‐1). Because the three mouse VEGF isoforms share the first five exons and the last exon, sequences located on these exons will target all three isoforms. The VEGF common forward primer is located on exon 4 (5′‐gccagcacatagagagaatgagc) and the VEGF120 reverse primer is located on the boundary of exon 5 and exon 7 (3′‐cggcttgtcacatttttctgg). VEGF164 reverse primer located on the boundary of exon 5 and exon 8 (3′‐caaggctcacagtgattttctgg). VEGF188 reverse primer located on the boundary of exon 5 and exon 6 (3′‐aacaaggctcacagtgaacgct).( 19 ) Ang‐1 (5′‐tggaagtgttatcacccagttctca and 3′‐acaatgggctgttccaactcaag). The expression of the housekeeping gene β‐actin was used to normalize for input cDNA. Real‐time monitoring of PCR products was carried out with fluorescence of SYBR green I (Takara Bio, Shiga, Japan) during every PCR cycle at the extension step.
Semiquantitative reverse transcription‐polymerase chain reaction for the VEGF family gene
Total RNA was extracted using Isogen. Single‐stranded cDNAs were synthesized from total RNA (2 µg) by reverse transcriptase using Super Script II. Semiquantitative RT‐PCR was carried out with specific primers for VEGF‐B, VEGF‐C, VEGF‐D, and placenta growth factor (PlGF). Primer sequences were as follows: VEGF‐B (449 bp), 5′‐gtcaaacaactagtgcccag and 3′‐tgtctgggttgagctctaag; VEGF‐C (623 bp), 5′‐ccatgcacttgctgtgcttc and 3′‐accggcaggaagtgtgattg; VEGF‐D (506 bp), 5′‐gtcctcattccaagaaactc and 3′‐ggtagtgggcaacagtga; PlGF (72 bp), 5′‐ttcagtccgtcctgtgtcctt and 3′‐gcacacagtgcagaccttca; β‐actin (360 bp): 5′‐cgtgacatcaaagagaagctggtcc and 3′‐gctcaggaggagcaatgatcttgat. PCR amplification of cDNA was performed under the following conditions: 94°C for 10 s, 62°C for 30 s, and 72°C for 30 s with 25 cycles. After amplification, PCR products were separated by electrophoresis on 2% agarose gel containing ethidium bromide and visualized by UV light illumination.
SiRNAs transfected NRS‐1 cells proliferation assay
NRS‐1 cells (2 × 105) transfected with the indicated siRNA expression plasmid were cultured in 96‐well round‐bottom plates (Coaster) at 200 µL/well. The cells were then pulsed with 1 µCi/well [3H] thymidine and incubated for 12 h at 37°C. Proliferation was measured by [3H] thymidine incorporation using an Aloka‐type LSC‐5100 liquid scintillation counter (Tokyo).
Injection schedule of NRS‐1 cell and cationized gelatin microspheres containing the siRNA expression plasmid into mice
Animal studies were performed in accordance with the Kanagawa Dental College guidelines for research animal care. Back tumors were generated by subcutaneously injecting 1 × 106 cells in 100 µL PBS into the backs of C3H/He mice (Japan SLC, Shizuoka, Japan) aged 8–10 weeks. Tumor volume was estimated using the formula: tumor volume (mm3) = length (mm) × width (mm)2 × 1/2. Five days after injection of tumor cells, the tumor‐bearing mice received intratumor injections of siVEGF‐5‐containing cationized gelatin microspheres. Cationized gelatin microspheres were prepared by chemically cross‐linking gelatin in the water‐in‐oil emulsion state.( 20 ) To impregnate siRNA expression plasmid DNA into cationized gelatin microspheres, 10 µL of PBS containing 10, 20 or 40 µg of siRNA expression plasmid DNA was dropped onto 1 mg of freeze‐dried cationized gelatin microspheres and then kept for 24 h at 4°C. The control group received intratumor injections of PBS or 20 µg of siEGFP‐containing cationized gelatin microspheres. Each treatment group contained 10 mice; each mouse received four intratumoral injections 5 days apart.
Fluorescent microscopic observation
Each animal in the experimental group received an intratumoral injection of cationized gelatin microspheres containing fluorescein‐labeled siVEGF‐5, while each animal in the control group received an intratumoral injection of siVEGF‐5 that was not carried by cationized gelatin microspheres. Tumors were removed 10 days after siVEGF‐5 injection and embedded in OCT compound (Miles, IN). Cryosections were prepared from the embedded sample to view the localization of fluorescein‐labeled siVEGF‐5. Images were obtained on a Zeiss microscope and analyzed using AxioImager software (Carl Zeiss, Thornwood, NY, USA).
Immunohistochemical analysis
The mice were killed and the implanted tumors were then surgically removed. Implanted tumor specimens were embedded in OCT compound, quickly frozen in dry ice, and stored at −80°C for immunohistochemical staining. Immunohistochemical staining for CD31 was carried out. We blocked 10‐µm sections by endogenous peroxidase using 3% hydrogen peroxidase, incubated them with rat antimouse CD31 mAb (clone MEC13.3, Pharmingen) overnight at 4°C, and then rinsed them three times. We detected bound primary Ab using the Simple Stain Mouse MAX‐PO (Nichirei, Tokyo, Japan) according to the manufacturer's instructions, and counted CD31‐positive vessels at 100× magnification.
We performed double immunohistochemical staining for CD31 with α‐smooth muscle actin (α‐SMA) or Ang‐1. After staining with CD31, the sections were rinsed three times in PBS and incubated a second time with anti‐α‐SMA (clone 1A4; DAKO, Carpinteria, CA, USA) or anti‐Ang‐1mAb (clone C‐19; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room temperature. Detection was carried out with the Histofine Mouse Stain Kit (Nichirei, Tokyo, Japan) according to the manufacturer's instructions. Mayer's hematoxylin was used as a counterstain.
Statistical analysis
All data were expressed as mean ± standard deviation of the mean. We analyzed the data with the unpaired Student's t‐test, and we considered results to be statistically significant if P < 0.01.
Results
Effects of VEGF siRNA on the expression of VEGF by NRS‐1 cells
Because the three mouse VEGF isoforms share exons 1–5 and exon 8, VEGF siRNA sequences located on these exons will target all three isoforms. We constructed five VEGF siRNA expression plasmids to target different regions on the first five exons. VEGF siRNA constructs containing each target sequence were transfected into NRS‐1 cells and total RNA was isolated 24 h post‐transfection. Expression of each VEGF isoform was analyzed by quantitative RT‐PCR. VEGF expression was significantly suppressed by siVEGF‐5 (Fig. 1). We observed that siVEGF‐5 reduced VEGF120 by 87.2%, VEGF164 by 80.5%, and VEGF188 by 80.4% (Fig. 1A). Enzyme‐linked immunosorbent assay (ELISA) for VEGF showed that siVEGF‐5 reduced VEGF production by 89% compared with control siEGFP‐transfected NRS‐1 cells (Fig. 1B). We also investigated the siVEGF‐5 transfection efficiency by FACS. The efficiency for transfection was 87.6% (Fig. 1C). To confirm the specificity of siVEGF‐5, we examined its effect on the expression of other VEGF family genes, including VEGF‐B, VEGF‐C, VEGF‐D and PlGF. In non‐transfected NRS‐1 cells we marginally detected VEGF‐D expression, but we did not detect VEGF‐B, VEGF‐C or PlGF. The expression level of VEGF‐D did not change upon transfection of siVEGF‐5 (Fig. 1D). These results show that siVEGF‐5 does not alter the gene expression of other VEGF family members. Next we determined if siVEGF is directly cytotoxic to NRS‐1 cells or if its effect is inhibitory. The amount of [3H] thymidine incorporated into siVEGF‐5‐transfected NRS‐1 cells was comparable to that of non‐transfected and control siEGFP plasmid‐transfected NRS‐1 cells (data not shown). These findings suggest that siVEGF has no direct cytotoxicity against NRS‐1 cells.
Figure 1.
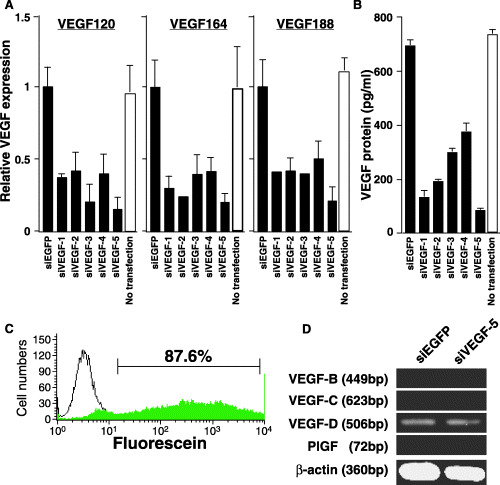
Effect of vector‐based small interfering RNA (siRNA) on vascular endothelial growth factor (VEGF) expression in NRS‐1 squamous cell carcinoma cells. (A) NRS‐1 cells were transfected with five different siRNA expression plasmids pU6‐siVEGF or pU6‐siEGFP. Twenty‐four hours post‐transfection, total RNA was isolated, and three isoforms of VEGF were quantified by real‐time polymerase chain reaction (PCR). mRNA levels of each isoform are shown. Data were normalized with the housekeeping gene β‐actin. Data are represented as mean ± SD of triplicate wells. Similar results were obtained in three independent experiments. (B) Cultured supernatants post‐transfection were collected and assayed for VEGF by enzyme‐linked immunosorbent assay (ELISA). Data are represented as mean ± SD of triplicate wells. Similar results were obtained in three independent experiments. (C) Twenty‐four hours post‐transfection. Efficiency of transfection to the NRS‐1 cells using fluorescein‐labeled siRNA was analyzed by FACS. (D) Expression of VEGF‐B, VEGF‐C, VEGF‐D, and placenta growth factor (PlGF) mRNA in cells transfected with siVEGF‐5 or siEGFP. Total RNA was extracted from the transfected cells and processed for semiquantitative reverse transcription (RT)‐PCR. β‐actin served as an internal control.
Inhibition of tumor growth by cationized gelatin microspheres containing VEGF siRNA expression plasmid
To determine whether or not suppression of VEGF by siRNA had an effect on tumor growth, NRS‐1 cells were injected subcutaneously into mice. NRS‐1 cells grew rapidly, resulting in palpable tumors 2–3 days following injection. Five days later, the palpable tumors were injected with cationized gelatin microspheres containing siVEGF‐5 expression plasmid or the control siEGFP expression plasmid. Intratumoral injections were repeated every 5 days for a total of four injections. The siVEGF‐5 expression plasmid gene therapy with cationized gelatin microspheres markedly inhibited tumor growth compared with the control siEGFP expression plasmid or PBS. The growth inhibitory effects of siVEGF‐5 were dose dependent. Moreover, we found fewer VEGF proteins in tumors treated with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid (Fig. 2).
Figure 2.
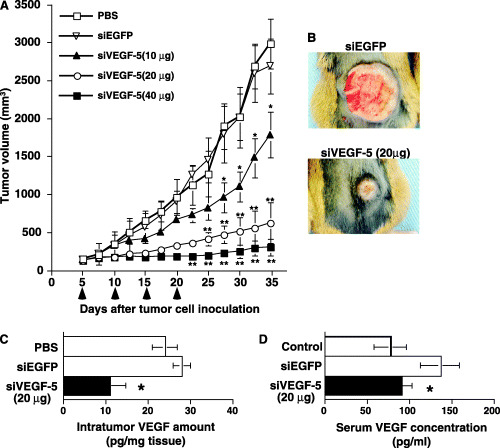
Inhibition of growth of subcutaneous NRS‐1 tumors by intratumoral injection of cationized gelatin microspheres containing the si vascular endothelial growth factor‐5 (siVEGF‐5) expression plasmid. (A) Tumor growth was monitored and tumor volume was calculated. On day 5, 10, 15 and 20, siVEGF‐5 or siEGFP was mixed with cationized gelatin microspheres and tumors were injected with each mixture. *P < 0.01; **P < 0.001, compared with phosphate‐buffered saline (PBS) or siEGFP injected tumor. (B) Photograph of xenograft tumors. siEGFP and siVEGF‐5 injected tumors were photographed on day 30. (C) Amount of VEGF protein detected in tumors. The excised xenograft tumors on day 30 were homogenized and VEGF protein levels were measured by enzyme‐linked immunosorbent assay (ELISA). Tumor samples were immersed and homogenized in a CelLytic‐M Mammalian cell lysis/extraction reagent (Sigma Chemical) at a buffer volume (µL)/sample weight (mg) ratio of 5:1 in order to normalize the influence of weight variance on the assay. The sample lysate was centrifuged and supernatant was applied to the well of the VEGF ELISA kit. Data are represented as the mean ± SD of three mice in each group. *P < 0.01 compared with PBS or siEGFP injected tumor. (D) Serum VEGF levels in control without tumor, siVEGF‐5, or siEGFP‐injected mice were measured by ELISA on day 30. Data are represented as the mean ± SD of three mice in each group. *P < 0.05 compared with siEGFP injected mouse.
Cationized gelatin microspheres prolong the period during which injected siVEGF‐5 exerts an influence
To characterize the degree to which cationized gelatin microspheres stabilize siVEGF‐5 in vivo, we injected tumors with fluorescein‐labeled siVEGF‐5 carried by cationized gelatin microspheres (Fig. 3). We observed strong fluorescence in tumors upon injecting them with fluorescein‐labeled siVEGF‐5 carried by cationized gelatin microspheres. On the contrary, fluorescence was not detected in tumors upon injecting them with fluorescein‐labeled siVEGF‐5 alone. These results suggest that cationized gelatin microspheres preserve the period during which siVEGF‐5 exerts an influence in vivo and control the release of siVEGF‐5.
Figure 3.
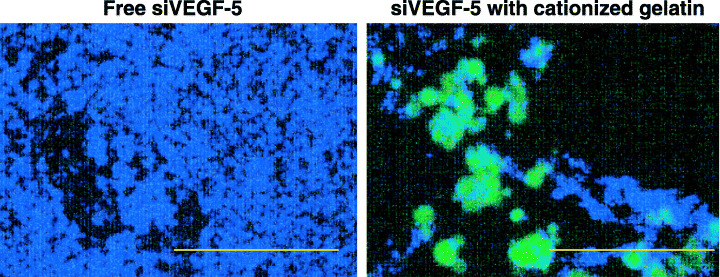
Effects of cationized gelatin microspheres on the stability of injected si vascular endothelial growth factor‐5 (siVEGF‐5). These fluorescent micrographs were taken 10 days after injection of fluorescein‐labeled siVEGF‐5. Fluorescein‐labeled siVEGF‐5 (green) with or without cationized gelatin microspheres was injected. Ten days after injection, tumors were excised and fluorescence was observed. Nuclei are stained with DAPI (blue). Scale bars, 200 µm.
Pathological and immunohistochemical descriptions
To study the relationship between the inhibition of tumor growth by the transfected siVEGF‐5 expression plasmid and tumor neovascularization, we stained intratumoral vessels with CD31 mAb. CD31‐positive vessels were abundant in tumors injected with the siEGFP expression plasmid. In contrast, significantly fewer microvessels were observed in tumors injected with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid compared with tumors injected with the control siEGFP expression plasmid, as determined by direct microvessel counting of CD31‐positive cells (Fig. 4A,B). Moreover, tumor vessels were highly dilated in siVEGF‐5 injected tumors. It has been reported that tumor vessels are structurally and functionally abnormal, with defective endothelium, basement membrane, and pericyte coverage.( 21 ) To assess the maturity of tumor vessels, we observed α‐SMA‐positive periendothelial cells in tumor tissue. Many peripheral blood vessels in the siEGFP‐injected tumors were poorly supported by smooth muscle cells. Numerous α‐SMA–negative periendothelial cells were detected in the majority of vessels in the control group of siEGFP‐injected tumors, indicating that immature blood vessels were present in the control tumors. In contrast, in the tumors injected with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid, some of the surviving vessels were α‐SMA‐positive and were supported by smooth muscle cells (Fig. 4C). We quantitatively determined the ratio of smooth muscle cells to endothelial cells in the walls of tumor vessels. This ratio was nearly six times higher in the vessels of tumors injected with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid than in control tumors (Fig. 4D). These results indicate that the increased fraction of α‐SMA‐positive vessels in tumors injected with the siVEGF‐5 expression plasmid supports normal vascular growth. A relevant research finding is that Ang‐1 induces smooth muscle to form new blood vessels and stabilizes vessel walls.( 22 ) Double immunostaining with CD31 and Ang‐1 antibodies showed that α‐SMA‐positive vessels were the most frequent Ang‐1‐positive cell type in tumors injected with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid (Fig. 5A). Next we determined levels of Ang‐1 mRNA in tumor tissues using quantitative RT‐PCR. We found that there was an approximately 150% increase of Ang‐1 mRNA in tumor tissue injected with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid (Fig. 5B). These results imply that Ang‐1 stabilizes the existing blood vessels in tumors injected with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid.
Figure 4.
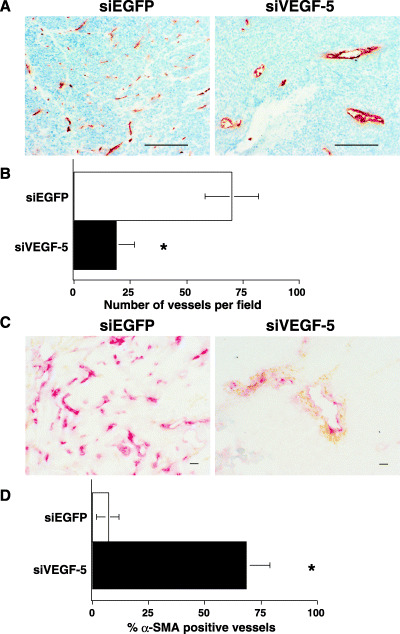
Immunohistochemical analysis of tumor blood vessels. (A) Tumor blood vessels were analyzed by staining with CD31 mAb. Few positively stained endothelial cells were detectable in si vascular endothelial growth factor‐5 (siVEGF‐5) injected tumors, whereas positively stained endothelial cells were readily detectable in siEGFP injected tumors (magnification 100×). Scale bars, 200 µm. (B) Quantitative assessment of the microvascularity index of siVEGF‐5‐injected tumors and siEGFP‐injected tumors. CD31‐positive endothelial cells were randomly counted at 100‐power magnification from five different fields. The count was significantly different from that of the control tumors. Data are the mean ± SD of three mice in each group. *P < 0.01 compared with a siEGFP‐injected tumor. (C) Double immunohistochemical staining of CD31 (red) and α‐smooth muscle actin (α‐SMA) (brown) was performed to identify blood vessel maturation. A large number of α‐SMA‐negative vessels were detectable in the siEGFP‐injected tumors, whereas surviving vessels were α‐SMA‐positive in the injected tumors (magnification 200×). Scale bars, 20 µm. (D) The vessel maturation index, defined as the percentage of vessels associated with α‐SMA‐positive pericytes, was significantly higher (*P < 0.01) for tumors injected with siVEGF‐5 than for tumors injected with siEGFP. Data are shown as the mean ± SD of three mice in each group.
Figure 5.
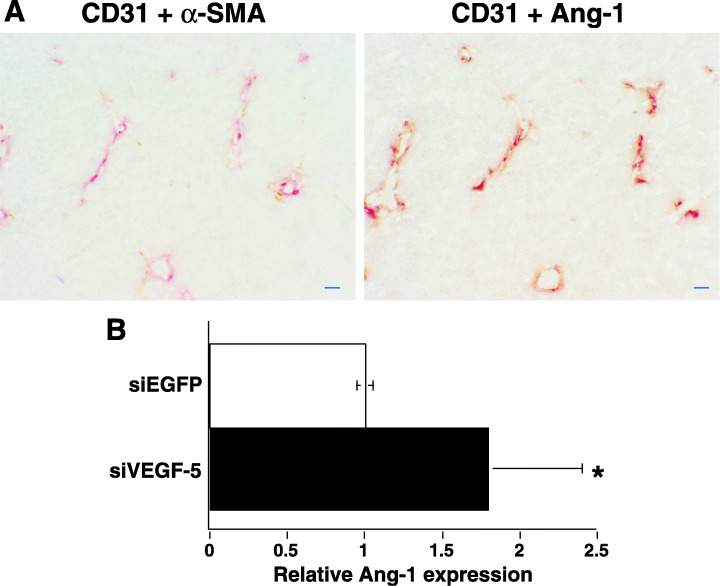
Immunohistochemical analysis of Ang‐1 expression in α‐smooth muscle actin (α‐SMA)‐positive blood vessels. Serial cryosections were fixed and stained. (A) Ang‐1 is expressed in the α‐SMA‐positive layer but not in the endothelial cells in the tumors injected with siVEGF‐5 (magnification 200×). CD31 (red) and α‐SMA (brown) staining (left panel) and CD31 (red) and Ang‐1 (brown) staining (right panel). α‐SMA‐positive layer also expressed Ang‐1 in the serial sections. Scale bars, 20 µm. (B) Ang‐1 expression increased in tumor tissue injected with siVEGF‐5. Quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) confirmed overexpression of Ang‐1 in vivo. Data were normalized with the housekeeping gene β‐actin. *P < 0.01 compared with an siEGFP‐injected tumor. Data are the mean ± SD of three mice in each group.
Discussion
For patients who have oral or esophageal squamous cell carcinoma, VEGF expression is associated with tumor progression and a poor prognosis because it stimulates angiogenesis.( 23 , 24 ) We have previously observed that VEGF is abundantly expressed in the primary tumors of patients who have oral squamous cell carcinoma (data not shown). In this study, we have sought to establish the efficacy of using siVEGF to suppress or turn off the expression of VEGF in murine squamous cell carcinoma. The different VEGF isoforms are associated with strikingly different vascularization.( 4 ) Tumors expressing VEGF120 effectively capture local vessels, which can be seen encircling the tumor and which do not possess extensive capillary beds in the tumors. In contrast, VEGF164 has the ability to induce tumors to expand both externally and internally. VEGF188 fails to adequately recruit the host vasculature around tumors, but induces a hypervascular network internal to the tumor. VEGF120 and VEGF164, because they are soluble isoforms, act at more distal sites to promote vascular recruitment, and VEGF188, because it is an isoform associated with the extracellular matrix, acts to promote the local expansion of capillary beds.
Because the three VEGF isoforms share exons 1–5 and exon 8, sequences located on these exons will target all three isoforms. Zhang et al. were able to target all mouse VEGF isoforms with siRNA, but could only decrease the level of these isoforms by 30%.( 19 ) We designed five different siVEGF plasmids targeting five different VEGF sequences in different regions of the VEGF mRNA. All were located between exons 1 and 5. Of these five, siVEGF‐5 strongly inhibited the expression of all isoforms in NRS‐1 cells in vitro. Furthermore, ELISA showed that VEGF protein secretion was similarly inhibited by siVEGF‐5. Tumors injected with siVEGF‐5 grew significantly more slowly in the mouse squamous cell carcinoma model, there was less VEGF expression, and blood vessel density was lower.
Taken together, these data confirm that siVEGF‐5 is highly functional and that all three isoforms of VEGF play a role in squamous cell carcinoma angiogenesis and tumor growth. These findings suggest that RNAi of VEGF can be an effective anti‐angiogenic strategy by which to treat squamous cell carcinoma. However, further studies are needed to unravel how VEGF isoforms participate in angiogenesis in tumors and to find potential applications for VEGF isoform‐specific siRNA therapy. Gene expression of VEGF is potently augmented under hypoxic conditions by increasing both mRNA transcription and stabilization.( 25 ) Transcriptional regulation of VEGF under hypoxic conditions is primarily mediated by hypoxia‐inducible factor 1 (HIF‐1), a heterodimeric protein transcription factor, composed of α and β subunits.( 26 ) During hypoxia, HIF‐1α is stabilized and binds to the VEGF promoter and transactivates gene expression. Detwiller et al. have shown that hypoxic upregulation of VEGF expression does not attenuate the efficacy of VEGF RNAi.( 27 )
Unlike normal blood vessels, tumor vessels are structurally and functionally abnormal.( 21 ) An imbalance of pro‐ and anti‐angiogenic factors causes endothelial cell migration and proliferation, and the resulting abnormal tumor vasculature is composed largely of immature vessels with increased permeability.( 28 , 29 ) In this study, the increased fraction of vessels positive for α‐SMA in tumors injected with cationized gelatin microspheres containing the siVEGF‐5 expression plasmid compared with the tumors injected with the control siEGFP expression plasmid shows that vasculature is normalized in the former case. Therefore, a decrease in microvessel density in tumors injected with siVEGF‐5 should cause a reduction in vascular surface area and a lowering of permeability.
Ang‐1 plays a role in the recruitment of pericytes and in vessel maturation, whereas the role of VEGF is to induce vessel formation.( 22 , 30 ) It has been reported that a VEGF/VEGF receptor‐2 blockade increases pericyte coverage of brain tumor vessels via upregulation of Ang‐1.( 31 ) We confirmed that expression of Ang‐1 was upregulated after siVEGF‐5 treatments by quantitative RT‐PCR. Furthermore, we observed immunohistochemically that Ang‐1 was expressed in α‐SMA positive layers in the siVEGF‐5 treated tumors. Other researchers have shown that treating rectal carcinoma patients with the VEGF‐specific antibody ‘bevacizumab’ increases the fraction of α‐SMA‐positive tumor vessels.( 32 ) Therefore, VEGF seems to be one of the key molecules that downregulates Ang‐1. However, the exact mechanisms by which Ang‐1 is downregulated after VEGF knockdown remains unknown.
Research has shown that Ang‐1 enhances tumor vessel maturation and inhibits tumor growth.( 33 , 34 ) Overexpression of Ang‐1 in human squamous cell carcinoma cells enhanced phosphorylation of the Tie2 receptor in vivo and significantly inhibited tumor growth.( 35 ) However, the mechanism by which Ang‐1‐mediated vessel maturation impairs tumor growth remains relatively unknown.
Although chemically synthesized siRNA is expensive, has a relatively short half‐life, and only transiently inhibits the target gene,( 36 ) our data shows that gene therapy targeting VEGF is a promising therapeutic strategy. In this study, to overcome these liabilities, we constructed vector‐based expression systems in which sense and antisense strands of short VEGF sequences were transcribed into the hairpin structure under the control of the U6 promoter. Furthermore, we introduced a system of prolonged gene expression based on the sustained release of siRNA plasmid DNA from cationized gelatin microspheres.( 16 , 17 , 20 ) The plasmid DNA in cationized gelatin microspheres is protected from degradation by several nucleases and proteases, thereby allowing the continuous release of plasmid DNA and prolonging its half‐life in vivo.
Takei et al. have shown that atelocollagen containing VEGF dsRNA stabilizes in tumors and effectively inhibits tumor angiogenesis.( 37 ) Atelocollagen can maintain its matrix structure and release plasmid DNA slowly in vivo for a long time without cross‐linking.( 38 ) However, the degradability of a collagen matrix (and consequently the time period of DNA release from the matrix) is difficult to control unless the collagen is chemically modified by a process such as cross‐linking or cationization. Compared with collagen, it is easy for gelatin to be chemically modified using processes such as derivation and cross‐linking, because gelatin is water‐soluble and has a random coil structure.( 39 ) On the contrary, collagen has a three‐dimensional helical structure and is water‐insoluble, making it more difficult to artificially modify its chemical nature and the extent of cross‐linking than it is to accomplish such modifications in gelatin.( 16 , 40 ) Although gelatin is required for cross‐linking by the glutaraldehyde used for microsphere preparation, the time period of plasmid DNA release from gelatin can be controlled by varying the extent of cross‐linking.( 41 ) The plasmid DNA‐cationized gelatin complex with its positive charge readily interacts with the cell surface and its negative charge.( 40 ) In our experiments, cationized gelatin microspheres stabilized the siVEGF‐5 in the tumors for at least 10 days (Fig. 3). Therefore, cationized gelatin microspheres that degrade more slowly can be used to lengthen the period during which plasmid DNA is released.
In conclusion, we have shown that inhibition of VEGF by a siVEGF‐cationized gelatin complex effectively suppresses tumor growth. The underlying concept of transfecting a siRNA expression vector complexed with cationized gelatin to block the VEGF signaling pathway holds promise as an anti‐angiogenic therapy for oral squamous cell carcinoma.
Acknowledgments
The Ministry of Education, Culture, Sports, Science and Technology of Japan supported this work with a Grant‐in‐Aid for the High‐Tech Research Center Project.
References
- 1. Fernando NH, Hurvitz HI. Inhibition of vascular endothelial growth factor in the treatment of colorectal cancer. Semin Oncol 2003; 30: 39–50. [DOI] [PubMed] [Google Scholar]
- 2. Yancopoulos GD, Davis S, Gale NW, Rudge JS, Weigand SJ, Holash J. Vascular‐specific growth factor and blood vessel formation. Nature 2000; 407: 242–8. [DOI] [PubMed] [Google Scholar]
- 3. Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci 2001; 114: 853–65. [DOI] [PubMed] [Google Scholar]
- 4. Grunstein J, Masbad JJ, Hickey R, Giordana F, Johnson RS. Isoforms of vascular endothelial growth factor act in a coordinated fashion to recruit and expand tumor vascular. Mol Cell Biol 2000; 20: 7282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shima DT, Kuroki M, Deutsch U, Ng YS, Adamis AP, D’Amore PA. The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post‐transcriptional regulatory sequences. J Biol Chem 1996; 271: 3877–83. [DOI] [PubMed] [Google Scholar]
- 6. Shintani S, Kiyota A, Mihara M et al. Association of preoperative radiation effect with tumor angiogenesis and vascular endothelial growth factor in oral squamous cell carcinoma. Cancer Sci 2000; 91: 1051–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moriyama M, Kumagai S, Kawashiri S, Kojima K, Kashihara K, Yamamoto E. Immunohistochemical study of tumor angiogenesis in oral squamous cell carcinoma. Oral Oncol 1997; 33: 369–74. [DOI] [PubMed] [Google Scholar]
- 8. McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet 2002; 3: 737–47. [DOI] [PubMed] [Google Scholar]
- 9. Hannon GJ. RNA interference. Nature 2002; 418: 244–51. [DOI] [PubMed] [Google Scholar]
- 10. Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short‐interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci USA 2002; 99: 6047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sui G, Soohoo B, Affar el F, Gay Y, Shi WC, Forrester A. DNA vector‐based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 2002; 99: 5515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyagishi M, Taira K. U6 promoter driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol 2002; 20: 497–500. [DOI] [PubMed] [Google Scholar]
- 13. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001; 411: 494–8. [DOI] [PubMed] [Google Scholar]
- 14. Sui G, Soohoo C, Affar el B et al. A DNA vector‐based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 2002; 99: 5515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tabata T, Ikada Y. Protein release from gelatin materials. Adv Drug Deliv Rev 1998; 31: 287–301. [DOI] [PubMed] [Google Scholar]
- 16. Kushibiki T, Tomishige R, Fukunaka Y, Kakemi M, Tabata T. In vivo release and gene expression of plasmid DNA by hydrogels of gelatin with different cationization extents. J Control Release 2003; 90: 207–16. [DOI] [PubMed] [Google Scholar]
- 17. Kushibiki T, Nagata‐Nakajima N, Sugai M, Shimizu A, Tabata Y. Delivery of plasmid DNA expressing small interference RNA for TGF‐beta type II receptors by cationized gelatin to prevent interstitial renal fibrosis. J Control Release 2005; 105: 318–31. [DOI] [PubMed] [Google Scholar]
- 18. Zhang XN, Xiong W, Wang JD, Hu YW, Xiang L, Yuan ZH. siRNA‐mediated inhibition of HBV replication and expression. World J Gastroenterol 2004; 10: 2967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L, Yang N, Mohamed‐Hadley A, Rubin SC, Coukos G. Vector‐based RNAi, a novel tool for isoform‐specific knock‐down of VEGF and anti‐angiogenesis gene therapy of cancer. Biochem Biophys Res Commun 2003; 303: 1169–78. [DOI] [PubMed] [Google Scholar]
- 20. Kushibiki T, Matsumoto K, Nakamura T, Tabata Y. Suppression of tumor metastasis by NK4 plasmid DNA released from cationized gelatin. Gene Ther 2004; 11: 1205–14. [DOI] [PubMed] [Google Scholar]
- 21. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000; 407: 249–57. [DOI] [PubMed] [Google Scholar]
- 22. Jain RK. Molecular regulation of vessel maturation. Nat Med 2003; 9: 685–93. [DOI] [PubMed] [Google Scholar]
- 23. Arora S, Kaur J, Sharma C et al. Stromelysin 3, Ets‐1, and vascular endothelial growth factor expression in oral precancerous and cancerous lesions: correlation with microvessel density, progression, and prognosis. Clin Cancer Res 2005; 11: 2272–84. [DOI] [PubMed] [Google Scholar]
- 24. Inoue K, Ozeki Y, Suganuma T, Sugiura Y, Tanaka S. Vascular endothelial growth factor expression in primary esophageal squamous cell carcinoma. Association with angiogenesis and tumor progression. Cancer 1997; 79: 206–13. [DOI] [PubMed] [Google Scholar]
- 25. Dvorak HF. Vascular permeability factor/vascular endothelial growth factor. a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol 2002; 20: 4368–80. [DOI] [PubMed] [Google Scholar]
- 26. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 2003; 3: 721–32. [DOI] [PubMed] [Google Scholar]
- 27. Detwiller KY, Fernando NT, Segal NH, Ryeom SW, D’Amore PA, Yoon SS. Analysis of hypoxia‐related gene expression in sarcomas and effect of hypoxia on RNA interference of vascular endothelial cell growth factor A. Cancer Res 2005; 65: 5881–9. [DOI] [PubMed] [Google Scholar]
- 28. Jain RK. Barriers to drug delivery in solid tumors. Sci Am 1994; 271: 58–65. [DOI] [PubMed] [Google Scholar]
- 29. Jain RK. Normalizing tumor vasculature with anti‐angiogenic therapy: a new paradigm for combination therapy. Nat Med 2001; 9: 987–9. [DOI] [PubMed] [Google Scholar]
- 30. Stoeltzing O, Ahmad SA, Liu W et al. Angiopoietin‐1 inhibits vascular permeability, angiogenesis, and growth of hepatic colon cancer tumors. Cancer Res 2003; 63: 3370–7. [PubMed] [Google Scholar]
- 31. Winkler F, Kozin SV, Tong RT et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin‐1, and matrix metalloproteinases. Cancer Cell 2004; 6: 553–63. [DOI] [PubMed] [Google Scholar]
- 32. Willett CG, Boucher Y, di Tomaso E et al. Direct evidence that the VEGF‐specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 2004; 10: 145–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hayes AJ, Haung WQ, Yu J et al. Expression and function of angiopoietin‐1 in breast cancer. Br J Cancer 2000; 83: 1154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian S, Hayes AJ, Metheny‐Barlow LJ, Li LY. Stabilization of breast cancer xenograft tumour neovasculature by angiopoietin‐1. Br J Cancer 2002; 86: 645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hawighorst T, Skobe M, Streit M et al. Activation of the tie2 receptor by angiopoietin‐1 enhances tumor vessel maturation and impairs squamous cell carcinoma growth. Am J Pathol 2002; 160: 1381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dave RS, Pomerantz RJ. RNA interference on the road to an alternate therapeutic strategy! Rev Med Virol 2003; 13: 373–85. [DOI] [PubMed] [Google Scholar]
- 37. Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S, Muramatsu T. A small interfering RNA targeting vascular endothelial growth factor as cancer therapeutics. Cancer Res 2004; 64: 3365–70. [DOI] [PubMed] [Google Scholar]
- 38. Ochiya T, Takahama Y, Nagahara S et al. New delivery system for plasmid DNA in vivo using atelocollagen as a carrier material: the Minipellet. Nat Med 1999; 5: 707–10. [DOI] [PubMed] [Google Scholar]
- 39. Fukunaka Y, Iwanaga K, Morimoto K, Kakemi M, Tabata Y. Controlled release of plasmid DNA from cationized gelatin hydrogels based on hydrogel degradation. J Control Release 2002; 80: 333–43. [DOI] [PubMed] [Google Scholar]
- 40. Kushibiki T, Tabata Y. A new gene delivery system based on controlled release technology. Curr Drug Deliv 2004; 1: 153–63. [DOI] [PubMed] [Google Scholar]
- 41. Truong‐Le VL, August JT, Leong KW. Controlled gene delivery by DNA‐gelatin nanospheres. Hum Gene Ther 1998; 9: 1709–17. [DOI] [PubMed] [Google Scholar]