

Abstract
Arsenic trioxide (ATO) is of limited therapeutic benefit for the treatment of solid tumors. Genistein exhibits anticancer and pro‐oxidant activities, making it a potential candidate to enhance the efficacy of ATO whose cytotoxicity is oxidation‐sensitive. This study sought to determine whether genistein synergizes with ATO to combat hepatocellular carcinoma (HCC). Three human HCC cell lines, namely HepG2, Hep3B, and SK‐Hep‐1, were incubated with ATO, genistein, or ATO + genistein. The cells were also pretreated with antioxidant agents N‐acetyl‐L‐cysteine (NAC) or butylated hydroxyanisole (BHA). Cell viability, apoptosis, intracellular reactive oxygen species (ROS), mitochondrial membrane potential (ΔΨm), expression of Bcl‐2, Bax, caspase‐9, and ‐3, and release of cytochrome c into the cytosol were examined. The synergistic effect of ATO and genistein was also assessed using HepG2 xenografts subcutaneously established in BALB/c nude mice. The results show that genistein synergized with ATO to reduce viability, induce apoptosis, and diminish the ΔΨm of cells. The combination therapy down‐regulated Bcl‐2 expression, up‐regulated Bax expression, enhanced the activation of caspase‐9 and ‐3, and increased the release of cytochrome c. The synergistic effect of ATO and genistein was diminished by pretreatment with NAC or BHA. Genistein increased the production of intracellular ROS, while ATO had little effect. Genistein synergized with a low dose of ATO (2.5 mg/kg) to significantly inhibit the growth of HepG2 tumors, and suppress cell proliferation and induce apoptosis in situ. There were no obvious side effects, as seen with a high dose of ATO (5 mg/kg). Combining genistein with ATO warrants investigation as a therapeutic strategy to combat HCC.
(Cancer Sci 2010; 101: 975–983)
Hepatocellular carcinoma (HCC) is the third leading cause of cancer death with an estimated worldwide incidence of over one million new cases per year.( 1 ) Despite extensive exploration for novel anticancer drugs and therapeutic strategies, there has been little success in improving the treatment of HCC. Only surgery offers a cure, but tumor resection is feasible for <15% of patients, and recurrence rates remain as high as 50% after tumor resection due to the aggressive features of HCC including rapid growth, resistance to chemotherapy, and lack of effective adjunct therapy after surgery.( 2 , 3 , 4 , 5 )
Arsenic trioxide (ATO) has been widely employed to treat acute promyelocytic leukemia (APL) since its original application at Harbin Medical University in China in the 1970s.( 6 ) Its therapeutic potential and antitumor activity have also been tested in a variety of solid tumors including HCC.( 7 , 8 , 9 , 10 , 11 , 12 ) The anticancer activities of ATO include inducing cell apoptosis and cell cycle arrest, and inhibiting tumor angiogenesis.( 7 , 8 , 9 , 10 , 11 , 12 ) However, the activity of ATO against solid tumors has not been as effective as against APL. ATO failed to show a therapeutic effect at an endurable dose in a recent phase II clinical trial.( 13 ) The doses of ATO required to exert detectable anticancer effects in solid tumors( 7 , 8 , 9 , 10 , 11 , 12 , 14 , 15 ) are much higher than those required to inhibit hematologic malignancies.( 16 , 17 ) We have previously demonstrated that ATO has a narrow window of therapeutic opportunity in respect of dosage.( 9 ) High doses are not clinically achievable without the risk of severe side effects due to toxicity. Therefore, new strategies to enhance the efficacy of ATO while reducing the dose of ATO to avoid the severe side effects are essential for the treatment of HCC.
Genistein, a soy‐derived isoflavone, that exhibits multiple biochemical effects,( 18 ) has been shown to inhibit the growth of breast cancer,( 19 ) prostate cancer,( 20 ) pancreatic cancer,( 21 ) and HCC( 22 ) cells, yet does not display cytotoxicity against normal cells.( 23 ) A recent study has shown that genistein induced the apoptosis of HCC cells by increasing intracellular reactive oxygen species (ROS), and inducing endoplasmic reticulum stress and mitochondrial injury.( 24 ) Genistein was shown to selectively potentiate ATO‐induced apoptosis in human leukemia cells.( 25 ) Therefore, we hypothesized that genistein may enhance the efficacy of ATO in treating HCC.
Materials and Methods
Mice, cell lines, and reagents. Male nude BALB/c mice (H‐2b), 4–6 weeks of age, were obtained from the Animal Research Center, The First Clinical Medical School of Harbin Medical University, China. The human HCC cell lines HepG2, Hep3B, and SK‐Hep‐1 were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were routinely cultured at 37°C in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal calf serum. An ATO solution was purchased from Yida Pharmaceutical, Harbin Medical University, China. Genistein, N‐acetyl‐L‐cysteine (NAC), butylated hydroxyanisole (BHA), and 2′,7′‐dichlorodihydrofluorescein‐diacetate (DCFHDA) were purchased from Sigma (St. Louis, MO, USA). Genistein was dissolved in DMSO to make a stock solution of 100 mm for in vitro assays. The PI (propidium iodide)/Annexin V‐FITC apoptosis detection kit was purchased from BD Biosciences (San Jose, CA, USA). 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazole carbocyanine iodide (JC‐1) was purchased from Molecular Probes (Eugene, OR, USA). The antibodies against Bcl‐2, Bax, caspase‐3 and ‐9, cytochrome c, and β‐actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti‐Ki67 Ab was purchased from Abcam (Cambridge, MA, USA).
Cell viability assay. We used a Cell Counting Kit‐8 (CCK‐8) kit (Dojindo Laboratories, Kumamoto, Japan) to determine the viability of cells. 3 × 103 cells in 200 μL of medium per well were seeded into 96‐well plates and cultured overnight, and the culture medium was replaced with fresh DMSO‐containing medium (control) or the same medium containing treatment agents. The final DMSO concentration was not >0.02%. Clinically achievable concentrations of ATO (1 to 2 μm) were chosen based on a previous report( 26 ) and our preliminary experiments. Genistein was used at 15 μm for HepG2 and SK‐Hep‐1 cells, and at 20 μm for Hep3B cells. The latter concentrations were selected based on the sensitivity of cells to genistein, as determined in our preliminary experiments, which showed that such concentrations led to 30–40% growth inhibition of cells after a 72‐h incubation. The cells were incubated for 72 h, the culture medium was replaced with 100 μL of fresh medium followed by the addition of 10 μL of CCK‐8 solution. The cells were incubated for 2 h at 37°C, and the optical density (OD) at 450 nm was recorded. The growth index (%) was calculated according to the formula: experimental OD value/control OD value × 100. The experiments and all the below in vitro assays were repeated thrice.
Detection of cell apoptosis. Cells (1 × 105) were washed with PBS, resuspended in 100 μL of binding buffer, and then incubated with 5 μL of Annexin V and 5 μL of PI for 15 min at room temperature in the dark according to the manufacturer’s instructions (BD Biosciences, San Jose, CA, USA). The cells were analyzed in a Beckman Coulter Epics Altra II cytometer (Beckman Coulter, Brea, CA, USA) to measure the apoptosis rate (%), and viewed by laser scanning confocal microscopy (LSM‐510; Carl Zeiss Jena, Jena, Germany).
Measurement of intracellular ROS. Intracellular ROS was measured using the DCFHDA method described previously.( 26 ) Briefly, the cells were incubated with 10‐μm DCFHDA for 20 min at 37°C in a 5% CO2 incubator, and then washed and resuspended in PBS at 1 × 106 cells/mL. The cells were analyzed by flow cytometry at an excitation wavelength of 514 nm, and the fluorescence intensity of dichlorofluorescein (DCF) was measured at an emission wavelength of 525 nm. Untreated cells served as controls. The intracellular ROS was expressed as fold‐increase of DCF fluorescence compared to control.
Measurement of mitochondrial membrane potential. The lipophilic, cationic dye, JC‐1, was used to measure changes in mitochondrial membrane potential (ΔΨm), as described previously.( 27 , 28 ) Cells were incubated with 10 μg/mL of JC‐1 for 20 min at 37°C in a 5% CO2 incubator, washed and resuspended in PBS at 1 × 106 cells/mL, and then analyzed by flow cytometry at an excitation wavelength of 514 nm. Data were collected at the emission wavelength of 529 nm (green fluorescence) of the JC‐1 monomer and at 585 nm (red fluorescence) for JC‐1 aggregates. The ratio of red/green fluorescence intensities was recorded, and the relative ΔΨm was calculated according to the formula: experimental ratio value/control ratio value × 100.
Animal model and treatments. All surgical procedures and care administered to the animals were in accordance with institutional animal ethic guidelines. Tumors were established by subcutaneous injection of 4 × 106 HepG2 tumor cells into the flanks of mice. Tumors volumes were estimated according to the formula: π/6 × a2 × b, where a is the short axis, and b the long axis. When tumors reached ∼100 mm3 at about 3 weeks, the mice were randomly assigned to five groups (each group had seven mice): control, genistein, ATO, ATO + genistein, and high dose of ATO. Mice received daily 200 μL i.p. injections of either PBS, genistein (50 mg/kg), ATO (2.5 mg/kg), 50 mg/kg genistein + 2.5 mg/kg ATO, or high dose of ATO (5.0 mg/kg), respectively. Genistein was suspended in PBS, and the ATO stock solutions were diluted with PBS. The doses and methods were based on our preliminary experiments and previous reports.( 22 , 29 ) The treatments lasted for 15 days and the size of tumors was recorded. The mice were euthanized 3 days after the last injection, blood samples were collected via cardiac puncture, and tumors were excised. The carcasses without tumors were weighed, and compared to original bodyweights before treatments to calculate bodyweight change (%) according to the formula: (carcass weight – original bodyweight)/original bodyweight × 100. Blood samples were divided and one aliquot was used to measure the white blood cell (WBC) count. The other aliquot was centrifuged at 300g for 10 min to collect sera, which were used to measure the levels of serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), urea nitrogen (BUN), and creatinine (Cr) with an auto‐biochemical analyzer (Toshiba, Tokyo, Japan). Each tumor was divided into two halves, one half was fixed with 10% buffered formalin, and the other stored at −80°C.
Quantitation of Ki‐67 proliferation index. The methodology has been previously described.( 30 ) Briefly, tumor sections were immunostained with an anti‐Ki‐67 Ab, counterstained with hematoxylin, and examined by microscopy. The Ki‐67 positive cells were counted in 10 randomly selected ×400 high‐power fields. The Ki‐67 proliferation index (%) was calculated according to the following formula: the number of Ki‐67‐positive cells/total cell count × 100.
In situ detection of apoptotic cells. The methodology has been described previously.( 31 ) Briefly, tumor sections were stained with the TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) agent (Roche, Shanghai, China), and examined by fluorescence microscopy. The total number of apoptotic cells in 10 randomly selected fields was counted. The apoptosis index (%) was calculated according to the formula: number of apoptotic cells/total number of nucleated cells × 100.
Western blotting. The methodology has been described previously.( 30 , 32 ) Briefly, cells were sonicated in RIPS buffer and homogenized; whereas tumor tissues were homogenized in protein lysate buffer. Debris was removed by centrifugation. Samples containing 50 μg of total protein were resolved on 12% polyacrylamide SDS gels, and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 3% BSA, incubated with a primary Ab, and subsequently incubated with an alkaline phosphatase‐conjugated secondary Ab. They were developed with 5‐bromo‐4‐chloro‐3‐indolyl phosphate/nitro blue tetrazolium (Tiangen Biotech, Beijing, China). Blots were stained with an anti‐β‐actin Ab to serve as an internal control. A Mitochondria/cytosol Fractionation Kit (Beyotime Inst. Biotech, Beijing, China) was used to prepare mitochondria‐free cytosolic fractions, which were used to detect expression of cytochrome c by Western blot analysis.
Statistical analysis. All the data are expressed as mean values ± SD. Comparisons among multiple groups were made with a one‐way ANOVA followed by an LSD (least significant difference) test. A value of <0.05 (P < 0.05) was used for statistical significance.
Results
Genistein synergizes with ATO to reduce the viability of HCC cells. HepG2, Hep3B, and SK‐Hep‐1 cells were incubated with ATO, genistein (15 μm for HepG2 and SK‐Hep‐1, 20 μm for Hep3B; the concentrations were consistent throughout the whole study), or ATO + genistein for 72 h, and cell viability was determined with a CCK‐8 kit to calculate the growth index. As shown in Figure 1, 1 μm ATO had little effect on the growth index, whereas the combination of 1 μm ATO and genistein led to 30–40% inhibition of the growth index compared to control. Furthermore, while incubation with 2 μm ATO resulted in 20–30% inhibition of the growth index, the combination of 2 μm ATO and genistein markedly reduced the growth index by up to 80%, compared to control. To investigate whether the effects of genistein and ATO were additive or synergistic, we calculated values for the coefficient of drug interaction (CDI) as described previously,( 19 , 33 ) based on the principles proposed by Chou and Talalay. ( 34 ) A CDI value less than, equal to, or >1 indicates that the drugs are synergistic, additive, or antagonistic, and <0.7 indicates a significantly synergistic effect.( 33 ) The CDI for HepG2, Hep3B, and SK‐Hep‐1 cells treated with genistein and 2 μm ATO was 0.407, 0.543, and 0.448, respectively, indicating that the two drugs had significantly synergistic effects in inhibiting the viability of HCC cells.
Figure 1.
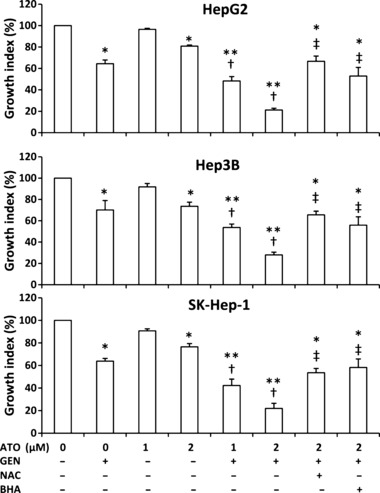
Cell growth in vitro. As indicated, HepG2, Hep3B, and SK‐Hep‐1 cells were incubated with arsenic trioxide (ATO), genistein (GEN), ATO + GEN, or pretreated with N‐acetyl‐L‐cysteine (NAC) or butylated hydroxyanisole (BHA) followed by ATO + GEN, for 72 h. Untreated cells served as the control. Cell viability was determined using a Cell Counting Kit‐8 (CCK‐8) assay to calculate the growth index. *Significant reduction in the growth index from control; **highly significant difference at P < 0.001 from control; †significant reduction from same dose ATO treatment; ‡significant increase from 2 μm ATO + GEN treatment.
Genistein synergizes with ATO to induce the apoptosis of HCC cells. HCC cells were incubated with ATO, genistein, or ATO + genistein for 48 h, stained with Annexin V/PI, and cell apoptosis was determined by flow cytometry. As shown in Figure 2(A), 1 μm ATO slightly increased the apoptosis rate of HepG2 cells compared to control, but the difference did not reach significance; whereas the combination of 1 μm ATO and genistein significantly increased the apoptosis rate by four‐fold compared to control. While incubation of cells with 2 μm ATO or genistein resulted in ∼1‐fold increase in the apoptosis rate of HepG2 cells, incubation with 2 μm ATO + genistein led to a ∼10‐fold increase in the apoptosis rate compared to control, and a ∼5‐fold increase compared to either 2 μm ATO or genistein alone,. Similar results were obtained for Hep3B and SK‐Hep‐1 cells (Fig. 2A). Representative histograms for the above cytometrically analyzed cells are shown in Figure 2(B). Annexin V/PI‐stained cells were viewed by confocal microscopy. As shown in Figure 2(C), apoptotic cells were almost undetectable amongst the untreated cells. However, late‐stage apoptotic cells were detected within 2 μm ATO + genistein‐treated cells, whose nuclei were stained red by PI.
Figure 2.
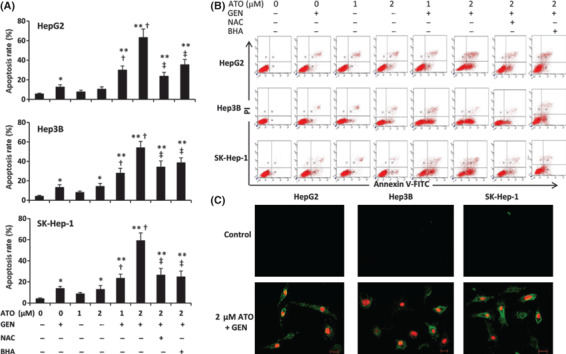
Cell apoptosis in vitro. (A) HepG2, Hep3B, and SK‐Hep‐1 cells were treated with arsenic trioxide (ATO), genistein (GEN) ATO + GEN, or pretreated with N‐acetyl‐L‐cysteine (NAC) or butylated hydroxyanisole (BHA) followed by ATO + GEN, for 48 h. Untreated cells served as the control. The cells were stained with Annexin V/PI, and subjected to flow cytometry to measure the apoptosis rate (%). *Significant increase in apoptosis rate from control; **highly significant difference at P < 0.001 from control; †significant increase from same dose ATO treatment; ‡significant reduction from 2 μm ATO + GEN treatment. (B) Representative histograms are shown for cytometrically analyzed cells. (C) Representative photographs of control and ATO + GEN‐treated cells stained with Annexin V/PI and viewed by laser scanning confocal microscopy (scale bar, 20 μm).
The role of ROS in the effects of genistein and ATO. HCC cells were incubated with 2 μm ATO, genistein, or ATO + genistein for 12 h, and then DCF fluorescence was recorded as a measure of intracellular ROS. As shown in Figure 3(A), the levels of intracellular ROS were highly significantly (P < 0.001) increased in genistein‐treated cells. ATO had little effect on intracellular ROS in HepG2 and SK‐Hep‐1 cells, and a weak though significantly increased intracellular ROS in Hep3B cells. The combination of ATO and genistein also elevated the levels of intracellular ROS, which were highly significantly (P < 0.001) increased compared to those of untreated cells, and were significantly (P < 0.01) higher than ROS levels following treatment with ATO, but were not significantly different from genistein alone. To confirm the role of ROS in the effects of genistein treatments, the cells were pretreated with 10 mm NAC, a well‐known inhibitor of ROS production,( 35 ) and then incubated with genistein alone or the combination of ATO and genistein. As shown in Figure 3(A), pretreatment with NAC significantly (P < 0.01) reduced the level of intracellular ROS in all three cell types, compared to genistein‐treated or ATO + genistein‐treated cells without NAC pretreatment. Similar results were obtained when the cells were pretreated with 50 μm BHA, another antioxidant (Fig. 3A). Representative histograms for the above cells are shown in Figure 3(B). Pretreatment with NAC or BHA also diminished the ability of ATO + genistein to inhibit cell viability (Fig. 1) and induce apoptosis (Fig. 2A,B).
Figure 3.
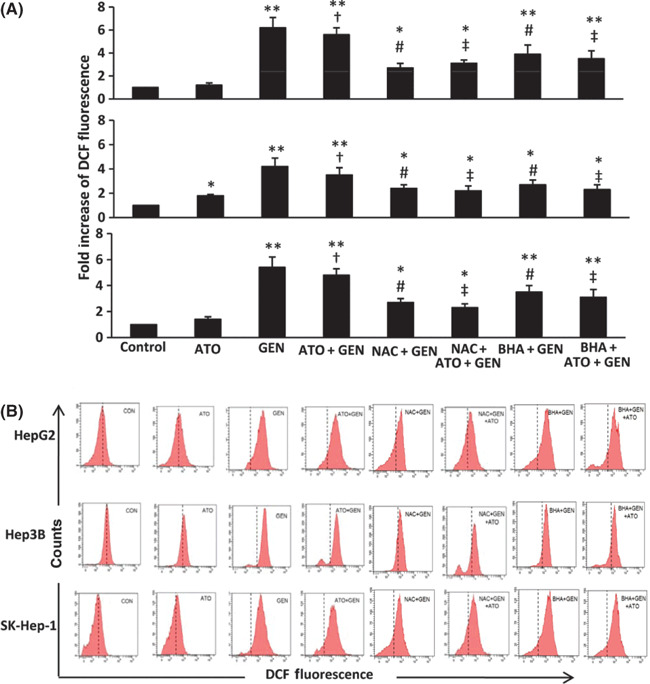
Levels of intracellular reactive oxygen species (ROS) in vitro. (A) HepG2, Hep3B, and SK‐Hep‐1cells were treated with 2 μm arsenic trioxide (ATO), genistein (GEN) (15 μm for HepG2 and SK‐Hep‐1, 20 μm for Hep3B), ATO + GEN, or pretreated with N‐acetyl‐L‐cysteine (NAC) or butylated hydroxyanisole (BHA) followed by GEN or ATO + GEN, for 12 h. Untreated cells served as the control. The cells were incubated with 2′,7′‐dichlorodihydrofluorescein‐diacetate (DCFHDA), then subjected to flow cytometry to measure levels of intracellular ROS, represented by dichlorofluorescein (DCF) fluorescence. *Significant increase in DCF fluorescence from control; **highly significant difference from control at P < 0.001; †significant increase from ATO treatment; #significant reduction from GEN treatment; ‡significant reduction from ATO + GEN treatment. (B) Representative histograms are shown for cytometrically analyzed cells stained with DCFHDA.
Genistein synergizes with ATO to diminish ΔΨm. The ΔΨm of cells was measured following a 48‐h incubation with 2 μm ATO, genistein, or ATO + genistein, given that ATO induces cell apoptosis via the mitochondrial intrinsic pathway, and ROS is closely related with ΔΨm. As shown in Figure 4(A), genistein significantly (P < 0.05) diminished the ΔΨm compared to control in all the three cell types, whereas ATO had little effect. The combination of ATO and genistein highly significantly (P < 0.001) diminished the ΔΨm compared to control, and also significantly (P < 0.01) diminished the ΔΨm compared with ATO alone. Pretreatment of the cells with NAC inhibited reductions of ΔΨm following the combinational treatment (Fig. 4A). Representative histograms for the above cells are shown in Figure 4(B).
Figure 4.
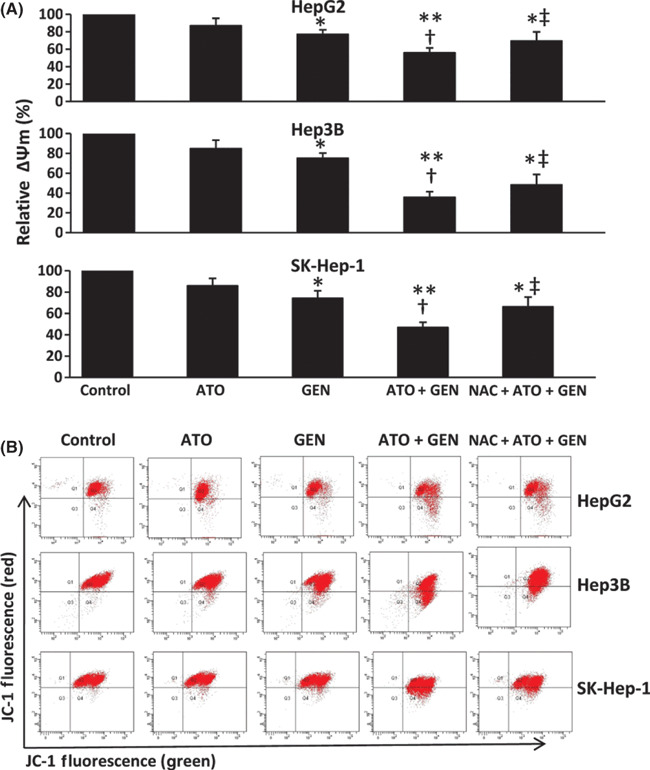
Changes in mitochondrial membrane potential in vitro. (A) HepG2, Hep3B, and SK‐Hep‐1 cells were treated with 2 μm arsenic trioxide (ATO), genistein (GEN) (15 μm for HepG2 and SK‐Hep‐1, 20 μm for Hep3B), ATO + GEN, or pretreated with N‐acetyl‐L‐cysteine (NAC) followed by ATO + GEN, for 48 h. Untreated cells served as the control. The cells were incubated with 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazole carbocyanine iodide (JC‐1), then subjected to flow cytometry to measure green and red fluorescence intensities, and the ratio of red/green fluorescence was recorded to calculate the relative ΔΨm. *Significant decrease in ΔΨm from control; **highly significant decrease from control at P < 0.001; †significant decrease from ATO treatment; ‡significant increase from ATO + GEN treatment. (B) Representative histograms are shown for cytometrically analyzed cells labeled with the JC‐1 dye.
Genistein synergizes with ATO to regulate apoptotic pro‐teins. As shown in Figure 5(A), both ATO and genistein up‐regulated Bax expression and down‐regulated Bcl‐2 expression, and activated caspase‐9 and ‐3. Genistein synergized with ATO to further up‐regulate Bax expression and down‐regulate Bcl‐2 expression, thus reducing the ratio of Bcl‐2/Bax. The combination also further increased the activation of caspase‐9 and ‐3 (Fig. 5A). The expression of cytochrome c in the cytosol was examined, given that the release of cytochrome c from mitochondria to the cytosol initiates the process of apoptosis. Both ATO and genistein increased the level of cytochrome c in the cytosol. The combination of ATO and genistein further increased the level of cytosolic cytochrome c, whereas the levels were diminished by pretreatment of cells with NAC (Fig. 5B).
Figure 5.
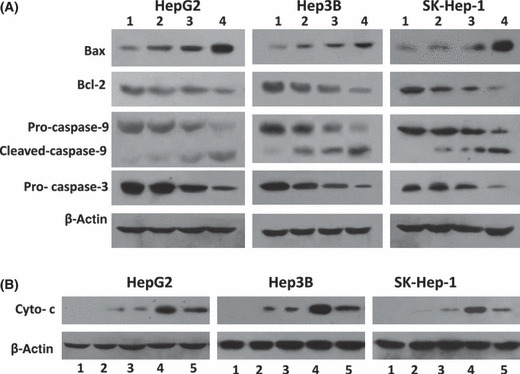
Expression of apoptosis‐related proteins in vitro. (A) HepG2, Hep3B, and SK‐Hep‐1 cells were treated with 2 μm arsenic trioxide (ATO) (lane 2), genistein (GEN) (15 μm for HepG2 and SK‐Hep‐1, 20 μm for Hep3B) (lane 3), or ATO + GEN (lane 4), for 48 h. Untreated cells served as the control (lane 1). The cells were homogenized and subjected to Western blot analysis to detect the expression of Bax, Bcl‐2, pro‐caspase‐9, cleaved caspase‐9, and pro‐caspase‐3. β‐Actin served as an internal control. (B) Cytoplasmic proteins from each of the treated cells as in (A) (lanes 1 to 4) were prepared using a Mitochondria/cytosol Fractionation Kit. As an additional control, cytoplasmic proteins were prepared from cells pretreated with N‐acetyl‐l‐cysteine (NAC) followed by ATO + GEN (lane 5). The expression of cytochrome c (Cyto‐c) in the mitochondria‐depleted cytosolic fractions was examined by Western blot analysis. β‐Actin served as an internal control.
Genistein and ATO synergistically suppress HCC tumors without obvious cytotoxicity. Tumors were established by subcutaneous injection of HepG2 cells, which have been demonstrated to be more resistant to ATO than other hepatocellular carcinoma cell lines.( 7 ) As shown in Figure 6(A), the tumors of mice treated with 2.5 mg/kg ATO reached 1680.3 ± 223.9 mm3 in volume 18 days after treatment, which was not significantly different compared to controls (1775.3 ± 289.4 mm3). Genistein alone significantly reduced the size of tumors (1368.0 ± 250.9 mm3). The tumors treated with 2.5 mg/kg ATO + genistein were highly significantly (P < 0.001) smaller than controls, reaching 407.7 ± 138.8 mm3 in volume. The CDI was 0.315, indicating that genistein and ATO have significant synergistic effects in suppressing the growth of HepG2 tumors. A high dose of ATO (5 mg/kg) also highly significantly (P < 0.001) suppressed the growth of tumors (958.0 ± 258.1 mm3 in volume) compared to controls. The cytotoxicity of the treatments was assessed by measuring bodyweight changes and WBC counts (Fig. 6B), examining liver function by measuring serum levels of AST and ALT (Fig. 6C), and examining renal function by measuring serum levels of BUN and Cr (Fig. 6D). Neither 2.5 mg/kg ATO or genistein alone, nor genistein + 2.5 mg/kg ATO in combination, significantly influenced any of the six health parameters. However, the high dose of 5 mg/kg ATO significantly reduced changes in bodyweight and WBC count, and elevated the serum levels of AST, ALT, and Cr, compared with control.
Figure 6.
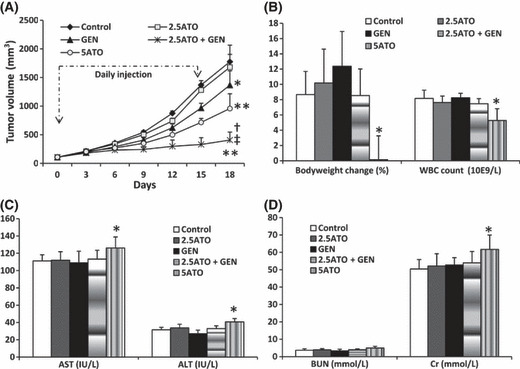
Genistein (GEN) synergizes with arsenic trioxide (ATO) to suppress HepG2 tumors in mice. When tumors reached ∼100 mm3 in volume, the mice received daily injections of 100 μL PBS (Control), or an equal volume of genistein at a dose of 50 mg/kg (GEN), ATO at doses of 2.5 (2.5ATO) or 5 (5ATO) mg/kg, or the combination of genistein + 2.5 mg/kg ATO (2.5ATO + GEN), for 15 days as indicated. (A) The sizes of tumors were recorded. *Significant difference in tumor volumes from control; **highly significant difference from control at P < 0.001; †significant difference from 2.5 mg/kg ATO; ‡significant difference from genistein. Blood samples were collected when the mice were euthanized on day 18, and tumors were removed. (B) The carcasses without tumors were weighed, and compared to the bodyweights on day 0 to calculate bodyweight change (%), and white blood cell (WBC) numbers were counted. (C) The serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), and (D) urea nitrogen (BUN) and creatinine (Cr) were measured. *Significant difference from control.
Genistein synergizes with ATO to inhibit cell proliferation in situ. As shown in Figure 7, there were fewer Ki‐67‐positive cells in the tumors of mice treated with 2.5 mg/kg ATO (Fig. 7B), genistein (Fig. 7C), genistein + 2.5 mg/kg ATO (Fig. 7D), or 5 mg/kg ATO (Fig. 7E), compared to controls. Ki‐67‐positive cells in tumor sections were counted to record the proliferation index. As shown in Figure 7F, treatment with ATO at the dose of 2.5 mg/kg resulted in a slight reduction in the proliferation index compared with control, but the difference did not reach significance. In contrast, genistein therapy significantly (P < 0.05) reduced the proliferation index. The proliferation index of tumors treated with genistein + 2.5 mg/kg ATO was highly significantly (P < 0.001) reduced by 56.9% compared to controls, and significantly lower than monotherapies with genistein or 2.5 mg/kg ATO (both P < 0.01). ATO at the dose of 5 mg/kg highly significantly (P < 0.001) reduced the proliferation index by 44.3% compared to control (Fig. 7F).
Figure 7.
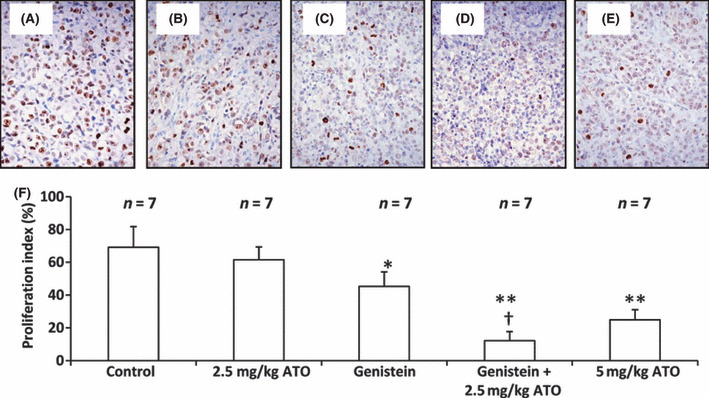
Genistein (GEN) synergizes with arsenic trioxide (ATO) to inhibit cell proliferation in situ. Illustrated are representative tumor sections prepared from mice that received daily injections of PBS (control) (A), 2.5 mg/kg ATO (B), genistein (C), genistein + 2.5 mg/kg ATO (D) or 5 mg/kg ATO (E), as in Figure 6. The sections were stained with an anti‐Ki‐67 Ab to detect proliferating cells. (F) Cells expressing Ki‐67 were counted to calculate the proliferation index. n, number of tumors assessed. *Significant difference in the proliferation index from control; **highly significant difference at P < 0.001 from control; †significant difference from ATO monotherapy.
Genistein synergizes with ATO to induce cell apoptosis in situ. As shown in Figure 8, a small number of TUNEL‐stained apoptotic cells were detected in the control tumors (Fig. 8A), whereas a greater number of apoptotic cells were detected in the tumors treated with 2.5 mg/kg ATO (Fig. 8B), genistein (Fig. 8C), genistein + 2.5 mg/kg ATO (Fig. 8D), and 5 mg/kg ATO (Fig. 8E). The numbers of apoptotic cells were counted to record the apoptosis index. As shown in 2, 8, 2.5 mg/kg ATO slightly increased the apoptosis index compared to control, but the difference did not reach significance. Genistein therapy significantly (P < 0.05) increased the apoptosis index compared to control. Treatment with the combination of genistein + 2.5 mg/kg ATO resulted in a highly significant (P < 0.001) four‐fold increase in the apoptosis index compared to control. The apoptosis index was also significantly increased (both P < 0.01) compared to that of mice treated with genistein or ATO monotherapy. Treatment with 5 mg/kg ATO also highly significantly (P < 0.001) increased the apoptosis index compared with control. As shown in Figure 8(G), genistein synergized with 2.5 mg/kg ATO to up‐regulate Bax expression and down‐regulate Bcl‐2 expression, thus reducing the ratio of Bcl‐2/Bax. The combination also increased the activation of caspase‐9 and ‐3, in accordance with the in vitro findings (Fig. 5A).
Figure 8.
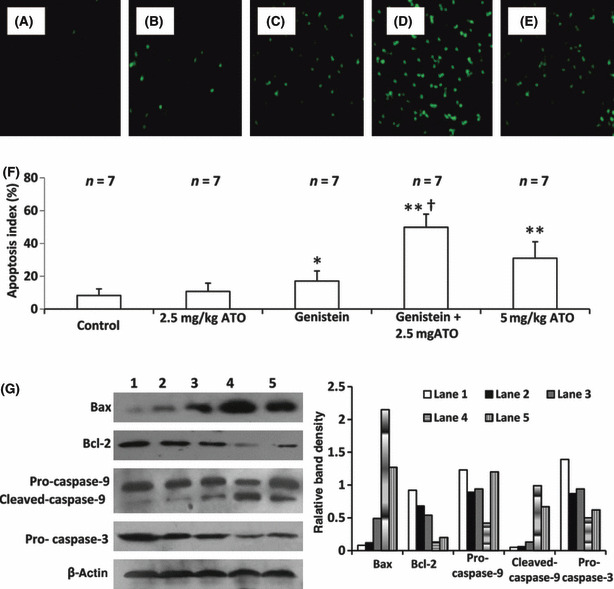
Genistein (GEN) synergizes with arsenic trioxide (ATO) to induce cell apoptosis in situ. Illustrated are representative tumor sections prepared from mice that received daily injections of PBS (control) (A), 2.5 mg/kg ATO (B), genistein (C), genistein + 2.5 mg/kg ATO (D), or 5 mg/kg ATO (E) as in Fig. 6. The sections were stained with the TUNEL agent to visualize apoptotic cells. (F) TUNEL‐positive cells were counted to calculate the apoptosis index. n, number of tumors assessed. *Significant difference in the apoptosis index from control; **highly significant difference at P < 0.001 from control; †significant difference from ATO monotherapy. (G) The tumor tissues were homogenized and subjected to Western blot analysis to detect expression of Bax, Bcl‐2, pro‐caspase‐9, cleaved caspase‐9, and pro‐caspase‐3 (left panel). The numbering of lanes is as in Figure 5(b). β‐Actin served as an internal control. The band density was measured and compared to that of β‐actin to calculate relative band density (right panel).
Discussion
The present study has demonstrated the synergistic effects of genistein and ATO in the treatment of HCC. Genistein synergized with ATO to inhibit tumor cell proliferation, and induce the apoptosis of HCC cells both in vitro and in vivo. This drug combination suppressed the growth of HCC tumors established in nude mice. A number of mechanisms appear to explain the synergistic effects of the two drugs including diminished ΔΨm, increased release of cytochrome c, up‐regulation of Bax expression, down‐regulation of Bcl‐2 expression, and activation of caspase‐9 and ‐3. This study demonstrated that ROS plays an important role in the synergistic effects of ATO and genistein in inhibiting proliferation and inducing apoptosis of HCC cells, as evidenced by the fact that the synergism was diminished by pretreating the cells with NAC or BHA. In accord, a recent report has shown that genistein selectively potentiates ATO‐induced apoptosis of human leukemia cells by virtue of its pro‐oxidant capacity.( 25 )
Genistein, a natural product, has displayed synergistic effects with chemotherapy in treating many types of cancer.( 19 , 20 , 21 , 23 ) Although genistein has previously been regarded as a ROS scavenging agent,( 36 , 37 , 38 ) several studies have demonstrated that genistein disrupts the respiratory chain, leading to the generation of ROS in isolated mitochondria and intact cells.( 39 , 40 , 41 ) A recent report showed that genistein elevated the intracellular levels of ROS in HCC cells.( 24 ) The results presented here confirm the latter finding, and demonstrate that elevated intracellular levels of ROS potentiate the cytotoxicity of ATO. This notion is also supported by the observation that anti‐oxidants diminished the enhanced therapeutic effects of the combinational treatment. NAC may directly reduce the activity of genistein( 24 ) or protect mitochondria from oxidation‐induced damage.( 42 ) Although not explored here, other possible mechanisms explaining the ability of genistein to enhance ATO cytotoxicity include inhibition of the expression or activity of multidrug resistance protein and P‐glycoprotein efflux pumps, thus decreasing ATO detoxification,( 43 ) or causing G2/M arrest as cells at the G2 phase are sensitive to the toxic actions of ATO.( 44 )
Cellular apoptosis is triggered by the death‐receptor‐induced extrinsic or mitochondrial‐apoptosome‐mediated intrinsic pathways.( 45 ) The released cytochrome c from mitochondria to the cytosol binds to Apaf‐1, resulting in proteolytic processing and activation of caspase‐9. Active caspase‐9 then activates caspase‐3, initiating a cascade of additional caspase activation that culminates in apoptosis.( 46 ) Both ATO and genistein have been regarded as mitochondria‐targeting drugs capable of inducing apoptosis via activation of the mitochondrial pathway.( 15 , 22 , 24 , 25 , 39 , 47 , 48 ) The present study has shown that genistein synergized with ATO to increase the release of cytochrome c into the cytosol, resulting in enhanced activation of caspase‐9 and ‐3. Hence, the synergistic effect of ATO and genistein in inducing the apoptosis of HCC cells may involve the mitochondrial pathway. The members of the Bcl‐2 family are the most prominent regulators of apoptosis in cancer cells, mediating their effects mainly through the mitochondrial pathway.( 49 ) Bcl‐2, located on the mitochondrial membrane, is an anti‐apoptotic protein that prevents changes in ΔΨm and inhibits the release of cytochrome c. Bax directly binds to Bcl‐2 and inhibits its function; hence the Bcl‐2/Bax ratio is negatively related to apoptosis.( 49 , 50 ) Although not investigated in the current study, it has been shown that targeting MEK/MAPK signal transduction potentiates ATO‐induced apoptosis in multiple myeloma cells through multiple signaling pathways,( 51 ) and genistein inhibits the activation of phosphatidylinositol 3‐kinase/AKT signal transduction pathways, which play a key role in the balance of cell survival and apoptosis.( 19 , 21 ) The synergistic effects of ATO and genistein may also be related to these signal pathways, which will require further investigation.
Finally, a preclinical animal model using HepG2 xenografts was employed to evaluate the potential clinical benefit of treatments. Both a high dose of ATO (5 mg/kg) and the combinational therapy of genistein and low dose ATO (2.5 mg/kg) markedly suppressed the growth of HepG2 tumors. In accord with our previous study,( 9 ) the high dose of ATO reduced the bodyweight of mice and the WBC count, and caused dysfunction of the liver and kidney in terms of increased serum levels of AST, ALT, and Cr. In contrast, the combination of genistein and low dose of ATO showed no obvious side effects, indicating that this combination may have clinical utility, and warranting future investigation of its ability to combat HCC in the clinic.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by grants from the Creative Foundation for Postgraduates in Harbin Medical University, China (HCXD2009005), and the National Natural Scientific Foundation of China (30872987 and 30973474). The experiments were carried out in the Key Laboratory of Hepatosplenic Surgery of Heilongjiang Province, the First Affiliated Hospital of Harbin Medical University. H. Jiang, Y. Ma, and X. Chen contributed equally to this work.
References
- 1. Parkin DM, Bray F, Ferlay J et al. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Roberts LR. Sorafenib in liver cancer – just the beginning. N Engl J Med 2008; 359: 420–2. [DOI] [PubMed] [Google Scholar]
- 3. Roayaie S, Blume IN, Thung SN et al. A system of classifying microvascular invasion to predict outcome after resection in patients with hepatocellular carcinoma. Gastroenterology 2009; 137: 850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Newell P, Villanueva A, Llovet JM. Molecular targeted therapies in hepatocellular carcinoma: from pre‐clinical models to clinical trials. J Hepatol 2008; 49: 1–5. [DOI] [PubMed] [Google Scholar]
- 5. Pang RW, Poon RT. From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology 2007; 72 (Suppl 1): 30–44. [DOI] [PubMed] [Google Scholar]
- 6. Soignet SL. Clinical experience of arsenic trioxide in relapsed acute promyelocytic leukemia. Oncologist 2001; 6 (Suppl 2): 11–6. [DOI] [PubMed] [Google Scholar]
- 7. Kito M, Akao Y, Ohishi N et al. Arsenic trioxide‐induced apoptosis and its enhancement by buthionine sulfoximine in hepatocellular carcinoma cell lines. Biochem Biophys Res Commun 2002; 291: 861–7. [DOI] [PubMed] [Google Scholar]
- 8. Luo L, Qiao H, Meng F et al. Arsenic trioxide synergizes with B7H3‐mediated immunotherapy to eradicate hepatocellular carcinomas. Int J Cancer 2006; 118: 1823–30. [DOI] [PubMed] [Google Scholar]
- 9. Liu B, Pan S, Dong X et al. Opposing effects of arsenic trioxide on hepatocellular carcinomas in mice. Cancer Sci 2006; 97: 675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oketani M, Kohara K, Tuvdendorj D et al. Inhibition by arsenic trioxide of human hepatoma cell growth. Cancer Lett 2002; 183: 147–53. [DOI] [PubMed] [Google Scholar]
- 11. Xu HY, Yang YL, Liu SM et al. Effect of arsenic trioxide on human hepatocarcinoma in nude mice. World J Gastroenterol 2004; 10: 3677–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang SH, Song JH, Kang HK et al. Arsenic trioxide‐induced apoptosis is independent of stress‐responsive signaling pathways but sensitive to inhibition of inducible nitric oxide synthase in HepG2 cells. Exp Mol Med 2003; 35: 83–90. [DOI] [PubMed] [Google Scholar]
- 13. Lin CC, Hsu C, Hsu CH et al. Arsenic trioxide in patients with hepatocellular carcinoma: a phase II trial. Invest New Drugs 2007; 25: 77–84. [DOI] [PubMed] [Google Scholar]
- 14. Maeda H, Hori S, Nishitoh H et al. Tumor growth inhibition by arsenic trioxide in the orthotopic metastasis model of androgen‐independent prostate cancer. Cancer Res 2001; 61: 5432–40. [PubMed] [Google Scholar]
- 15. Kito M, Matsumoto K, Wada N et al. Antitumor effect of arsenic trioxide in murine xenograft model. Cancer Sci 2003; 94: 1010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen ZX, Shi ZZ, Fang J et al. All‐trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 2004; 101: 5328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niu C, Yan H, Yu T et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow‐up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 1999; 94: 3315–24. [PubMed] [Google Scholar]
- 18. Banerjee S, Li Y, Wang Z et al. Multi‐targeted therapy of cancer by genistein. Cancer Lett 2008; 269: 226–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mai Z, Blackburn GL, Zhou JR. Soy phytochemicals synergistically enhance the preventive effect of tamoxifen on the growth of estrogen‐dependent human breast carcinoma in mice. Carcinogenesis 2007; 28: 1217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burich RA, Holland WS, Vinall RL et al. Genistein combined polysaccharide enhances activity of docetaxel, bicalutamide and Src kinase inhibition in androgen‐dependent and independent prostate cancer cell lines. BJU Int 2008; 102: 1458–66. [DOI] [PubMed] [Google Scholar]
- 21. El‐Rayes BF, Ali S, Ali IF et al. Potentiation of the effect of erlotinib by genistein in pancreatic cancer: the role of Akt and nuclear factor‐kappaB. Cancer Res 2006; 66: 10553–9. [DOI] [PubMed] [Google Scholar]
- 22. Gu Y, Zhu CF, Iwamoto H et al. Genistein inhibits invasive potential of human hepatocellular carcinoma by altering cell cycle, apoptosis, and angiogenesis. World J Gastroenterol 2005; 11: 6512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, Ahmed F, Ali S et al. Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res 2005; 65: 6934–42. [DOI] [PubMed] [Google Scholar]
- 24. Yeh TC, Chiang PC, Li TK et al. Genistein induces apoptosis in human hepatocellular carcinomas via interaction of endoplasmic reticulum stress and mitochondrial insult. Biochem Pharmacol 2007; 73: 782–92. [DOI] [PubMed] [Google Scholar]
- 25. Sánchez Y, Amrán D, Fernández C et al. Genistein selectively potentiates arsenic trioxide‐induced apoptosis in human leukemia cells via reactive oxygen species generation and activation of reactive oxygen species‐inducible protein kinases (p38‐MAPK, AMPK). Int J Cancer 2008; 123: 1205–14. [DOI] [PubMed] [Google Scholar]
- 26. Sturlan S, Baumgartner M, Roth E et al. Docosahexaenoic acid enhances arsenic trioxide‐mediated apoptosis in arsenic trioxide‐resistant HL‐60 cells. Blood 2003; 101: 4990–7. [DOI] [PubMed] [Google Scholar]
- 27. Keshavan P, Schwemberger SJ, Smith DL et al. Unconjugated bilirubin induces apoptosis in colon cancer cells by triggering mitochondrial depolarization. Int J Cancer 2004; 112: 433–45. [DOI] [PubMed] [Google Scholar]
- 28. De Proost I, Pintelon I, Brouns I et al. Functional live cell imaging of the pulmonary neuroepithelial body microenvironment. Am J Respir Cell Mol Biol 2008; 39: 180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou JR, Mukherjee P, Gugger ET et al. Inhibition of murine bladder tumorigenesis by soy isoflavones via alterations in the cell cycle, apoptosis, and angiogenesis. Cancer Res 1998; 58: 5231–8. [PubMed] [Google Scholar]
- 30. Chen H, Sun B, Pan S et al. Dihydroartemisinin inhibits growth of pancreatic cancer cells in vitro and in vivo. Anticancer Drugs 2009; 20: 131–40. [DOI] [PubMed] [Google Scholar]
- 31. Liu F, Wang P, Jiang X et al. Antisense hypoxia‐inducible factor 1 α gene therapy enhances the therapeutic efficacy of doxorubicin to combat hepatocellular carcinomas. Cancer Sci 2008; 99: 2055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma L, Luo L, Qiao H et al. Complete eradication of hepatocellular carcinomas by combined vasostatin gene therapy and B7H3‐mediated immunotherapy. J Hepatol 2007; 46: 98–106. [DOI] [PubMed] [Google Scholar]
- 33. Wang D, Wang Z, Tian B et al. Two hour exposure to sodium butyrate sensitizes bladder cancer to anticancer drugs. Int J Urol 2008; 15: 435–41. [DOI] [PubMed] [Google Scholar]
- 34. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 35. Dodd S, Dean O, Copolov DL et al. N‐acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther 2008; 8: 1955–62. [DOI] [PubMed] [Google Scholar]
- 36. Barnes S. Effect of genistein on in vitro and in vivo models of cancer. J Nutr 1995; 125: 777S–83S. [DOI] [PubMed] [Google Scholar]
- 37. Wei H, Cai Q, Rahn RO. Inhibition of UV light‐ and Fenton reaction induced oxidative DNA damage by the soybean isoflavone genistein. Carcinogenesis 1996; 17: 73–7. [DOI] [PubMed] [Google Scholar]
- 38. Ruiz‐Larrea MB, Mohan AR, Paganga G et al. Antioxidant activity of phytoestrogenic isoflavones. Free Radic Res 1997; 26: 63–70. [DOI] [PubMed] [Google Scholar]
- 39. Pelicano H, Feng L, Zhou Y et al. Inhibition of mitochondrial respiration. A novel strategy to enhance drug‐induced apoptosis in human leukaemia cells by a reactive oxygen species‐mediated mechanism. J Biol Chem 2003; 89: 37832–9. [DOI] [PubMed] [Google Scholar]
- 40. Salvi M, Brunati AM, Clari G et al. Interaction of genistein with the mitochondrial electron transport chain results in the opening of the membrane transition pore. Biochim Biophys Acta 2002; 1556: 187–96. [DOI] [PubMed] [Google Scholar]
- 41. Hwang JT, Park IJ, Shin JI et al. Genistein, EGCG, and capsaicin inhibit adipocyte differentiation process via activating. AMP‐activated protein kinase. Biochem Biophys Res Commun 2005; 338: 694–9. [DOI] [PubMed] [Google Scholar]
- 42. Zafarullah M, Li WQ, Sylvester J et al. Molecular mechanisms of N‐acetylcysteine actions. Cell Mol Life Sci 2003; 60: 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castro AF, Altenberg GA. Inhibition of drug transport by genistein in multidrug‐resistant cells expressing P‐glycoprotein. Biochem Pharmacol 1997; 53: 89–93. [DOI] [PubMed] [Google Scholar]
- 44. McCollum G, Keng PC, States JC et al. Arsenite delays progression through each cell cycle phase and induces apoptosis following G2/M arrest in U937 myeloid leukaemia cells. J Pharmacol Exp Ther 2005; 313: 877–87. [DOI] [PubMed] [Google Scholar]
- 45. Cossarizza A, Franceschi C, Monti D et al. Protective effect of N‐acetylcysteine in tumor necrosis factor‐a‐induced apoptosis in U937 cells: the role of mitochondria. Exp Cell Res 1995; 220: 232–40. [DOI] [PubMed] [Google Scholar]
- 46. Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407: 770–6. [DOI] [PubMed] [Google Scholar]
- 47. Li P, Nijhawan D, Budihardjo I et al. Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–89. [DOI] [PubMed] [Google Scholar]
- 48. Larochette N, Decaudin D, Jacotot E et al. Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp Cell Res 1999; 249: 413–21. [DOI] [PubMed] [Google Scholar]
- 49. Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl‐2 family. J Cell Sci 2009; 122: 437–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shore GC, Nguyen M. Bcl‐2 proteins and apoptosis: choose your partner. Cell 2008; 135: 1004–6. [DOI] [PubMed] [Google Scholar]
- 51. Lunghi P, Giuliani N, Mazzera L et al. Targeting MEK/MAPK signal transduction module potentiates ATO‐induced apoptosis in multiple myeloma cells through multiple signaling pathways. Blood 2008; 112: 2450–62. [DOI] [PubMed] [Google Scholar]