

Abstract
The TEL (ETV6)–AML1 (RUNX1) chimeric gene fusion is the most common genetic abnormality in childhood acute lymphoblastic leukemias. Evidence suggests that this chimeric gene fusion constitutes an initiating mutation that is necessary but insufficient for the development of leukemia. In a search for additional genetic events that could be linked to the development of leukemia, we applied a genome‐wide array‐comparative genomic hybridization technique to 24 TEL–AML1 leukemia samples and two cell lines. It was found that at least two chromosomal imbalances were involved in all samples. Recurrent regions of chromosomal imbalance (>10% of cases) and representative involved genes were gain of chromosomes 10 (17%) and 21q (25%; RUNX1) and loss of 12p13.2 (87%; TEL), 9p21.3 (29%; p16INK4a/ARF), 9p13.2 (25%; PAX5), 12q21.3 (25%; BTG1), 3p21 (21%; LIMD1), 6q21 (17%; AIM1 and BLIMP1), 4q31.23 (17%; NR3C2), 11q22‐q23 (13%; ATM) and 19q13.11‐q13.12 (13%; PDCD5). Enforced expression of TEL and to a lesser extent BTG1, both single genes known to be located in their respective minimum common region of loss, inhibited proliferation of the TEL–AML1 cell line Reh. Together, these findings suggest that some of the genes identified as lost by array‐comparative genomic hybridization may partly account for the development of leukemia. (Cancer Sci 2007; 98: 698–706)
TEL (ETV6)–AML1 (RUNX1), generated by the chromosomal translocation t(12;21) (p13;q22), is the most prevalent fusion gene in pediatric cancer, occurring in some 25% of childhood acute lymphoblastic leukemias (ALL).( 1 , 2 ) Twin studies and retrospective analysis of archived neonatal blood spots of patients with ALL indicate that the chimeric gene arises predominantly during fetal hemopoiesis at a rate that considerably exceeds that of overt clinical ALL, and that TEL–AML1‐harboring fetal clones expand and can persist in a clinically covert state for more than a decade.( 1 , 2 ) These observations indicate that additional ‘secondary’ and postnatal genetic events are required for overt clinical leukemia. Findings obtained with animal models of the TEL–AML1 translocation are consistent with this notion.( 3 , 4 , 5 )
Fluorescence in situ hybridization (FISH), comparative genomic hybridization (CGH) and loss of heterozygosity (LOH) studies have all been used in efforts to identify the genetic events required for TEL–AML1‐harboring clones to become overtly leukemic.( 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ) Genomic loss of untranslocated TEL and gain of the AML1 gene have often been found; however, the methods used were not broadly applied to the genome at large, or the analyses met with limited resolution.
In the study presented here, we used the array‐CGH technique, which covers genome‐wide chromosomal imbalances (gains/losses) with high resolution, to analyze a total of 24 TEL–AML1 leukemia clinical samples and two cell lines. The results obtained revealed that all the samples, in addition to the TEL–AML1 translocation, harbored at least two chromosomal imbalances, including previously unidentified ones.
Materials and Methods
Patient material and cell lines. Leukemia cell samples were collected from the National Nagoya Hospital in Japan (16 cases), and the Japanese Red Cross Nagoya First Hospital (eight cases). Informed consent was obtained from parents or guardians of all patients to have their samples used for banking and molecular analysis. Leukemia cells accounted for at least 70% of all clinical samples. All samples featured the TEL–AML1 chimeric transcript as detected by reverse transcription–polymerase chain reaction (PCR) analysis. The ALL cell lines harboring the TEL–AML1 translocation, Reh and KOPN41, were obtained from the American Type Culture Collection (Manassas, VA, USA), Dr K. Sugita and Dr S. Nakazawa (University of Yamanashi, Nakakoma, Japan), respectively. Cells were maintained in RPMI‐1640 medium (Gibco‐BRL, New York, NY, USA), supplemented with 10% fetal calf serum, at 37°C under 5% CO2/95% air.
Comparative genomic hybridization. CGH analysis was carried out as described previously,( 15 ) on DNA obtained from bone marrow cells of 24 patients and the various cell lines. At least 10 metaphases were visualized using a BX‐60‐RF microscope (Olympus, Tokyo, Japan) equipped with IP Laboratory Scientific Imaging Software (Scanalytics, Fairfax, VA, USA). The threshold sets corresponded to a mean hybridization ratio between tumor and normal of >1.2:1 for gain and <0.80:1 for loss.( 15 ) Overrepresentation was interpreted as high‐level amplification when the ratio exceeded 2.0. Heterochromatic regions in the centromeric and paracentromeric parts of chromosomes, the short arm of acrocentric chromosomes, and all regions adjacent to telomeres were not included in the evaluation. Aberrations of chromosomes 19 and 22 were not evaluated as they have been reported to show false‐positive results in negative controls.( 16 ) The cut‐off value for the X chromosome of female patients was less than two copies (Table 1).
Table 1.
Summary of patient characteristics and chromosomal imbalances detected by a conventional comparative genomic hybridization (CGH) method
| Case/cell line | Age (years) | Sex | WBC counts at Dx | NCI risk | CR | Relapse | Survival | Losses by CGH | Gains by CGH |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | M | 14 700 | SR | Y | Y | N | 6q21‐q22.1 | 10, 162 |
| 2 | 4 | M | 3 600 | SR | Y | Y | Y | None | None |
| 3 | 2 | M | 11 000 | SR | Y | N | Y | 12p11 | None |
| 4 | 5 | F | 7 400 | SR | Y | N | Y | None | None |
| 5 | 3 | M | 34 200 | SR | Y | Y | Y | None | 10 |
| 6 | 5 | F | 3 900 | SR | Y | N | Y | 12p11.2‐p13.2 | 10, 21 |
| 7 | 5 | F | 77 800 | HR | Y | N | Y | None | None |
| 8 | 13 | F | 700 | HR | Y | N | Y | 12p11.2‐pter | None |
| 9 | 4 | M | 14 800 | SR | Y | Y | Y | None | 21 |
| 10 | 2 | M | 86 000 | HR | Y | N | Y | None | None |
| 11 | 3 | M | 14 000 | SR | Y | U | N | 6q22 | 21 |
| 12 | 3 | F | 39 200 | SR | Y | Y | Y | 11q13.3‐qter, 12p, 6q26‐qter | None |
| 13 | 2 | F | 159 000 | HR | N | NE | N | 12p | Xp |
| 14 | 7 | M | 8 400 | SR | Y | N | Y | 7q32‐qter | 10 |
| 15 | 6 | F | 37 500 | HR | Y | N | Y | None | Xq26‐qter |
| 16 | 2 | M | 5 100 | SR | Y | N | Y | None | None |
| 17 | 8 | M | 11 700 | HR | Y | N | Y | 11q21‐qter | 21q21‐qter, Xq21.3‐qter |
| 18 | 4 | F | 36 400 | HR | Y | N | Y | 12p | None |
| 19 | 8 | F | 10 100 | HR | Y | N | Y | 6q21‐q23 | 21 |
| 20 | 3 | M | 10 800 | HR | Y | N | Y | None | None |
| 21 | 4 | F | 8 100 | SR | Y | N | Y | 12p11‐p12 | None |
| 22 | 6 | M | 51 800 | HR | Y | N | Y | None | 21q22‐qter |
| 23 | 2 | F | 12 900 | HR | Y | N | Y | 1p34‐pter, 9p, 11q12, 12p12.1‐per, 16p, 17p | None |
| 24 | 3 | F | 16 700 | HR | Y | N | Y | 12p13 | None |
| Reh | NA | NA | NA | NA | NA | NA | NA | 3p21.1‐p21.3 | 21q21‐qter |
| KOPN41 | NA | NA | NA | NA | NA | NA | NA | 12p13.2‐pter | 21q21‐qter |
All cases were confirmed to have TEL–AML1 transcripts by reverse transcription–polymerase chain reaction. CR, complete remission; Dx, diagnosis; HR, high risk; NA, not applicable; NCI, National Cancer Institute; NE, not evaluable; SR, standard risk; U, unknown; WBC, white blood cell count.
Array‐CGH. A genome‐wide scanning array with 2304 bacteria artificial chromosome (BAC) and P1‐derived artificial chromosome (PAC) clones, covering the whole human genome at a resolution of roughly 1.3 Mb, was used in this study,( 17 ) whereas in some experiments a contig BAC array for chromosome 12p13.2 encompassing the TEL gene was used. The contig array contained 19 BAC clones that included four BAC for TEL and its adjacent genes covering 2 Mb. The location of all clones used for the array‐CGH were confirmed by standard FISH analyses. BAC/PAC clones were amplified by degenerate oligonucleotide primer (DOP)‐PCR and spotted on glass slides, as described previously.( 17 , 18 ) DNA preparation from cells, labeling, hybridization and scanning analyses were carried out according to the protocol previously described,( 19 ) with minor modifications.( 20 ) The data obtained were processed as described elsewhere to detect chromosomal imbalances.( 21 )
Oligonucleotide array‐CGH analyses of some DNA samples were conducted according to the manufacturer's instructions. Briefly, 3 µg of genomic DNA from the reference (46, XY male) and the corresponding experimental sample were digested with AluI and RsaI, and then filtered with the aid of QIAprep spin columns (Qiagen, Hilden, Germany). A genomic DNA labeling kit (Agilent Technologies, Santa Clara, CA, USA) was used to label the resultant DNA with Cy3‐dUTP or Cy5‐dUTP, and Microcon YM‐30 filter units (Millipore, Billeria, MA, USA) were used for filtering. Experimental and reference DNAs thus labeled were then mixed and hybridized with Human Genomic CGH 244K Microarrays (Agilent) in the presence of Cot‐1 DNA at 65°C for 40 h. After washing, the microarray slides were scanned with an Agilent Microarray Scanner, and the data were processed with Future Extraction software (Agilent).
Retroviral transduction and cell proliferation assays. cDNA for INK4a and ARF were kindly provided by Dr Scott Lowe (Cold Spring Harbor Laboratory, Cold Springs Harbor, NY, USA). cDNAs for TEL and BTG1 were cloned by the PCR method using primer sets harboring restriction sites that allowed for subsequent subcloning; TTTAGATCTAAAGAA‐TTCTCTGGGTTGGGGAGAGGAA/TTTGTCGACTTCCCGGGTCTCTTCCTTTA for TEL, and TTTTCTCGAGAATTC TCCCCTAGAACCAGTAGCC/TTTTCTCGAGACCTGATAC‐AGTCATCATATTG for BTG1. The resulting products were, respectively, digested with BglII/SalI and XhoI, and then inerted into the BamHI/XhoI or XhoI site of the pcDNA‐Flag vector (Invitrogen, Carlsbad, CA, USA). Respective cDNAs tagged with Flag at the carboxyl terminus were then isolated with EcoRI and inserted into the EcoRI site upstream of the ires‐green fluorescent protein (GFP) cassette of the pMXs‐iresGFP retroviral vector (a kind gift from Dr T. Kitamura, University of Tokyo). Retrovirus was produced by cotransfection into 293T cells of the respective retroviral vector and the packaging plasmid with VSV‐G envelope, pCGCGP (a kind gift from Dr A. Abe, Nagoya University). Reh cells were infected with the virus on retronectin‐coated dishes (Takara, Otsu, Japan), according to the manufacturer's instruction.
Retrovirally infected cells were monitored for GFP expression by flow‐cytometry on a FACS Caliber (BD Bioscience, San Jose, CA, USA). In some experiments, infected cells were stained with PKH‐26 dye (Sigma, St Louis, MI, USA) and monitored for red fluorescence, along with GFP to analyze cell division.
Results
CGH analyses of TEL–AML1 clinical samples. We first applied a conventional CGH technique to 24 TEL–AML1 clinical samples to identify chromosomal imbalances. The results are summarized in Table 1. Eighteen cases showed chromosomal imbalances, including loss of 12p11‐p13 (nine cases) and 6q22 (three cases), and gain of chromosome 10 (four cases), chromosome 21 (four cases) and 21q22‐qter (three cases), whereas six cases showed no alterations. These frequently altered genomic regions have been reported previously.( 6 , 7 , 22 , 23 )
Genomic profile of patient samples and TEL–AML1 cell lines. To analyze genomic alterations in more detail, we next used the array‐CGH technique. Our initial analysis was conducted on cell lines with the TEL–AML1 translocation. Array‐CGH profiles of KOPN41 (Fig. 1a) and Reh (Fig. 1b) cells revealed several common regions of chromosomal imbalance, including gain of regions on chromosome 21q22‐qter and loss of regions on 9p21.1, 12p13 and 12q21. Among these, gain of 21q22‐qter and loss of regions on 3p21.31, 9p21.3, 12p13.2 and 12q21.3 were subsequently confirmed in Reh cells by FISH analysis (Fig. 1c). Of particular note, loss of the region on 12q21.3 (the region where the BTG1 gene resides) was marginally detected by array‐CGH (the log2 ratio was −0.27, whereas the normal limit was within ±0.2), but the loss was subsequently confirmed by FISH analysis as a partial deletion of BAC, thus demonstrating the high‐resolution capacity of the array‐CGH method used.
Figure 1.
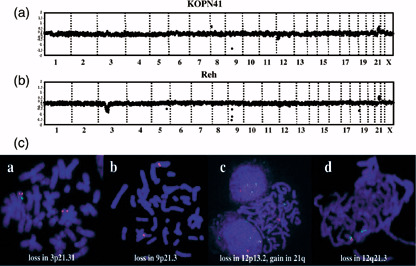
Array‐comparative genomic hybridization (CGH) profiles and fluorescent in situ hybridization (FISH) analysis of TEL–AML1 cell lines. Log2 ratios of signals of cell lines/sample versus normal control are plotted for all clones based on their chromosomal position, with chromosomes separated by vertical lines. Normal range of the log2 ratios are within ±0.2. Clones are arranged from chromosome 1–22 and X within each chromosome on the basis of the Sanger Center Mapping Position, July 2004 version. (a) In the KOPN41 cell line, chromosomal imbalances detected were loss of 9p21.3, 12p12.3‐pter and 12q21.3, and gain of 8p23.2 and 21q22.12‐qter. (b) In the Reh cell line, loss of 3p14.3‐p22.3, 5q23.2, 9p21.3, 12p13.2, 12q21.3 and 18q23, and gain of 21q22.12‐qter were detected. (c) FISH analyses of Reh cells for the indicated chromosomal imbalances. BAC clones used were (a) RP11‐509I21 (green) and RP11‐335I9 (red), (b) RP11‐149I2 (green) and RP11‐30O14 (red), (c) RP11‐71L1 (green) and RP11‐525I3 (red), and (d) RP11‐887P2 (green) and RP11‐796E2 (green). Loss of signals by RP11‐509I21 (3p21.31) in (a), RP11‐149I2 (9p21.3) in (b), RP11‐525I3 (12p13.2) in (c), a diminished signal by RP11‐796E2 (12q21.3) in (d), and an extra signal by RP11‐71L1 in (c) were clearly detected. Note that BAC RP11‐525I3 (12p13.2) used in (c) is not included in the array‐CGH analysis shown in (b), but is included in the contig‐array in Fig. 3, where the corresponding region of loss is presented.
We then analyzed the 24 TEL–AML1 clinical samples using array‐CGH and found that all cases exhibited at least two chromosomal imbalances. A representative array‐CGH profile of a patient (case no. 12) is shown in Fig. 2a. Loss of regions on chromosomes 3q11.2‐q13.13, 6q22.1, 6q25‐qter, 9p13.3, 9p21.3, 9p23, 11q14.1‐qter, 12p11.22‐p13.2, 12q21.33 and 19q13.11‐q13.12 were clearly detected. Among these, loss of 3q11.2‐q13.13, 6q22.1, 9p13.3, 9p21.3, 9p23, 12q21.33 and 19q13.11‐q13.12 were not detected using conventional CGH (Table 1).
Figure 2.
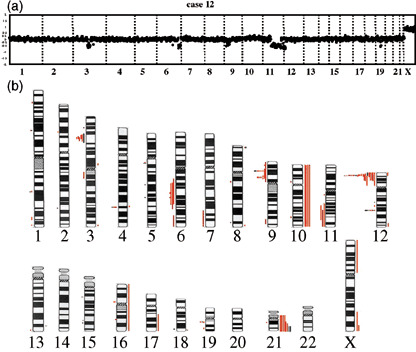
Summary of chromosomal imbalances. (a) A representative array‐comparative genomic hybridization (CGH) profile of patient samples (case 12) is presented. Detected were loss of 3q11.2‐q13.13, 6q22.1, 6q25‐qter, 9p13.3, 9p21.3, 9p23, 11q14.1‐qter, 12p11.22‐p13.2, 12q21.33 and 19q13.11‐q13.12. (b) Chromosomal imbalances detected in all patient samples (red lines) and two cell lines (black lines) are presented. Regions of loss and gain are represented by vertical lines, on the left (loss) and right (gain) side of each ideogram.
A summary of chromosomal gains and losses identified with the array‐CGH in the 24 TEL–AML1 cases and two cell lines is presented in Fig. 2. Recurrent chromosomal imbalances found in no less than three patient samples included a gain of 21q (six cases), 10 (four cases), and loss of 12p13.2 (21 cases), 9p21.3 (seven cases), 9p13.2 (six cases), 12q21.3 (six cases), 3p21 (five case), 6q21 (four cases), 4q31.23 (four cases), 11q22‐q23 (three cases) and 19q13.11‐q13.12 (three cases). These findings suggest that a limited number of non‐random genomic alterations could be causally related to the development of TEL–AML1 leukemia.
Array analysis of the 12p13.2 chromosome region in TEL–AML1 cases. We next sought to identify minimal common regions within clustered regions representing genomic deletions. Because deletion at 12p13 is known to be the most prevalent, occurring in some 60–80% of TEL–AML1 cases,( 1 ) we initially focused on this region with special attention to the TEL gene. Our initial arrays did not contain BAC for TEL, but 10 out of 24 cases (case 3, 6, 7, 8, 10, 13, 18, 21, 23 and 24) showed deletion of BAC encompassing a minimum of 25 Mb (BAC RP11‐277P12, RP11‐267J23 and RP11‐180‐M15). This suggests the occurrence of large‐scale deletions including TEL and its neighboring genes at both the telomeric and centromeric sides (Fig. 3). To analyze cases that did not show such large deletion areas surrounding the TEL gene (14 out of 24 cases: cases 1, 2, 4, 5, 9, 11, 12, 14, 15, 16, 17, 19, 20 and 22, and the Reh and KOPN41 cell lines), we constructed a new array that included two BAC for the TEL gene and one BAC each directly adjacent to the TEL gene at both the telomeric and centromeric sides. Results of the array analysis of the deleted regions in each of the patient samples are summarized in Fig. 3. In all, 21 out of 24 cases (87.5%), as well as the Reh and KOPN41 cell lines, showed deletion at 12p13.2. Case 19 showed gain of TEL and its telomeric regions. It is known that chromosomal translocations such as TEL–AML1 fusion often accompany deletion of the chromosomal regions adjacent to the break point on the allele involved in the translocation. This type of deletion therefore conceivably coincides with the TEL–AML1 translocation that represents the ‘first hit’ event associated with the development of leukemia. Deletion of the TEL gene, or part of it, found in our contig array could therefore be located on the allele involved in the TEL–AML1 translocation. However, given that deletion of the TEL gene or part of it is on the allele involved in the TEL–AML1 translocation, exons 1–5 should be retained as these exons are contained in the TEL–AML1 fusion gene. Because BAC RP11‐434C1 and RP11‐418C2 contain exon 1 and exons 3–8, respectively, RP11‐434C should be retained in this case. Cases 9 and 16, as well as the Reh and KOPN41 cell lines, which exhibit deletion of BAC‐434C1, can thus be assumed to have lost the TEL gene or part of it on the allele not involved in the TEL–AML1 translocation. Along the same lines, the cases mentioned earlier that showed large‐scale deletions (cases 3, 6, 7, 8, 10, 13, 18, 21, 23 and 24) were judged to have lost the region including TEL on the allele not involved in the TEL–AML1 translocation. Altogether, 12 of the 24 cases (cases 3, 6, 7, 8, 9, 10, 13, 16, 18, 21, 23 and 24) and the Reh and KOPN41 cell lines exhibited loss of the region including the TEL gene on the allele not involved in the TEL‐AML1 translocation. The loss, ipso facto, is unlikely to have coincided with the translocation. It should be noted that the KOPN41 cell line, unlike cases 9 and 16 and Reh, did not show deletion of BAC RP11‐418C2, which contains exons 3–8 of the TEL gene, but exhibited deletion of BAC RP11‐434C1 and its telomeric region (Fig. 3). Thus, the minimum region of deletion at 12p13 occurring independently of TEL–AML1 translocation in both the samples and cell lines analyzed was most likely the TEL gene. Cases 2, 4 and 5 exhibited loss of a region at BAC RP11‐418C2, but not at adjacent regions (BAC RP11‐434C1 and RP11‐525I3). These cases may represent intragenic deletion of the TEL gene on the allele not involved in the TEL–AML1 translocation,( 8 ) and could thus be included in the cases with TEL deletion not accompanying the translocation.
Figure 3.
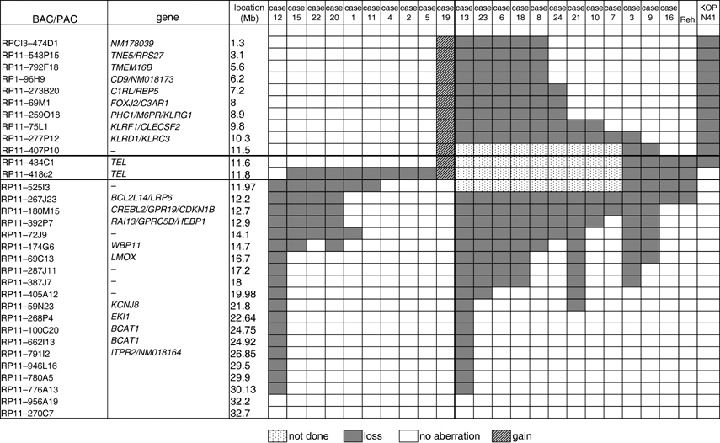
Common region of loss at 12p13.2 region. Bacteria artificial chromosome and P1‐derived artificial chromosome (BAC/PAC) clones, their location, and the absence or presence of loss are shown in each case and cell line. Filled‐in and hatched boxes represent losses and gains, respectively. Dotted boxes indicate clones not tested.
Consensus regions deleted at 3p21.3, 4q31.23, 6q21, 9p13.2, 9p21.3, 11q22.1‐q23.3, 12q21.33 and 19q13.11‐q13.12. Our BAC‐array‐CGH analysis showed, in addition to the TEL region, common regions of loss at 3p21.3, 4q31.23, 6q21, 9p13.2, 9p21.3, 11q22.1‐q23.3, 12q21.33 and 19q13.11‐q13.12.
3p21.3. Loss of the 3p21.31 region has been reported in many solid tumors.( 24 , 25 , 26 ) In our array‐CGH analysis, the minimum common region of loss was flanked by BAC RPCI5‐1053D16 and RP5‐1034C14 (Fig. 4a). This region contains a variety of genes, including LIMD1 (LIM domains‐containing protein 1), LZTFL1 (leucine zipper transcription factor‐like 1) and FYCO1 (FYVE and coiled‐coil domain containing 1), in addition to genes that code for several chemokines, SMARCC1 (SWI/SNF‐related matrix associated actin‐dependent regulator of chromatin subfamily member 1), CDC25A, ZNF589 (zinc finger protein 589), LRRC2 (leucine rich repeat containing 2) and TDGF1 (teratocarcinoma‐derived growth factor 1 presursor). Among these, the LIMD1 product is capable of inhibiting tumor growth by interacting with the retinoblastoma protein and suppressing E2F1‐mediated transcription.( 27 ) LRRC2 ( 28 ) is a relative of ras suppressor protein 1 (RSP‐1), which inhibits ras‐mediated transformation of NIH3T3 cells. These findings suggest that the 3p21.31 region deleted in our patient samples contained tumor suppressors.
Figure 4.
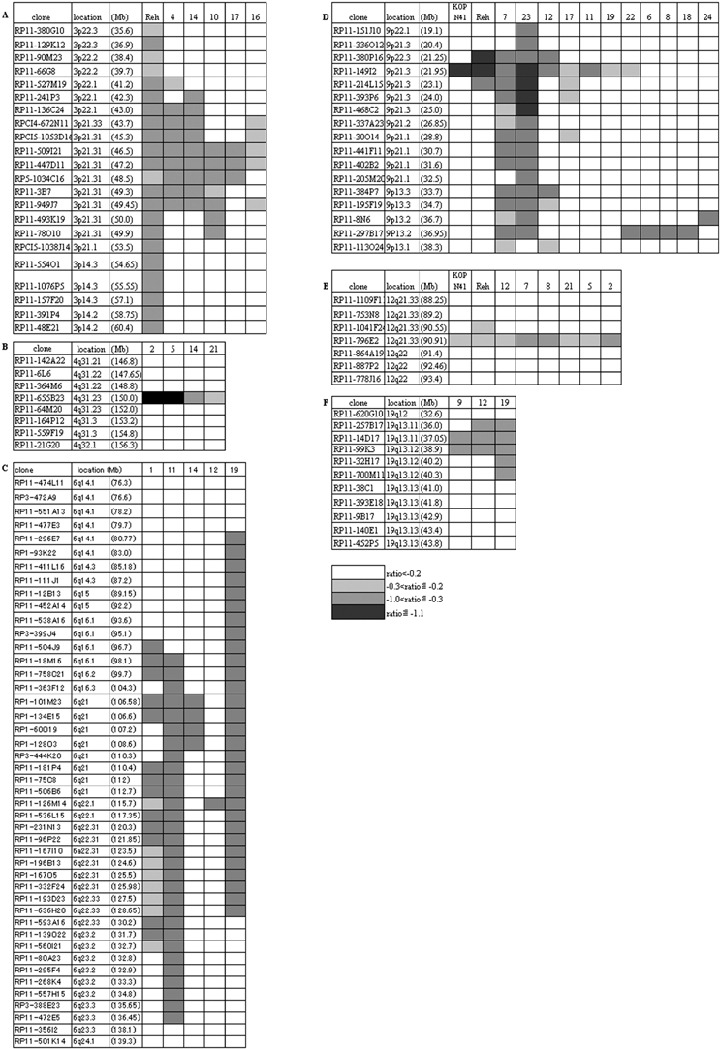
Common region of loss at 3p21.3, 4q31.23, 6q21, 9p13.2, 9p21.3, 12q21.33 and 19q13.11‐q13.12 regions. Bacteria artificial chromosome and P1‐derived artificial chromosome (BAC/PAC) clones, their location, and the log2 ratios of signals of case/cell line versus normal control are presented as indicated.
4q31.23. Loss of the 4q31 region has been reported in solid tumors.( 29 , 30 ) Four of the cases included in our study exhibited loss of the region flanked by BAC RP11‐364M6 and RP11‐64M20 (Fig. 4b). This region contains genes including EDNRA (endothelin‐1 receptor precursor), TMEM34 (transmembrane protein 34), ARHGAP10 (Rho GTPase activating protein 10), NR3C2 (mineralocorticoid receptor), DCAK2‐HUMAN (serine/threonine‐protein kisase DCAMKL2) and LRBA (lipopolysaccharide‐responsive and beige‐like anchor protein). It is speculated that deregulated intracellular signaling through loss of these genes may facilitate the development of leukemia.
6q21. Deletion of the 6q21 region has been found in lymphoid malignancies,( 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 ) and contains genes including AF6q21 (FOXO3A) implicated in cell cycle and apoptosis,( 39 ) a putative tumor suppressor AIM1 (absent in melanoma protein 1),( 38 ) and BLIMP1 (B lymphocyte‐induced maturation protein 1),( 40 ) implicated in plasma cell differentiation. Among these, AIM1 and BLIMP1 were included in the region commonly lost in our patient samples and flanked by BAC RP11‐363F12 and RP1‐60O19 (Fig. 4c). Case 12 showed a loss only at BAC RP11‐126M14, which was shared by cases 1, 11 and 19; however, this BAC contains no known cancer‐related genes, and does not fall in the reported regions of loss or LOH in lymphoid leukemia.( 32 , 33 , 34 , 35 , 36 )
9p13.2. Results of an LOH study of small lung cancers suggest that the 9p13 region contains tumor suppressors.( 41 , 42 ) Our array‐CGH analysis revealed that six cases showed loss of the region flanked by BAC RP11‐8N6 and RP11‐113O24 (Fig. 4d). This region contains the PAX5 gene that encodes the transcription factor B cell‐specific activating protein (BSAP), essential for B cell lineage commitment,( 40 ) whereas overexpression of Pax5 has been linked to the development of lymphoma.( 43 , 44 ) In our study, this region containing PAX5 was unexpectedly deleted. The region also contains miscellaneous genes including ZCCHC7 (zinc finger CCHC domain containing 7), ZBTB5 (zinc finger and BTB domain containing protein 5), FRMPD1 (FERM and PDZ domain containing 1), RG9MTD3 (RNA guanine‐9‐methyl transferase domain containing 3), WDR32 (WD repeat domain 32) and SHB (Src homology 2 domain containing adoptor protein B), although their involvement in relation to leukemia remains unknown.
9p21.3. Seven cases and two cell lines exhibited loss of 9p21.3, and the common region of loss was flanked by BAC RP11‐380P16 and RP11‐214L15 (Fig. 4d). This region contains genes for the interferon alpha precursors CDKN2A (cyclin dependent kinase 4 inhibitor A; INK4a) and CDKN2B (cyclin dependent kinase 4 inhibitor B; INK4b). The latter two genes are frequently deleted or inactivated in many tumors.( 45 )
11q22.1‐q23.3. Three cases in our study exhibited loss of the 11q22.1‐q23.3 region spanning approximately 15 Mb. The limited number of cases available for analysis made it impossible to identify the respective genes in this large chromosomal area, although this region overlaps with the region deleted in some lymphoid malignancies,( 46 , 47 ) and contains the ATM gene as a candidate tumor suppressor. Mutations of the ATM gene have also been implicated in the development of acute lymphoblastic leukemia.( 48 , 49 , 50 )
12q21.33. Six cases and two cell lines exhibited loss of the region flanked by BAC RP11‐1041F24 and RP11‐864A19 (Fig. 4e). This region contains BTG1 as the only known gene. BTG1 belongs to the anti‐proliferative (APRO) gene family, the product of which is capable of inhibiting cell proliferation.( 51 , 52 , 53 )
19q13.11‐q13.12. Loss of the 19q13 region has been reported in some cancers.( 54 , 55 ) Three of our cases showed loss of this region flanked by BAC RP11‐257B17 and RP11‐32H17 (Fig. 4f). This region contains miscellaneous genes including PDCD5 (programmed cell death protein 5), the product of which is capable of enhancing apoptosis,( 56 ) ANKRD27 (ankyrin repeat domain 27), ULE1B‐HUMAN (ubiquitin‐like 2 activating enzyme E1B) and RHPN2 (Rhophilin 2). Loss of these genes may lead to resistance to apoptosis and deregulated cellular signaling.
Regions of loss presented here are relatively large in size, thus precluding accurate delineation with the BAC array‐CGH method of the genes responsible in their respective regions. We therefore next used the oligonucleotide array‐CGH method for selected samples (cases 2, 5, 11, 12, 14, 17, and cell lines Reh and KOPN41) for further narrowing down of genes responsible. Because 3p21.31, 6q16 and 19q13.11 exhibited large regions of loss, it was unlikely that the genes responsible could be singled out there. We therefore focused on the 4q21.23, 9p21.3 and 12q21.33 regions. Figure 5 illustrates the oligonucleotide array‐CGH profiles of each of these regions. The results showed that NR3C and ARHGAP10 in 4q31.23, BTG1 in 12q21.33, and CDNK2A, CDNK2B and MTAP in 9p21.3 were the genes responsible for the respective region of loss. Among these, deletion of NR3C in cases 2 and 5, and MTAP/CDNK2A/CDNK2B in Reh and KOPN41, were homozygous, as indicated by their respective log2 ratios.
Figure 5.
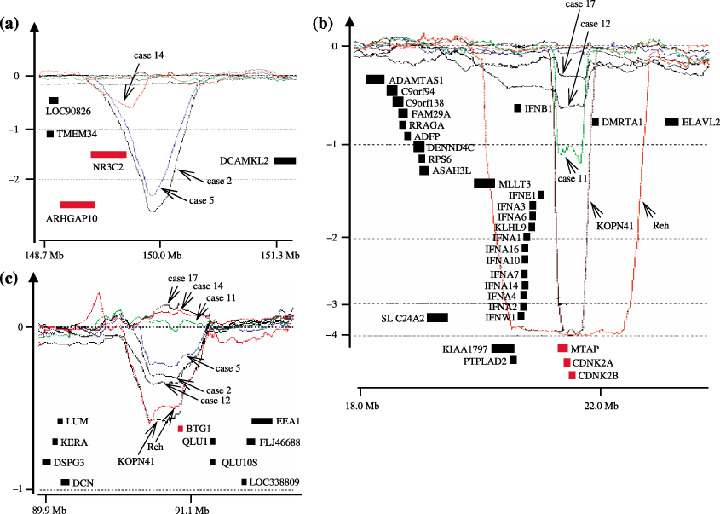
Oligonucleotide array‐comparative genomic hybridization (CGH) profiles. DNA from cases 2, 5, 11, 12, 14, 17, and cell lines Reh and KOPN41 were used for the oligonucleotide array‐CGH analysis of the 4q21.23, 9p21.3 and 12q21.33 regions. Horizontal and vertical axes, respectively, represent the positions of the oligonucleotide probes and log2 ratios of the hybridized signals. Locations of genes are presented as solid bars, with the genes contained in the minimum region of loss shown in red.
Enforced expression of TEL and BTG1 is capable of limiting proliferation of Reh cells. Finally, we sought to determine which of the deleted genes are likely to be associated with the development of leukemia. To this end, the products of the genes involved were expressed using a retroviral expression system. We focused on TEL and BTG1, as these genes were found to be single genes commonly deleted in 12p13.2 and 12q21.3, respectively. Reh cells were infected with retrovirus for TEL or BTG1 that coexpress GFP by virtue of the ires‐GFP cassette (Fig. 6a), and the percentage GFP expression in the bulk culture was then monitored by flow cytometry; we were unsuccessful in our efforts to infect KOPN41 cells. Arf and Ink4a, often deleted in many cancers including TEL–AML1 leukemia, were also included in the analysis. Expression of exogenously transduced genes was confirmed by western blotting (not shown). Figure 6b shows typical FACS data immediately following infection and 4 weeks thereafter. Reh cells infected with GFP‐only control virus displayed 18.5% GFP immediately following infection, and remained largely unchanged over 4 weeks. In contrast, Reh cells infected with Arf‐ and Ink4a‐virus, as expected, resulted in decreased GFP expression over time, suggesting that enforced expression of Arf and Ink4a compromise cell growth. Of particular note, enforced expression of TEL, and to a lesser extent of BTG1, also compromised cell growth. The time‐course of GFP expression is summarized in Fig. 6c. Because Arf and Ink4a are well‐established tumor suppressors, we next focused on TEL to further examine its effect on cell growth. Reh cells were infected with TEL‐ and GFP‐only control viruses, cell membranes were stained with PKH‐26 dye, and GFP expression monitored together with dye fluorescence (Fig. 6d). Reh cells infected with the GFP‐only control virus displayed equal PKH‐26 fluorescence intensity over time when comparing GFP‐positive and GFP‐negative fractions, suggesting equivalent cell division in both fractions. In contrast, in the case of TEL‐virus‐infected Reh cells, the GFP‐positive fraction displayed higher PKH‐26 fluorescence compared with the GFP‐negative fraction, although it was noted that fluorescence levels were equivalent immediately following infection. Cell death as detected by 7‐AAD (amino‐actinomycin D) staining was similar over time in TEL‐virus‐infected cells (not shown). These findings suggest that inhibited cell proliferation, but not enhanced cell death, is the primary cause of the compromised cell growth observed.
Figure 6.
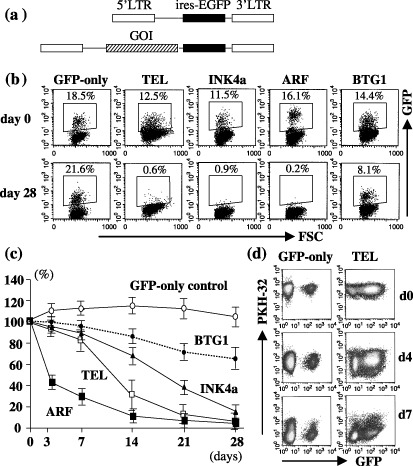
Effect of enforced expression of TEL and BTG1 on growth of Reh cells. (a) Schematic drawing of the retroviral vectors used (not to scale). (b) Typical FACS profile showing green fluorescent protein (GFP) expression of Reh cells infected with the indicated virus. Note that the GFP‐control essentially displayed constant GFP expression over 28 days, in contrast to TEL‐, INK4a‐, Arf‐ and BTG1‐virus infected cells, where GFP expression decreased with time. (c) Time‐course of GFP expression. % GFP expression relative to that of day 0 is shown. Typical data of five independent experiments is presented together with the mean and SD of triplicate samples. (d) Inhibited cell division of TEL‐expressing Reh cells. Reh cells were infected with GFP‐only control‐viruses or TEL‐viruses, and immediately stained with PKH26 fluorescent dye. FACS profiles for GFP and PKH‐26 fluorescence of infected cells are shown. Two independent experiments yielded similar results.
Discussion
Postnatal secondary genetic events have been postulated as being crucial for TEL–AML1‐harboring clones to become overtly leukemic.( 1 , 2 ) FISH analysis using probes for TEL and AML1 has frequently detected deletion of non‐translocated TEL and amplification of AML1, suggesting that these genomic alterations are candidates for the secondary genetic events.( 6 , 14 )
In a search for additional genomic abnormalities, detailed cytogenetic studies and CGH analyses have been conducted, albeit with low resolution, thus making the detection of chromosomal imbalances limited. Here we used a genome‐wide array‐CGH method with proven higher resolution to identify genomic alterations in 24 TEL–AML1 patient samples. Our findings showed that: (1) all patients had, in addition to the TEL–AML1 translocation, at least two chromosomal imbalances, including gain of chromosomes 10 and 21q and loss of chromosomes 3p21, 4q31, 9p13, 9p21, 12p13, 12q21 and 19q13; (2) 12 out of 24 patients (50%) showed deletion of the TEL gene on the allele not involved in the TEL–AML1 translocation; and (3) genes responsible for cell cycle regulation were also frequently deleted (45.8%).
Using a conventional CGH method, Ma et al. (two cases)( 6 ) and Kanerva et al. (eight cases)( 7 ) demonstrated the loss of 6q, 8p, 9p, 11q21qter and 12p, and gain of 7p15‐pter, 8q22‐qter, 10p12‐pter, Xq23‐q26 and 21 in TEL–AML1 leukemia samples. CGH analysis of our patient samples also identified loss of 6q (four cases), 9p (one case), 11q (three cases), 12p (nine cases), and gain of 21q (six cases) and Xq25‐qter (two cases). Loss of 7q32‐qter (one case), 1p34 (one case), 16p (one case) and 17p (one case), and gain of 16 (one cases), 10 (four cases) and Xp (one case) were additionally identified in our CGH study, although some of these were reported by cytogenetic analyzes.( 22 , 23 ) Six cases had no abnormalities, as indicated by the CGH method used.
Our array‐CGH analysis revealed chromosomal imbalances in more detail than was possible using conventional CGH. The region encompassing the TEL gene on the allele not involved in the TEL–AML1 translocation is reportedly deleted in TEL–AML1 leukemia. The findings of LOH studies using PCR‐mediated detection of microsatellite markers and expression analysis of genes involved in the deleted region of leukemia samples suggest that TEL is most likely the gene in the minimum common region of deletion, although these studies are not necessarily confined to TEL–AML1 leukemia.( 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ) In the study presented here, we used TEL–AML1 leukemia samples and cell lines exclusively for the array‐CGH analysis and found that TEL is indeed the gene in question. It is known that chromosomal translocations such as the TEL–AML1 fusion often accompany deletion of the chromosomal regions adjacent to the break point on the allele involved in the translocation. Indeed, through detailed FISH analyses of Reh and KOPN41 cell lines (S. Tsuzuki and S. Karnan, unpublished observations, May 2005), we found that in the case of the Reh cell line, TEL on the allele not involved in the TEL–AML1 translocation was translocated to chromosome 5, which then became associated with the partial deletion of TEL. The other TEL allele in Reh cells was translocated to the AML1 gene, and was accompanied by deletion of regions centromeric to the TEL gene (RP11‐525I3 and RP11‐267J23). Similarly, in the case of KOPN41, TEL not involved in the TEL–AML1 translocation was found to be fused to material of chromosome 21 (and did not generate a TEL–AML1 chimera), which accompanied deletion of regions telomeric to the break point, although the TEL gene and its neighboring genes on the allele involved in the TEL–AML1 translocation remained undeleted. Thus, both the Reh and KOPN41 cell lines showed partial deletion of the TEL gene and its adjacent chromosomal regions on the allele not involved in the TEL–AML1 translocation. The overlapping region of the deletion was in BAC RP11‐434C1, which contained exon 1 of the TEL gene exclusively. When combined with the findings of our patient sample analysis, these results suggest that the minimum common region of loss at 12p13.3 is the TEL gene. Cases 2, 4 and 5 may feature intragenic deletions of the TEL gene on the allele not involved in the TEL–AML1 translocation, as previously reported.( 8 ) Importantly, loss of TEL is not confined to leukemia, but has also been reported for a subset of solid tumors, such as prostate cancer.( 57 , 58 ) These findings suggest that TEL might act as a tumor suppressor.
Along the same lines, it should be noted that most of the chromosomal losses found in our study are also found in non‐hematopoietic malignancies.( 59 ) In addition to the INK4a/ARF and ATM regions, the 3p21.3, 4q31.23, 6q21, 9p21.2 and 19q13.12 regions are often found to be deleted in many tumors. It is not clear which genes are responsible for tumor development in these regions, but LIMD1 ( 27 ) and LRRC2 ( 28 ) in 3p21.31, AIM ( 38 ) in 6q21 and PDCD5 in 19q13.11‐q13.12( 56 ) are plausible candidates. Detailed analyses to determine the minimum common region of deletion should provide a better understanding of the genes and mechanisms involved in the development of leukemia.
The development of leukemia is thought to require at least two mutations comprising class I mutations, which deregulate cell proliferation, and class II mutations, which restrict cell differentiation.( 60 ) The TEL–AML1 translocation has been classified as a class II mutation with inhibitory effects on B cell differentiation.( 4 ) As for class I mutations, gain‐of‐function mutations of tyrosine kinases, such as c‐kit and flt‐3, have frequently been found in myeloid leukemia,( 60 ) but not in TEL–AML1 leukemia.( 61 ) Thus, loss of cell cycle inhibitor genes observed in our study may represent a class I mutation; LIMD1, INK4a/ARF, BTG1 and TEL are included in the region of loss. Of these, we focused on TEL and BTG1, which are both single genes known to be located in the minimum common region of loss, and set out to examine effects on the cell growth of TEL–AML1‐harboring Reh cells. Results show that TEL and BTG1 inhibit cell growth, just as the well‐established tumor suppressors Ink4a/Arf; the ability of BTG1 to inhibit cell growth was less marked compared with other inhibitors, but was reproducible.( 62 , 63 ) These findings suggest that deletion of TEL and BTG1 could partly account for the class I mutation in TEL–AML1 leukemia.
In conclusion, the study presented here demonstrated that, in addition to previously described abnormalities such as deletion of TEL and amplification of the AML1 gene, ALL with t(12;21) contains a limited number of non‐random chromosomal imbalances. The chromosomal imbalances found in our study suggest pathways for tumor development that are also involved in cancers other than TEL–AML1 leukemia. Identification of some of the genes located in each region associated with the chromosomal imbalances was possible using array‐CGH, with its attendant high resolution, coupled with functional analysis. The data presented here should help to further our understanding of the molecular mechanisms leading to the development of leukemia.
Acknowledgments
We are grateful to Dr H. Asou (Hiroshima University) for information concerning the KOPN41 cell line. This work was supported in part by a Grant‐in Aid for Scientific Research from the Japanese Society for the Promotion of Science (S. T. and M. S.), a Grant‐in‐aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (M. S.), the Japan Leukemia Research Fund (S. T.), and the Aichi Cancer Research Foundation (S. T.).
References
- 1. Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood 2003; 102: 2321–33. [DOI] [PubMed] [Google Scholar]
- 2. Zelent A, Greaves M, Enver T. Role of the TEL–AML1 fusion gene in the molecular pathogenesis of childhood acute lymphoblastic leukaemia. Oncogene 2004; 23: 4275–83. [DOI] [PubMed] [Google Scholar]
- 3. Bernardin F, Yang Y, Cleaves R et al. TEL‐AML1, expressed from t(12;21) in human acute lymphocytic leukemia, induces acute leukemia in mice. Cancer Res 2002; 62: 3904–8. [PubMed] [Google Scholar]
- 4. Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first‐hit functions of the t(12;21) TEL–AML1 translocation in mice. Proc Natl Acad Sci USA 2004; 101: 8443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL–AML1 promotes development of specific hematopoietic lineages consistent with preleukemic activity. Blood 2004; 103: 3890–6. [DOI] [PubMed] [Google Scholar]
- 6. Ma SK, Wan TS, Cheuk AT et al. Characterization of additional genetic events in childhood acute lymphoblastic leukemia with TEL/AML1 gene fusion: a molecular cytogenetics study. Leukemia 2001; 15: 1442–7. [DOI] [PubMed] [Google Scholar]
- 7. Kanerva J, Niini T, Vettenranta K et al. Loss at 12p detected by comparative genomic hybridization (CGH): association with TEL–AML1 fusion and favorable prognostic features in childhood acute lymphoblastic leukemia (ALL): A multi‐institutional study. Med Pediatr Oncol 2001; 37: 419–25. [DOI] [PubMed] [Google Scholar]
- 8. Cave H, Cacheux V, Raynaud S et al. ETV6 is the target of chromosome 12p deletions in t(12;21) childhood acute lymphocytic leukemia. Leukemia 1997; 11: 1459–64. [DOI] [PubMed] [Google Scholar]
- 9. Montpetit A, Larose J, Boily G, Langlois S, Trudel N, Sinnett D. Mutational and expression analysis of the chromosome 12p candidate tumor suppressor genes in pre‐B acute lymphoblastic leukemia. Leukemia 2004; 18: 1499–504. [DOI] [PubMed] [Google Scholar]
- 10. Raimondi SC, Shurtleff SA, Downing JR et al. 12p abnormalities and the TEL gene (ETV6) in childhood acute lymphoblastic leukemia. Blood 1997; 90: 4559–66. [PubMed] [Google Scholar]
- 11. Raynaud S, Cave H, Baens M et al. The 12;21 translocation involving TEL and deletion of the other TEL allele: two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood 1996; 87: 2891–9. [PubMed] [Google Scholar]
- 12. Patel N, Goff LK, Clark T et al. Expression profile of wild‐type ETV6 in childhood acute leukaemia. Br J Haematol 2003; 122: 94–8. [DOI] [PubMed] [Google Scholar]
- 13. Wlodarska I, Baens M, Peeters P et al. Biallelic alterations of both ETV6 and CDKN1B genes in a t(12;21) childhood acute lymphoblastic leukemia case. Cancer Res 1996; 56: 2655–61. [PubMed] [Google Scholar]
- 14. Attarbaschi A, Mann G, Konig M et al. Austrian Berlin‐Frankfurt‐Munster cooperative Study Group. Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia 2004; 18: 1611–16. [DOI] [PubMed] [Google Scholar]
- 15. Hu J, Khanna V, Jones M, Surti U. Chromosomal imbalances in uterine leiomyosarcomas: potential markers for clinical diagnosis and prognosis. Genes Chromosomes Cancer 2001; 31: 117–24. [DOI] [PubMed] [Google Scholar]
- 16. Franke S, Wlodarska I, Maes B et al. Lymphocyte predominance Hodgkin disease is characterized by recurrent genomic imbalances. Blood 2001; 97: 1845–53. [DOI] [PubMed] [Google Scholar]
- 17. Tagawa H, Karnan S, Suzuki R et al. Genome‐wide array‐based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM . Oncogene 2005; 24: 1348–58. [DOI] [PubMed] [Google Scholar]
- 18. Kameoka Y, Tagawa H, Tsuzuki S et al. Contig array CGH at 3p14.2 points to the FRA3B/FHIT common fragile region as the target gene in diffuse large B‐cell lymphoma. Oncogene 2004; 23: 9148–54. [DOI] [PubMed] [Google Scholar]
- 19. Pinkel D, Segraves R, Sudar D et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 1998; 20: 207–11. [DOI] [PubMed] [Google Scholar]
- 20. Ota A, Tagawa H, Karnan S et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31‐q32 amplification in malignant lymphoma. Cancer Res 2004; 64: 3087–95. [DOI] [PubMed] [Google Scholar]
- 21. Tagawa H, Tsuzuki S, Suzuki R et al. Genome‐wide array‐based comparative genomic hybridization of diffuse large B‐cell lymphoma: comparison between CD5‐positive and CD5‐negative cases. Cancer Res 2004; 64: 5948–55. [DOI] [PubMed] [Google Scholar]
- 22. Raynaud SD, Dastugue N, Zoccola D, Shurtleff SA, Mathew S, Raimondi SC. Cytogenetic abnormalities associated with the t(12;21): a collaborative study of 169 children with t(12;21)‐positive acute lymphoblastic leukemia. Leukemia 1999; 13: 1325–30. [DOI] [PubMed] [Google Scholar]
- 23. Martineau M, Jalali GR, Barber KE et al. ETV6/RUNX1 fusion at diagnosis and relapse: some prognostic indications. Genes Chromosomes Cancer 2005; 43: 54–71. [DOI] [PubMed] [Google Scholar]
- 24. Mosse YP, Greshock J, Margolin A et al. High‐resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes Chromosomes Cancer 2005; 43: 390–403. [DOI] [PubMed] [Google Scholar]
- 25. Senchenko VN, Liu J, Loginov W et al. Discovery of frequent homozygous deletions in chromosome 3p21.3 LUCA and AP20 regions in renal, lung and breast carcinomas. Oncogene 2004; 23: 5719–28. [DOI] [PubMed] [Google Scholar]
- 26. Tischoff I, Markwarth A, Witzigmann H et al. Allele loss and epigenetic inactivation of 3p21.3 in malignant liver tumors. Int J Cancer 2005; 115: 684–9. [DOI] [PubMed] [Google Scholar]
- 27. Sharp TV, Munoz F, Bourboulia D et al. LIM domains‐containing protein 1 (LIMD1), a tumor suppressor encoded at chromosome 3p21.3, binds pRB and represses E2F‐driven transcription. Proc Natl Acad Sci USA 2004; 101: 16 531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kiss H, Yang Y, Kiss C et al. The transcriptional map of the common eliminated region 1 (C3CER1) in 3p21.3. Eur J Hum Genet 2002; 10: 52–61. [DOI] [PubMed] [Google Scholar]
- 29. Knosel T, Schluns K, Stein U et al. Genetic imbalances with impact on survival in colorectal cancer patients. Histopathology 2003; 43: 323–31. [DOI] [PubMed] [Google Scholar]
- 30. Dave BJ, Nelson M, Pickering DL et al. Cytogenetic characterization of diffuse large cell lymphoma using multi‐color fluorescence in situ hybridization. Cancer Genet Cytogenet 2002; 132: 125–32. [DOI] [PubMed] [Google Scholar]
- 31. Jackson A, Carrara P, Duke V et al. Deletion of 6q16‐q21 in human lymphoid malignancies: a mapping and deletion analysis. Cancer Res 2000; 60: 2775–9. [PubMed] [Google Scholar]
- 32. Takeuchi S, Koike M, Seriu T et al. Frequent loss of heterozygosity on the long arm of chromosome 6: identification of two distinct regions of deletion in childhood acute lymphoblastic leukemia. Cancer Res 1998; 58: 2618–23. [PubMed] [Google Scholar]
- 33. Merup M, Moreno TC, Heyman M et al. 6q deletions in acute lymphoblastic leukemia and non‐Hodgkin's lymphomas. Blood 1998; 91: 3397–400. [PubMed] [Google Scholar]
- 34. Gerard B, Cave H, Guidal C, Dastugue N, Vilmer E, Grandchamp B. Delineation of a 6 cM commonly deleted region in childhood acute lymphoblastic leukemia on the 6q chromosomal arm. Leukemia 1997; 11: 228–32. [DOI] [PubMed] [Google Scholar]
- 35. Sherratt T, Morelli C, Boyle JM, Harrison CJ. Analysis of chromosome 6 deletions in lymphoid malignancies provides evidence for a region of minimal deletion within a 2‐megabase segment of 6q21. Chromosome Res 1997; 5: 118–24. [DOI] [PubMed] [Google Scholar]
- 36. Menasce LP, Orphanos V, Santibanez‐Koref M, Boyle JM, Harrison CJ. Deletion of a common region on the long arm of chromosome 6 in acute lymphoblastic leukaemia. Genes Chromosomes Cancer 1994; 10: 26–9. [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y, Matthiesen P, Harder S et al. A 3‐cM commonly deleted region in 6q21 in leukemias and lymphomas delineated by fluorescence in situ hybridization. Genes Chromosomes Cancer 2000; 27: 52–8. [DOI] [PubMed] [Google Scholar]
- 38. Ray ME, Wistow G, Su YA, Meltzer PS, Trent JM. AIM1, a novel non‐lens member of the betagamma‐crystallin superfamily, is associated with the control of tumorigenicity in human malignant melanoma. Proc Natl Acad Sci USA 1997; 94: 3229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burgering BM, Kops GJ. Cell cycle and death control: long live forkheads. Trends Biochem Sci 2002; 27: 352–60. [DOI] [PubMed] [Google Scholar]
- 40. Schebesta M, Heavey B, Busslinger M. Transcriptional control of B‐cell development. Curr Opin Immunol 2002; 14: 216–23. [DOI] [PubMed] [Google Scholar]
- 41. Kim SK, Ro JY, Kemp BL et al. Identification of three distinct tumor suppressor loci on the short arm of chromosome 9 in small cell lung cancer. Cancer Res 1997; 57: 400–3. [PubMed] [Google Scholar]
- 42. Park SY, Kim YH, In KH, Chun YH, Park SH. Chromosomal aberrations in Korean nonsmall cell lung carcinomas: degenerate oligonucleotide primed polymerase chain reaction comparative genomic hybridization studies. Cancer Genet Cytogenet 2004; 152: 153–7. [DOI] [PubMed] [Google Scholar]
- 43. Busslinger M, Klix N, Pfeffer P, Graninger PG, Kozmik Z. Deregulation of PAX‐5 by translocation of the Emu enhancer of the IgH locus adjacent to two alternative PAX‐5 promoters in a diffuse large‐cell lymphoma. Proc Natl Acad Sci USA 1996; 93: 6129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iida S, Rao PH, Nallasivam P et al. The t(9;14) (p13;q32) chromosomal translocation associated with lymphoplasmacytoid lymphoma involves the PAX‐5 gene. Blood 1996; 88: 4110–17. [PubMed] [Google Scholar]
- 45. Sherr CJ. Principles of tumor suppression. Cell 2004; 116: 235–6. [DOI] [PubMed] [Google Scholar]
- 46. Eclache V, Caulet‐Maugendre S, Poirel HA et al. Cryptic deletion involving the ATM locus at 11q22.3 approximately q23.1 in B‐cell chronic lymphocytic leukemia and related disorders. Cancer Genet Cytogenet 2004; 152: 72–6. [DOI] [PubMed] [Google Scholar]
- 47. Cuneo A, Bigoni R, Rigolin GM et al. Late appearance of the 11q22.3‐23.1 deletion involving the ATM locus in B‐cell chronic lymphocytic leukemia and related disorders: Clinico‐biological significance. Haematologica 2002; 87: 44–51. [PubMed] [Google Scholar]
- 48. Boultwood J. Ataxia telangiectasia gene mutations in leukaemia and lymphoma. J Clin Pathol 2001; 54: 512–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gumy Pause F, Wacker P, Maillet P, Betts D, Sappino AP. ATM gene alterations in childhood acute lymphoblastic leukemias. Hum Mutat 2003; 21: 554. [DOI] [PubMed] [Google Scholar]
- 50. Bullrich F, Rasio D, Kitada S et al. ATM mutations in B‐cell chronic lymphocytic leukemia. Cancer Res 1999; 59: 24–7. [PubMed] [Google Scholar]
- 51. Rouault JP, Rimokh R, Tessa C et al. BTG1, a member of a new family of antiproliferative genes. EMBO J 1992; 11: 1663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kuo ML, Duncavage EJ, Mathew R et al. Arf induces p53‐dependent and ‐independent antiproliferative genes. Cancer Res 2003; 63: 1046–53. [PubMed] [Google Scholar]
- 53. Matsuda S, Rouault J, Magaud J, Berthet C. In search of a function for the TIS21/PC3/BTG1/TOB family. FEBS Lett 2001; 497: 67–72. [DOI] [PubMed] [Google Scholar]
- 54. Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M. Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150‐kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin Cancer Res 2005; 11: 1119–28. [PubMed] [Google Scholar]
- 55. Smith JS, Tachibana I, Lee HK et al. Mapping of the chromosome 19 q‐arm glioma tumor suppressor gene using fluorescence in situ hybridization and novel microsatellite markers. Genes Chromosomes Cancer 2000; 29: 16–25. [DOI] [PubMed] [Google Scholar]
- 56. Liu H, Wang Y, Zhang Y et al. TFAR19, a novel apoptosis‐related gene cloned from human leukemia cell line TF‐1, could enhance apoptosis of some tumor cells induced by growth factor withdrawal. Biochem Biophys Res Commun 1999; 254: 203–10. [DOI] [PubMed] [Google Scholar]
- 57. Kibel AS, Freije D, Isaacs WB, Bova GS. Deletion mapping at 12p12‐13 in metastatic prostate cancer. Genes Chromosomes Cancer 1999; 25: 270–6. [PubMed] [Google Scholar]
- 58. Latil A, Guerard M, Berthon P, Cussenot O. 12p12‐13 deletion in prostate tumors and quantitative expression of CDKN1B and ETV6 candidate genes. Genes Chromosomes Cancer 2001; 31: 199–200. [DOI] [PubMed] [Google Scholar]
- 59. Knuutila S, Aalto Y, Autio K et al. DNA copy number losses in human neoplasms. Am J Pathol 1999; 155: 683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Speck NA, Gilliland DG. Core‐binding factors in haematopoiesis and leukaemia. Nat Rev Cancer 2002; 2: 502–13. [DOI] [PubMed] [Google Scholar]
- 61. Armstrong SA, Mabon ME, Silverman LB et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 2004; 103: 3544–6. [DOI] [PubMed] [Google Scholar]
- 62. Fenrick R, Wang L, Nip J et al. TEL, a putative tumor suppressor, modulates cell growth and cell morphology of ras‐transformed cells while repressing the transcription of stromelysin‐1. Mol Cell Biol 2000; 20: 5828–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rompaey LV, Potter M, Adams C, Tel Grosveld G. induces a G1 arrest and suppresses Ras‐induced transformation. Oncogene 2000; 19: 5244–50. [DOI] [PubMed] [Google Scholar]