

Abstract
For the development of cancer vaccine therapies, we have searched for possible epitope peptides that can elicit cytotoxic T lymphocytes (CTL) to the TTK protein kinase (TTK), lymphocyte antigen 6 complex locus K (LY6K) and insulin‐like growth factor (IGF)‐II mRNA binding protein 3 (IMP‐3), which were previously identified to be transactivated in the majority of lung and esophageal cancers. We screened 31, 17 and 17 candidate human leukocyte antigen (HLA)‐A*2402‐binding peptides to parts of TTK, LY6K and IMP‐3, respectively. As a result, we successfully established strong CTL clones stimulated by TTK‐567 (SYRNEIAYL), LY6K‐177 (RYCNLEGPPI) and IMP‐3‐508 (KTVNELQNL) that have specific cytotoxic activities against the HLA‐A24‐positive target cells pulsed with the candidate peptides. Subsequent analysis of the CTL clones also revealed their cytotoxic activities against lung and esophageal tumor cells that endogenously express TTK, LY6K or IMP‐3. A cold target inhibition assay further confirmed that the CTL cell clones specifically recognized the MHC class I–peptide complex. Our results strongly imply that TTK, LY6K and IMP‐3 are novel tumor‐associated antigens recognized by CTL, and TTK‐567 (SYRNEIAYL), LY6K‐177 (RYCNLEGPPI) and IMP‐3‐508 (KTVNELQNL) are HLA‐A24‐restricted epitope peptides that can induce potent and specific immune responses against lung and esophageal cancer cells expressing TTK, LY6K and IMP‐3. (Cancer Sci 2007; 98: 1803–1808)
CD8+ cytotoxic T lymphocytes (CTL) have been demonstrated to recognize peptide epitopes derived from tumor‐associated antigens (TAA) that are presented on MHC class I molecules, and to kill tumor cells. After the discovery of the melanoma antigen‐associated gene (MAGE) family as the first example of TAA, other TAA have been discovered using immunological approaches,( 1 , 2 , 3 , 4 , 5 ) and some of them, such as MAGE, gp100, squamous cell carcinoma antigen recognized by T cells (SART) and NY‐ESO‐1,( 3 , 5 , 6 , 7 ) are now in the process of clinical development as targets for immunotherapy. Although other molecules like p53, HER2/neu and carcinoembryonic antigen (CEA) are not TAA, they have also been used as targets for cellular immune responses due to their overexpression or mutation in tumor cells.( 8 , 9 , 10 ) Although progress in immunological therapies has been significant( 11 , 12 , 13 ) the number of candidate TAA is still very limited to cover the large number of cancer patients.
Recent developments in cDNA microarray technologies have enabled us to obtain comprehensive gene expression profiles of malignant cells compared with normal cells.( 14 , 15 , 16 ) This approach uncovers the activity of the genes underlying the complex nature of cancer cells and leads to identification of genes whose expression is deregulated in tumor cells.( 17 ) This approach combined with the expression profiles of normal tissues has great advantages for identifying TAA. We have been examining the expression profiles of more than 1000 clinical cancer tissues as well as more than 30 normal tissues or organs( 18 , 19 ) and accumulating comprehensive expression data sets of more than 30 000 transcripts.
Among the transcripts we examined, we identified TTK protein kinase (TTK), lymphocyte antigen 6 complex locus K (LY6K) and IGF‐II mRNA binding protein 3 (IMP‐3), which showed three‐fold or higher expression in cancer cells compared with normal control cells in more than 40% of the lung or esophageal cancers analyzed.( 20 ) However, the transcripts of these genes were hardly detectable in normal organs by Northern blot analysis, except the testis and placenta.( 18 , 19 ) Thus, TTK, LY6K and IMP‐3 are considered novel targets for cancer treatments, including immunological therapy. To validate the possibility of developing a cancer vaccine therapy using these molecules as targets, we screened candidate peptides derived from these gene products by examining the capabilities of their stimulation on peripheral blood mononuclear cells (PBMC) from healthy volunteers. We then successfully identified peptides that could stimulate CTL that recognized and killed lung and esophageal tumor cells endogenously expressing these proteins.
Materials and Methods
Cell lines. A24LCL cells (HLA‐A24/24) and EHM (HLA‐A3/3) human B‐lymphoblastoid cell lines were generous gifts from Takara Shuzo. The A24LCL cells were used for peptide‐mediated cytotoxicity assays. Lung and esophageal carcinoma cell lines TE1 (HLA‐A2402+) and TE13 (HLA‐A2402‐), and lung cancer PC9 (HLA‐A2402‐) cells were provided by Tohoku University. Expression levels of TTK, LY6K and IMP‐3 in the lung and esophageal carcinoma cell lines were determined by semiquantitative reverse transcription–polymerase chain reaction. We observed strong expression of all three genes in TE1, strong TTK and IMP‐3 expression in PC9, and strong LY6K expression in TE13.
Candidate selection of peptides derived from TTK, LY6K and IMP‐3. Nine‐mer and 10‐mer peptides derived from TTK, LY6K and IMP‐3 that bound to the HLA‐A24 molecule were predicted using the binding‐prediction software program BIMAS (http://bimas.dcrt.nih.gov/cgi‐bin/molbio/ken_parker_comboform). These peptides were synthesized by metallopanstimulin (MPS) according to the standard solid‐phase synthesis method and purified by reverse‐phase high‐performance liquid chromatography (HPLC). The purity (>90%) and identity of the peptides were determined by analytical HPLC and mass spectrometry analysis, respectively. Peptides were dissolved in dimethylsulfoxide at 20 mg/mL and stored at –80°C.
In vitro CTL induction. Monocyte‐derived dendritic cells (DC) were used as antigen‐presenting cells to induce CTL responses against peptides presented on HLA. DC were generated in vitro as described elsewhere.( 10 , 21 ) Briefly, PBMC isolated from a normal volunteer (HLA‐A*2402) using Ficoll‐Plaque (Pharmacia) solution were separated by adherence to a plastic tissue culture flask (Becton Dickinson) so as to enrich the monocyte fraction. The monocyte‐enriched population was cultured in the presence of 1000 U/mL granulocyte–macrophage colony‐stimulating factor (GM‐CSF; provided by Kirin Brewery) and 1000 U/mL interleukin (IL)‐4 (Genzyme) in AIM‐V (Invitrogen) containing 2% heat‐inactivated autologous serum (AS). After 7 days in culture, the cytokine‐generated DC were pulsed with 20 µg/mL HLA‐A24‐binding peptides in the presence of 3 µg/mL β2‐microglobulin (Sigma) for 4 h at 20°C in AIM‐V. These peptide‐pulsed DC were then irradiated (5500 rad) and mixed at a 1:20 ratio with autologous CD8+ T cells, obtained by positive selection with Dynabeads M‐450 CD8 (Dynal) and Detacha beads (Dynal). These cultures were set up in 48‐well plates (Corning); each well contained 1.5 × 104 peptide‐pulsed DC, 3 × 105 CD8+ T cells and 10 ng/mL IL‐7 (Genzyme) in 0.5 mL AIM‐V with 2% AS. After 3 days, these cultures were supplemented with IL‐2 (Chiron) to a final concentration of 20 IU/mL. On days 7 and 14, the T cells were further restimulated with the autologous peptide‐pulsed DC. The DC were prepared each time in the same way described above. Cytotoxicity was tested against peptide‐pulsed A24LCL cells after the third round of peptide stimulation on day 21.
Cytotoxic T lymphocyte expansion procedure. Cytotoxic T lymphocytes were expanded in culture using a method similar to that described by Riddell et al. 22 , 23 . A total of 5 × 104 CTL were resuspended in 25 mL AIM‐V with 5% AS with 2.5 × 106 irradiated (3300 rad) PBMC and 5 × 106 irradiated (8000 rad) EHM cells in the presence of 40 ng/mL anti‐CD3 monoclonal antibody (Pharmingen). One day after initiating the cultures, 120 IU/mL IL‐2 was added to the cultures. The cultures were fed with fresh AIM‐V with 5% AS containing 30 IU/mL IL‐2 on days 5, 8 and 11.
Establishment of CTL clones. The dilutions were made to have 0.3, 1 and 3 CTL/well in a 96‐well round‐bottomed microtiter plate (Nalge Nunc International). CTL were cultured with 7 × 104 cells/well allogenic PBMC, 1 × 104 cells/well EHM, 30 ng/mL anti‐CD3 antibody and 125 U/mL IL‐2 in a total of 150 µL/well of AIM‐V containing 5% AS. IL‐2 (50 µL/well) was added to the medium 10 days later so that IL‐2 became 125 U/mL at the final concentration. The cytotoxic activity of CTL was tested on the 14th day, and CTL clones were expanded using the same method as above.
Cytotoxicity assay. Target cells were labeled with 3.7 MBq Na2 51CrO4 (Perkin Elmer Life Sciences) for 1 h at 37°C in a CO2 incubator. Peptide‐pulsed targets were prepared by incubating the cells with 20 µg/mL peptide for 16 h at 37°C before labeling. Labeled target cells were rinsed and mixed with effector cells in a final volume of 0.2 mL in round‐bottomed microtiter plates. The plates were centrifuged (4 min at 800g) to increase cell‐to‐cell contact and placed in a CO2 incubator at 37°C. After 4 h of incubation, 0.1 mL of the supernatant was collected from each well and the radioactivity was determined with a gamma counter.
The percentage of specific cytotoxicity was determined by calculating the percentage of specific 51Cr release using the following formula:
| ([cpm of the test sample release – cpm of the spontaneous release]/[cpm of the maximum release – cpm of the spontaneous release]) × 100. |
Spontaneous release was determined by incubating the target cells alone, in the absence of effector cells, and the maximum release was obtained by incubating the targets with 1 mol/L HCl. All measurements were carried out in duplicate, and the standard errors of the means were consistently below 10% of the value of the mean.
Antigen specificity was confirmed by the cold target inhibition assay, which utilized unlabeled A24LCL cells that were pulsed with or without peptide (20 µg/mL for 16 h at 37°C) to compete for the recognition of 51Cr‐labeled tumor cells.
Results
Stimulation of T cells using candidate HLA‐A24‐binding peptides derived from TTK, LY6K or IMP‐3. Table 1 shows the HLA‐A*2402‐binding candidate peptides for TTK, LY6K and IMP‐3 in order of highest binding affinity as predicted by the binding‐prediction software BIMAS. Candidate peptides consisting of nine and 10 amino acids are shown. Stimulation of CTL by each of these peptides was carried out according to the method described in ‘Materials and Methods’. CTL that showed detectable cytotoxic activity to the cells pulsed with each peptide were expanded, and the CTL clones were further validated for their cytotoxic activities.
Table 1.
Human leukocyte antigen‐A24 binding peptides derived from TTK, LY6K and IMP‐3
| TTK | LY6K | IMP‐3 | ||||||
|---|---|---|---|---|---|---|---|---|
| Start position | Amino acid sequence (mer) | Binding score | Start position | Amino acid sequence (mer) | Binding score | Start position | Amino acid sequence (mer) | Binding score |
| 90 | RYSQAIEAL (9) | 400 | 154 | KPEEKRFLL (9) | 14.4 | 350 | SYENDIASM (9) | 37.5 |
| 567 | SYRNEIAYL (9) | 200 | 48 | RADPPWAPL (9) | 9.6 | 141 | GFQLENFTL (9) | 30 |
| 549 | IYAIKYVNL (9) | 200 | 205 | LWLAILLLL (9) | 8.4 | 508 | KTVNELQNL (9) | 14.4 |
| 590 | DYEITDQYI (9) | 90 | 57 | GTMALLALL (9) | 7.2 | 26 | KIPVSGPFL (9) | 12 |
| 652 | NFLIVDGML (9) | 42 | 203 | GGLWLAILL (9) | 7.2 | 192 | KPCDLPLRL (9) | 11.5 |
| 141 | KFAFVHISF (9) | 28 | 62 | LALLLVVAL (9) | 7.2 | 433 | RFAGASIKI (9) | 11 |
| 214 | SFSGSLGHL (9) | 20 | 53 | WAPLGTMAL (9) | 6 | 505 | KGGKTVNEL (9) | 10.6 |
| 28 | KNEDLTDEL (9) | 19 | 214 | ASIAAGLSL (9) | 6 | 190 | KQKPCDLPL (9) | 9.6 |
| 111 | RIQVRFAEL (9) | 15.8 | 54 | APLGTMALL (9) | 6 | 152 | AYIPDEMAA (9) | 9 |
| 108 | SFARIQVRF (9) | 14 | 177 | RYCNLEGPPI (10) | 100 | 320 | LYNPERTIT (9) | 9 |
| 71 | KLEKNSVPL (9) | 12 | 159 | RFLLEEPMPF (10) | 30 | 4 | LYIGNLSEN (9) | 8.25 |
| 810 | KYVLGQLVGL (10) | 600 | 152 | RPKPEEKRFL (10) | 9.6 | 470 | IYGKIKEENF (10) | 100 |
| 725 | YYMTYGKTPF (10) | 150 | 211 | LLLASIAAGL (10) | 8.4 | 272 | KFTEEIPLKI (10) | 18.5 |
| 598 | IYMVMECGNI (10) | 75 | 172 | KCCKIRYCNL (10) | 8 | 290 | RLIGKEGRNL (10) | 12 |
| 728 | TYGKTPFQQI (10) | 72 | 169 | FYLKCCKIRY (10) | 7.5 | 309 | KITISPLQEL (10) | 10.6 |
| 755 | EFPDIPEKDL (10) | 36 | 57 | GTMALLALLL (10) | 7.2 | 350 | SYENDIASMN (10) | 10.5 |
| 490 | CFQQQQHQIL (10) | 36 | 53 | WAPLGTMALL (10) | 6 | 192 | KPCDLPLRLL (10) | 9.6 |
| 143 | AFVHISFAQF (10) | 18 | ||||||
| 569 | RNEIAYLNKL (10) | 15.8 | ||||||
| 359 | KTESSLLAKL (10) | 15.8 | ||||||
| 553 | KYVNLEEADN (10) | 15 | ||||||
| 168 | KAVERGAVPL (10) | 14.4 | ||||||
| 232 | RGQTTKARFL (10) | 12 | ||||||
| 185 | RNLNLQKKQL (10) | 12 | ||||||
| 777 | KQRISIPELL (10) | 11.2 | ||||||
| 573 | AYLNKLQQHS (10) | 10.8 | ||||||
| 373 | EYQEPEVPES (10) | 9.9 | ||||||
| 74 | KNSVPLSDAL (10) | 9.6 | ||||||
| 315 | KPSGNDSCEL (10) | 8.8 | ||||||
| 61 | NPEDWLSLLL (10) | 8.6 | ||||||
| 763 | DLQDVLKCCL (10) | 8.6 | ||||||
Start position indicates the number of amino acids from the N‐terminus of TTK, LY6K and IMP‐3. Binding score was obtained using the BIMAS binding‐prediction program.
The CTL clones stimulated with the HLA‐A24‐binding peptide TTK‐567 (SYRNEIAYL) (Fig. 1a), LY6K‐177 (RYCNLEGPPI) (Fig. 1b) and IMP‐3‐508 (KTVNELQNL) (Fig. 1c) showed potent cytotoxic activity against the peptide‐pulsed target cells, whereas they showed no significant cytotoxic activity against cells without the peptide pulse.
Figure 1.
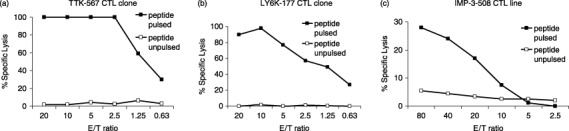
Established clones showing peptide‐specific cytotoxicity. (a) The cytotoxic T lymphocyte (CTL) clone raised against TTK‐567 had peptide‐specific cytotoxicity. It showed high cytotoxic activity against target cells (A24LCL) pulsed with TTK‐567 (—▪—), whereas it did not show significant cytotoxic activity against the same target cells (A24LCL) pulsed with no peptides (—□—). (b) The CTL clone raised against LY6K‐177 had peptide‐specific cytotoxicity. The CTL clone showed high cytotoxic activity against target cells (A24LCL) pulsed with LY6K‐177 (—▪—), whereas it did not show significant cytotoxic activity against the same target cells (A24LCL) pulsed with no peptides (—□—). (c) The CTL clone raised against IMP‐3‐508 had peptide‐specific cytotoxicity. The CTL clone showed high cytotoxic activity against target cells (A24LCL) pulsed with IMP‐3‐508 (—▪—), whereas it did not show significant cytotoxic activity against the same target cells (A24LCL) pulsed with no peptides (—□—). This demonstrates that all three CTL clones had peptide‐specific cytotoxicity.
Cytotoxic activity against lung and esophageal cancer cell lines endogenously expressing TTK, LY6K or IMP‐3. The established CTL clones raised by each of these peptides were examined for their ability to recognize and for their cytotoxic activity against lung and esophageal tumor cells that endogenously express TTK, LY6K or IMP‐3. Cytotoxic activity against TE1 cells, which endogenously express TTK and possess the HLA‐A24 allele, was examined using the TTK‐567‐specific CTL clone as effector cells. PC9 cells were used as the control because they endogenously express TTK but did not have the HLA‐A24 allele. The CTL clone showed high cytotoxic activity against TE1 cells but it showed no significant cytotoxic activity against PC9 cells (Fig. 2a), indicating that the CTL clone has cytotoxic activity restricted to HLA‐A24.
Figure 2.
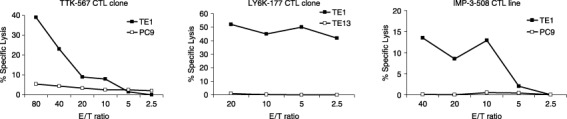
Established cytotoxic T lymphocyte (CTL) clones recognized and lysed tumor cells endogenously expressing the appropriate target in a human leukocyte antigen (HLA)‐restricted fashion. (a) The CTL clone raised against TTK‐567 recognized and lysed tumor cells endogenously expressing TTK in a HLA‐restricted fashion. Cytotoxic activity against TE1 cells, which endogenously express TTK and HLA‐A24, was tested using the CTL clone raised against TTK‐567 as effector cells. PC9 cells, which endogenously express TTK but do not express HLA‐A24, were used as the target cells. The CTL clone showed high cytotoxic activity against TE1 cells that express both TTK and HLA‐A24 (—▪—). However, it did not show significant cytotoxic activity against PC9 cells, which express TTK but not HLA‐A24 (—□—). (b) The CTL clone raised against LY6K‐177 recognized and lysed tumor cells endogenously expressing LY6K in a HLA‐restricted fashion. Cytotoxic activity against TE1 cells, which endogenously express LY6K and HLA‐A24, was tested using the CTL clone raised against LY6K‐177 as effector cells. TE13 cells, which endogenously express LY6K but do not express HLA‐A24, were used as the target cells. The CTL clone showed high cytotoxic activity against TE1 cells that express both LY6K and HLA‐A24 (—▪—). However, it did not show significant cytotoxic activity against TE13 cells, which express LY6K but not HLA‐A24 (—□—). (c) The CTL clone raised against IMP‐3‐508 recognized and lysed tumor cells endogenously expressing IMP‐3 in a HLA‐restricted fashion. Cytotoxic activity against TE1 cells, which endogenously express IMP‐3 and HLA‐A24, was tested using the CTL clone raised against IMP‐3‐508 as effector cells. PC9 cells, which endogenously express IMP‐3 but do not express HLA‐A24, were used as target cells. The CTL clone showed high cytotoxic activity against TE1 cells that express both IMP‐3 and HLA‐A24 (—▪—). However, it did not show significant cytotoxic activity against PC9 cells, which express IMP‐3 but not HLA‐A24 (—□—).
Cytotoxic activity against TE1 cells, which endogenously express LY6K and have the HLA‐A24 allele, were also examined using the LY6K‐177‐specific CTL clone as effector cells. TE13 cells, which endogenously express LT6K but lack HLA‐A24 expression, served as the control. The CTL clone showed high cytotoxic activity against TE1 cells but did not show significant cytotoxic activity against TE13 cells (Fig. 2b), implying that the CTL clone has cytotoxic activity restricted to HLA‐A24.
Because TE1 cells also endogenously express IMP‐3, they were used to examine cytotoxic activity using the IMP‐3‐508‐specific CTL line as effector cells. The PC9 cells were used as the control as they endogenously express IMP‐3 but do not express the HLA‐A24 allele. The CTL line showed potent cytotoxic activity against TE1 cells. However, the cell line did not show any significant cytotoxic activity against PC9 cells (Fig. 2c), indicating that the cells have HLA‐A24‐restricted cytotoxicity.
These CTL clones also showed no cytotoxic activity against A24LCL cells, which do not express TTK, LY6K or IMP‐3 but do express HLA‐A24 (Fig. 1). These results clearly demonstrate that cytotoxic activities were specific to cells expressing either TTK, LY6K or IMP‐3. These results also suggest that TTK‐567, LY6K‐177 and IMP‐3‐508 are naturally presented on HLA‐A24 of tumor cells and can be recognized by CTL.
Cold target inhibition assay. To confirm the specificity of the CTL clones, we carried out a cold target inhibition assay as described in ‘Materials and Methods’. The TE1 cells labeled with Na2 51CrO4 were prepared as the hot target and the A24LCL cells pulsed with TTK‐567 peptide were used as the cold target (inhibitor). The cytotoxic activity of the TTK‐567‐specific CTL clone against TE1 cells was specifically inhibited with the addition of A24LCL cells pulsed with the TTK‐567 peptide (Fig. 3a).
Figure 3.
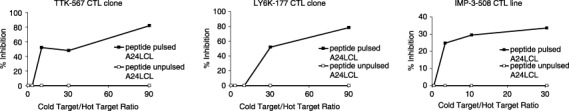
Established cytotoxic T lymphocyte (CTL) clones specifically recognized the appropriate targets in a human leukocyte antigen (HLA)‐restricted manner. (a) The CTL clone raised against TTK‐567 specifically recognized TTK‐567 in a HLA‐A24 restricted manner. For the cold target inhibition assay, TE1 cells labeled with Na2 51CrO4 were prepared as the hot target, whereas TTK‐567 peptide‐pulsed A24LCL cells were used as the cold target (inhibitor). The effector/target (E/T) ratio was fixed at 20. The cytotoxic activity of the TTK‐567 CTL clone against TE1 cells was inhibited by the addition of A24LCL cells pulsed with the identical peptide (—▪—), whereas the cytotoxic activity was not inhibited by the addition of A24LCL cells without any peptides (—□—). (b) The CTL clone raised against LY6K‐177 specifically recognized LY6K in a HLA‐A24 restricted manner. For the cold target inhibition assay, TE1 cells labeled with Na2 51CrO4 were prepared as the hot target, whereas LY6K‐177 peptide‐pulsed A24LCL cells were used as the cold target (inhibitor). The E/T ratio was fixed at 20. The cytotoxic activity of the LY6K‐177 CTL clone against TE1 was inhibited by the addition of A24LCL cells pulsed with the identical peptide (—▪—), whereas the cytotoxic activity was not inhibited by the addition of A24LCL cells without any peptides (—□—). (c) The CTL clone raised against IMP‐3‐508 specifically recognized IMP‐3 in a HLA‐A24 restricted manner. For the cold target inhibition assay, TE1 cells labeled with Na2 51CrO4 were prepared as the hot target, whereas IMP‐3‐508 peptide‐pulsed A24LCL cells were used as the cold target (inhibitor). The E/T ratio was fixed at 20. The cytotoxic activity of the IMP‐3‐508 CTL line against TE1 cells was inhibited by the addition of A24LCL cells pulsed with the identical peptide (—▪—), whereas the cytotoxic activity was not inhibited by the addition of A24LCL cells without any peptides (—□—).
Regarding LY6K, TE1 cells labeled with Na2 51CrO4 were prepared as the hot target, and A24LCL cells pulsed with the LY6K‐177 peptide were used as the cold target (inhibitor). The cytotoxic activity of the LY6K‐177‐specific CTL clone against TE1 was specifically inhibited by the addition of A24LCL cells pulsed with the peptide (Fig. 3b).
Similarly, TE1 cells labeled with Na2 51CrO4 were prepared as the hot target and A24LCL cells pulsed with IMP‐3‐508 peptide were used as the cold target (inhibitor). The cytotoxic activity of the IMP‐3‐508‐specific CTL line against TE1 cells was specifically inhibited by the addition of A24LCL cells pulsed with the peptide (Fig. 3c). Specific cytotoxicity against TE1 cells was significantly inhibited when peptide‐pulsed cold target was added to the assay at various ratios. However, it was not inhibited when the cold target without the peptide pulse was added. These results are indicated as a percentage of specific lysis at the effector/target (E/T) ratio of 20.
Homology analysis of the antigen peptides. To examine whether there are other molecules that include amino acid sequences corresponding to these three peptides, we carried out a homology search using the BLAST algorithm (http://www.ncbi.nlm.nih.gov/blast/blast.cgi). As we found no significant similarity to other human amino acid sequences deposited in the database, there is little possibility that the immune responses we observed were associated with other molecules.
Discussion
The development of peptide vaccinations could be further promoted if new and promising TAA could be discovered.( 2 , 3 , 4 , 5 , 6 , 7 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 ) Because cDNA microarray technologies can disclose comprehensive gene expression profiles of malignant cells,( 14 , 15 , 16 ) this approach could be very useful for the identification of potential TAA. We have identified epitope peptides derived from novel TAA, taking advantage of our huge set of expression profile data for more than 1000 human cancers.( 32 , 33 , 34 ) In the present study, we applied the same strategy for three proteins, TTK, LY6K and IMP‐3, that were upregulated in the majority of lung and esophageal cancers, and successfully identified three new peptide epitopes that induce potent CTL activity. Because the expression levels of TTK, LY6K and IMP‐3 were very low or hardly detectable in normal tissues, these peptides induced significant and specific immune responses against cancer cells without inducing autoimmune responses against the normal cells.
Recently, it has been reported that TTK is strongly expressed in another kind of tumor.( 35 ) Furthermore, it is associated with cell proliferation and cell cycle kinases.( 36 , 37 ) The expression pattern of TTK is ideal for immunotherapy, and its function is essential for tumors. LY6K has been reported to be highly expressed in head and neck squamous cell carcinoma and breast cancer.( 38 , 39 ) IMP‐3 has been reported as a clinically significant biomarker for cervical cancer and renal cell carcinoma.( 40 , 41 ) Furthermore, it is strongly associated with cell adhesion and tumor invasion.( 42 ) From these reports, TTK, LY6K and IMP‐3 were thought to be good molecular targets for immunotherapy. Therefore, we examined whether these molecules were recognized by T cells to determine the epitope peptides.
Because the HLA‐A24 allele is one of the most common HLA‐A alleles, especially in the Japanese population,( 43 , 44 , 45 ) we predicted the candidate HLA‐A24‐binding peptides corresponding to parts of the TTK, LY6K and IMP‐3 proteins using the binding‐prediction software BIMAS. After the in vitro stimulation of T cells by DC loaded with each of these peptides, CTL were successfully established using TTK‐567 (SYRNEIAYL), LY6K‐177 (RYCNLEGPPI) and IMP‐3‐508 (KTVNELQNL). We confirmed their potent cytotoxic activities against peptide‐pulsed A24LCL cells. Furthermore, the CTL clones and lines derived by stimulation with each of these peptides showed specific cytotoxicity against the HLA‐A24‐positive lung and esophageal carcinoma cell lines in which TTK, LY6K and IMP‐3 are expressed endogenously at high levels. The cytotoxic activities of these CTL clones and lines were not observed to cell lines lacking the expression of either HLA‐A24 or a target TAA. We also confirmed significant inhibition of the cytotoxic activities of these CTL clones using the cold target assay. These results strongly suggest that TTK, LY6K and IMP‐3 are novel TAA of lung and esophageal cancer cells and that TTK‐567, LY6K‐177 and IMP‐3‐508 are promising HLA‐A24‐restricted epitope peptides that can enhance antigen‐specific immune responses in a HLA‐A24‐restricted manner. Because these antigens are highly expressed in the majority of lung and esophageal cancers, they appear to be good targets for use as immunotherapies against these types of cancers.
References
- 1. Boon T. Tumor antigens recognized by cytotoxic T lymphocytes: present perspectives for specific immunotherapy. Int J Cancer 1993; 54: 177–80. [DOI] [PubMed] [Google Scholar]
- 2. Boon T, Van Der Bruggen P. Human tumor antigen recognized by T lymphocytes. J Exp Med 1996; 183: 725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van Der Bruggen P, Traversari C, Chomez P, Lurquin C, De‐Plaen E, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254: 1643–7. [DOI] [PubMed] [Google Scholar]
- 4. Brichard V, Van Pel A, Wolfel T et al . The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA‐A2 melanomas. J Exp Med 1993; 178: 489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawakami Y, Eliyahu S, Sakaguchi K et al . Identification of the immunodominant peptides of the MART‐1 human melanoma antigen recognized by the majority of HLA‐A2‐restricted tumor infiltrating lymphocytes. J Exp Med 1994; 180: 347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shichijo S, Nakao M, Imai Y et al . A gene encoding antigenic peptides of human squamous cell carcinoma recognized by cytotoxic T lymphocytes. J Exp Med 1998; 187: 277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen YT, Scanlan MJ, Sahin U et al . A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acd Sci USA 1997; 94: 1914–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Umano Y, Tsunoda T, Tanaka H, Matsuda K, Yamaue H, Tanimura H. Generation of cytotoxic T cell responses to an HLA‐A24 restricted epitope peptide derived from wild type p53. Br J Cancer 2001; 84: 1052–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanaka H, Tsunoda T, Nukaya I et al . Mapping the HLA‐A24‐restricted T‐cell epitope peptide from a tumor‐associated antigen HER2/neu: Possible immunotherapy for colorectal carcinomas. Br J Cancer 2001; 84: 94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nukaya I, Yasumoto M, Iwasaki T et al . Identification of HLA‐A24 epitope peptide of carcinoembryonic antigen which induces tumor reactive cytotoxic T lymphocyte. Int J Cancer 1999; 80: 92–7. [DOI] [PubMed] [Google Scholar]
- 11. Rosenberg SA, Yang JC, Schwartzentruber DJ et al . Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nature Med 1998; 4: 321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mukherji B, Chakraborty NG, Yamasaki S et al . Induction of antigen‐specific cytolytic T cells in situ in human melanoma by immunization with synthetic peptide‐pulsed autologous antigen presenting cells. Proc Natl Acad Sci USA 1995; 92: 8078–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu X, Chakraborty NG, Sporn JR, Kurtzman SH, Ergin MT, Mukherji BH. Enhancement of cytolytic T lymphocyte precursor frequency in melanoma patients following immunization with the MAGE‐1 peptide loaded antigen presenting cell‐based vaccine. Cancer Res 1996; 56: 2479–83. [PubMed] [Google Scholar]
- 14. Okabe H, Satoh S, Kato T et al . Genome‐wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res 2001; 61: 2129–37. [PubMed] [Google Scholar]
- 15. Lin Y‐M, Furukawa Y, Tsunoda T, Yue C‐T, Yang K‐C, Nakamura Y. Molecular diagnosis of colorectal tumors by expression profiles of 50 genes expressed differentially in adenomas and carcinomas. Oncogene 2002; 21: 4120–8. [DOI] [PubMed] [Google Scholar]
- 16. Hasegawa S, Furukawa Y, Li M et al . Genome‐wide analysis of gene expression in intestinal‐type gastric cancers using a complementary DNA microarray representing 23 040 genes. Cancer Res 2002; 62: 7012–17. [PubMed] [Google Scholar]
- 17. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell 2000; 103: 311–20. [DOI] [PubMed] [Google Scholar]
- 18. Saito‐Hisaminato A, Katagiri T, Kakiuchi S, Nakamura T, Tsunoda T, Nakamura Y. Genome‐wide profiling of gene expression in 29 normal human tissues with a cDNA microarray. DNA Res 2002; 9: 35–45 [DOI] [PubMed] [Google Scholar]
- 19. Ochi K, Daigo Y, Katagiri T et al . Expression profiles of two types of human knee joint cartilage. J Hum Genet 2003; 48: 177–82. [DOI] [PubMed] [Google Scholar]
- 20. Kikuchi T, Daigo Y, Katagiri T et al . Expression profiles of non‐small cell lung cancers on cDNA microarrays: identification of genes for prediction of lymph‐node metastasis and sensitivity to anti‐cancer drugs. Oncogene 2003; 22: 2192–205. [DOI] [PubMed] [Google Scholar]
- 21. Tsai V, Southwood S, Sidney J et al . Identification of subdominant CTL epitopes of the GP100 melanoma‐associated tumor antigen by primary in vitro immunization with peptide‐pulsed dendritic cells. J Immunol 1997; 158: 1796–802. [PubMed] [Google Scholar]
- 22. Riddell SR, Elliott M, Lewinsohn DA et al . T‐cell mediated rejection of gene‐modified HIV‐specific cytotoxic T lymphocytes in HIV‐infected patients. Nat Med 1996; 2: 216–23. [DOI] [PubMed] [Google Scholar]
- 23. Walter EA, Greenberg PD, Gilbert MJ et al . Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T‐cell clones from the donor. N Engl J Med 1995; 333: 1038–44. [DOI] [PubMed] [Google Scholar]
- 24. Harris CC. Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies. J Natl Cancer Inst 1996; 88: 1442–5. [DOI] [PubMed] [Google Scholar]
- 25. Butterfield LH, Koh A, Meng W et al . Generation of human T cell response to an HLA‐A2.1 restricted peptide epitope derived from αfetoprotein. Cancer Res 1999; 59: 3134–42. [PubMed] [Google Scholar]
- 26. Vissers JL, De Vries IJ, Schreurs MW et al . The renal cell carcinoma associated antigen G250 encodes a human leukocyte antigen HLA‐A2.1 restricted epitope recognized by cytotoxic T lymphocyte. Cancer Res 1999; 59: 5554–9. [PubMed] [Google Scholar]
- 27. Van Der Burg SH, Visseren MJ, Brandt RM, Kast WM, Melief CJ. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC peptide complex stability. J Immunol 1996; 156: 3308–14. [PubMed] [Google Scholar]
- 28. Tanaka F, Fujie T, Tahara K et al . Induction of antitumor cytotoxic T lymphocytes with a MAGE 3 encoded synthetic peptide presented by human leukocytes antigen A24. Cancer Res 1997; 57: 4465–8. [PubMed] [Google Scholar]
- 29. Fujie T, Tahara K, Tanaka F, Mori M, Takesako K, Akiyoshi T. A MAGE 1 encoded HLA‐A24 binding synthetic peptide induces specific anti tumor cytotoxic T lymphocytes. Int J Cancer 1999; 80: 169–72. [DOI] [PubMed] [Google Scholar]
- 30. Kikuchi M, Nakao M, Inoue Y et al . Identification of a SART 1 derived peptide capable of inducing HLA‐A24 restricted and tumor specific cytotoxic T lymphocytes. Int J Cancer 1999; 81: 459–66. [DOI] [PubMed] [Google Scholar]
- 31. Oiso M, Eura M, Katsura F et al . A newly identified MAGE3 derived epitope recognized by HLA‐A24 restricted cytotoxic T lymphocytes. Int J Cancer 1999; 81: 387–94. [DOI] [PubMed] [Google Scholar]
- 32. Uchida N, Tsunoda T, Wada S, Furukawa Y, Nakamura Y, Tahara H. Ring finger protein (RNF) 43 as a new target for cancer immunotherapy. Clin Cancer Res 2004; 10: 8577–86. [DOI] [PubMed] [Google Scholar]
- 33. Watanabe T, Suda T, Tsunoda T et al . Identification of Immunoglobulin superfamily 11 (IGSF11) as a novel target for cancer immunotherapy of gastro‐intestinal and hepatocellular carcinomas. Cancer Sci 2005; 96: 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suda T, Tsunoda T, Uchida N et al . Identification of secernin 1 (SCRN1) as a novel immunotherapy target for gastric cancer using the expression profiles of cDNA microarray. Cancer Sci 2006; 97: 411–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan B, Xu Y, Woo JH et al . Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res 2006; 12: 405–10. [DOI] [PubMed] [Google Scholar]
- 36. Mills GB, Schmandt R, McGill M et al . Expression of TTK, a novel human protein kinase, is associated with cell proliferation. J Biol Chem 1992; 267: 16 000–6. [PubMed] [Google Scholar]
- 37. De Carcer G, De Castro IP, Malumbres M. Targeting cell cycle kinases for cancer therapy. Curr Med Chem 2007; 14: 969–85. [DOI] [PubMed] [Google Scholar]
- 38. De Nooij‐van Dalen AG, Van Dongen GA, Smeets SJ et al . Characterization of the human Ly‐6 antigens, the newly annotated member Ly‐6K included, as molecular markers for head‐and‐neck squamous cell carcinoma. Int J Cancer 2003; 103: 768–74. [DOI] [PubMed] [Google Scholar]
- 39. Lee JW, Lee YS, Yoo KH et al . LY‐6K gene: a novel molecular marker for human breast cancer. Oncol Rep 2006; 16: 1211–14. [PubMed] [Google Scholar]
- 40. Li C, Rock KL, Woda BA, Jiang Z, Fraire AE, Dresser K. IMP3 is a novel biomarker for adenocarcinoma in situ of the uterine cervix: an immunohistochemical study in comparison with p16(INK4a) expression. Mod Pathol 2007; 20: 242–7. [DOI] [PubMed] [Google Scholar]
- 41. Jiang Z, Chu PG, Woda BA, Rock KL, Liu Q. Analysis of RNA‐binding protein IMP3 to predict metastasis and prognosis of renal‐cell carcinoma: a retrospective study. Lancet Oncol 2006; 7: 556–64. [DOI] [PubMed] [Google Scholar]
- 42. Vikesaa J, Hansen TV, Jonson L et al . RNA‐binding IMPs promote cell adhesion and invadopodia formation. EMBO J 2006; 25: 1456–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Date Y, Kimura A, Kato H, Sasazuki T. DNA typing of the HLA‐A gene: population study and identification of four new alleles in Japanese. Tissue Antigens 1996; 47: 93–101. [DOI] [PubMed] [Google Scholar]
- 44. Kondo A, Sidney J, Southwood S et al . Prominent roles of secondary anchor residues in peptide binding to HLA‐A24 human class II T molecules. J Immunol 1995; 155: 4307–12. [PubMed] [Google Scholar]
- 45. Kubo RT, Sette A, Grey HM et al . Definition of specific peptide motifs for four major HLA‐A alleles. J Immunol 1994; 152: 3913–24. [PubMed] [Google Scholar]