

Abstract
Mongolian gerbils are an ideal animal model to explore the role of H. pylori on cancer development. However, there have been no established adenocarcinoma cell lines from this model animal. In the present study, we have established cancer cell lines from a primary gastric cancer tissue of a Mongolian gerbil. The derived cells could be stably attached with H. pylori, revealed under a scanning electron microscope, and easily transplanted to the nude mice. Rapid phosphorylation of IκB, Erk1/2, and AKT of these cells was observed by Interleukin‐1 beta stimulation, and luciferase reporter gene assay on transcriptional activation of Nuclear Factor kappa B after challenging with either H. pylori NCTC11637 or its isogenic cagE‐knockout mutant, H. pylori revealed the cagE‐dependent NF‐κB transcriptional activation. The newly established cancer cell lines from the in vivo gastric carcinogenesis model animal, the Mongolian gerbil, can be used to develop effective therapeutic strategies against gastric cancer, especially in exploring the effect of H. pylori, and thus might greatly contribute to gastric cancer prevention and treatment in humans. (Cancer Sci 2005; 96: 170–175)
Helicobacter pylori (H. pylori) infection has been proven to facilitate gastric disorders 1 , 2 , 3 and gastric cancer development through epidemiological studies and in vitro experimental studies. 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 As for in vivo experimental animal models, Mongolian gerbils (Meriones unguiculatus) have been used to study the virulence of H. pylori infection upon gastric carcinogenesis 12 , 13 , 14 , 15 , 16 , 17 , 18 because of their high susceptibility to infection with H. pylori. After the infection, the glandular stomachs of gerbils exhibit acute gastritis such as erosion, foveolar cell hyperplasia, and varying degrees of multifocal cystic dilatation and infiltration of inflammatory cells. Long‐term persistent infection in gerbils results in chronic active gastritis including intestinal metaplasia and glandular atrophy, generating severe dysplastic proliferation of the gastric glands into submucosa with interruptions of the lamina muscularis mucosa. The histological changes and gastric disorders induced by H. pylori infection in Mongolian gerbils resemble those in humans very well. (15)
Though the precise mechanism of generating histological changes after H. pylori infection has not been satisfactory clarified, one of the typical features of H. pylori infection has been focused on the activation of signaling pathways in the target host cell. Some virulent factors of H. pylori are documented to link with the cag pathogenicity islands in the genome, (19) of which CagA protein is translocated from bacteria into host cells through the type IV secretion system, 20 , 21 comprised of transporters including CagE protein encoded by cagE. Tyrosine residues of translocated CagA are phosphorylated in host cells, which can perturb signal transduction interacting with a host cell protein SHP‐2 that connects receptor tyrosine kinases and activates Ras/MAPK signaling, resulting in deregulation of the pathway activation. 11 , 20 , 21 In addition, it has also been documented that H pylori adhesion can directly activate TRAF2 or TRAF6 to induce nuclear localization of NF‐κB, resulting in transcriptional activation. (22) Taken together, these results suggest that deregulation of these signaling pathways is associated with the generation of gastric disorders. However, the actual meaning of these alterations in transcriptional activities in gastric carcinogenesis remains unclear. The concept of responsible virulence depending on CagA function is supported by epidemiological studies including a meta‐analysis of the positive relationship between H. pylori CagA seropositivity and gastric cancer. (10) The majority of the Japanese population have the ESS‐type (11) of CagA, a strong virulence factor of H. pylori. Nevertheless, gastric cancer occurs in 0.5% of the H. pylori‐infected Japanese population per year. (9) Epidemiological studies indicate that not only the strain diversity of H. pylori, but also the effects of overproduction of pro‐inflammatory cytokine and environmental factors might serve to develop gastric cancers.
A series of studies using Mongolian gerbils with chemical carcinogen and H. pylori infection has demonstrated that H. pylori infection apparently enhances gastric carcinogenesis. 12 , 13 , 14 , 15 Persistent infection with H. pylori can enhance to produce all histological types (well‐differentiated, poorly differentiated, and signet ring cell type) of gastric carcinomas in the glandular stomach of this animal, while subsequent eradication at an early stage of inflammation reduced such enhancing effects. 14 , 15 The promotion and progression of gastric cancers were apparently related to the time course of the inflammatory status after infection with virulent H. pylori. However, it is not known how the initiation or progression of cancers is caused by such infection, because there have been no established cancer cell lines from an in vivo experimental carcinogenesis model with apparent histories of H. pylori infection, available to explore the mechanism of its virulence.
In this study, we first present the establishment of new gastric cancer cell lines with different invasive activities, designated MGC1 and MGC2, from one original cancer tissue induced in the Mongolian gerbil, an analogous model of H. pylori infection to humans, to explore the essential biological features and the mechanism for enhancement and progression of H. pylori‐induced gastric carcinomas.
Materials and Methods
Tumor development. The protocol for gastric cancer induction was previously described in detail. (14) In brief, specific pathogen‐free male, 7‐week‐old Mongolian gerbils (Meriones unguiculatus; MGS/Sea, Seac Yoshitomi, Ltd, Fukuoka, Japan) were housed in steel cages on hardwood chip bedding in an air‐conditioned biohazard room with a 12‐h light/12‐h dark cycle. They were given rodent food CRF‐1 (Oriental Yeast Co. Ltd, Tokyo, Japan) irradiated with 30 Gy gamma rays and autoclaved distilled water ad libitum. MNU (Sigma Chemical Co. St. Louis, MO, USA) was dissolved in distilled water at a concentration of 40 p.p.m. and freshly prepared three times per week or administered in drinking water in light‐shielded bottles ad libitum. MNU was thus given in the drinking water for 10 weeks. At week 11, Hp strain ATCC 43504 (cagA +, vacA +) samples containing 1.0 × 108 colony‐forming units were delivered intragastrically using an oral catheter. Finally, at week 50, gerbils were sacrificed under deep ether anesthesia, laparotomized, and tissue samples were taken from occurring tumors. The animal care committee of the Aichi Cancer Center Research Institute approved the experimental design, and the animals were cared for in accordance with institutional guidelines.
Primary culture procedure. For primary culture, an advanced gastric cancer tissue with perineural invasion occurring in a gerbil was subjected to primary culture. The fresh tumor tissue was washed extensively with phosphate‐buffered saline, cut into approximately 1‐mm3 cubes and minced in RPMI 1640 medium (Nissui Pharmaceutical, Tokyo, Japan). Small pieces of the tumor were then treated with Dispase (50 units/mL, Godo Shusei, Tokyo, Japan) for 30 min at 37°C, and large tumor pieces were allowed to settle. Supernatant fluid containing cell clumps was collected after centrifugation at 100× g , and cell pellets were re‐suspended in RPMI 1640 containing 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA) plus serum expander MITO (0.1%, Collaborative Biomedical Products Bedford, MA, USA), 100 units/mL penicillin, and 100 µg/mL streptomycin (Gibco), then cultured on dishes coated with collagen type I (Iwaki, Tokyo, Japan) or non‐coated plastic dishes in a humidified 5% CO2 incubator at 37°C with weekly changes of the medium. One month thereafter, growing colonies were harvested with trypsin/EDTA and passaged several times. Fibroblasts were removed with mechanical scraping and the differential attachment selection method.
The absence of contamination with Mycoplasma pulmonis or the two viruses, mouse hepatitis virus and sendai virus, was confirmed with the assistance of the Experimental Animal Research Center of Japan (Tokyo) with a direct agar method or a PCR method. For counting growth activities, Cells were plated at 1 × 105 cells/dish in 35‐mm plastic dishes in RPMI 1640 supplemented with 10% FBS in the absence of MITO. The cell number was counted in triplicate with a hemocytometer 1, 3, 5, 7, 9, and 11 days after seeding.
Tumorigenicity in nude mice. Exponentially growing cells were harvested with trypsin‐EDTA, washed with RPMI 1640, and resuspended in Hank's balanced salt solution (HBSS, Sigma, St. Louis, MO). For subcutaneous inoculation, a total of 1 × 106 cells suspended in 0.2 mL of HBSS were injected into the subcutaneous tissue of the left femurs of seven‐week‐old athymic nude mice (BALB/c nu/nu) weighing 18–20 g, to investigate the tumorigenicity of MGC1 or MGC2 cells. The mice were killed 5 weeks after inoculation, and the tumors were excised and subjected to histological examination.
Histochemical & immunohistochemical analysis. Original primary tumors and transplanted tissues were fixed in 10% buffered formalin, embedded in paraffin and sectioned at 4 µm. Cells harvested with trypsin/EDTA on slide glasses, original tumor tissue slices, and transplanted tissue slices were examined for hematoxylin and eosin (H&E) staining, Alcian blue‐periodic acid Schiff staining (AB‐PAS) as well as immunohistochemistry for cytokeratin.
Scanning electron microscope analysis. Cell samples were fixed in 1% glutaraldehyde (in 0.05 M cacodylate buffer, pH 7.2), and dehydrated through a graded ethanol series. Following dehydration in 100% ethanol, the alcohol was replaced with isoamyl acetate. Specimens were dried in 100% isoamyl acetate in a critical point drying apparatus (HCP‐1, HITACHI, Tokyo), coated with gold, and observed under a scanning electron microscope (S‐2250 N, HITACHI, Tokyo) at 20 kV.
Phosphorylation after cellular stimulation with recombinant IL‐1 beta. Anti‐phospho I kappa B (pIκB) (Ser32), antiphospho Akt (Ser473) Ab, and anti phospho‐p44/42 MAPK (Thr 202/Tyr204) mAbs, were purchased from Cell Signal (Beverly, MA), anti‐α Tubulin (Clone DM1A) from Sigma (Amersham, Uppsala, Sweden).
MGC cell lines (1 × 106) were incubated for different time periods with recombinant human IL‐1β 10 ng/mL at 37°C in RPMI. Cells were washed once with cold PBS, and then lyzed at 4°C in lysis buffer (10 mM Tris‐HCl, pH 7.5, 150 mM NaCl, 1% Triton X‐100, 1 mM EDTA, and complete Protease Inhibitor Cocktail from Roche Applied Science, Indianapolis, IN). 10‐µg whole cell lysates in Laemmli sample buffer were separated by 12% SDS‐PAGE under reducing conditions, transferred to polyvinylidene difluoride membranes (Amersham, Uppsala, Sweden) and immunoblotted with indicated Abs. Bound Abs were visualized using Western Lightning Chemiluminescence Plus (PerkinElmer Life Sciences, Boston, MA).
Luciferase assay on transcriptional activation of Nuclear Factor kappa B. 2 × 105 cells were cotransfected in a 24‐well culture dish with two expression plasmids, a luciferase reporter gene under the transcriptional control of NF‐κB element (pNF‐κB luciferase; BD Science, San Diego, CA), and a transfection efficiency indicator (pRL‐CMV; Promega, Madison, WI) using Lipofectamin 2000 (Invitrogen). After 24‐h incubation in RPMI containing 10% FCS, cells were challenged to be infected with 1 × 105 of NCTC11637 or its isogenic cagE knockout mutant (cagE KO) H. pylori for 12 h. For constructing cagE KO, a portion of the genes encoding the cagE gene was amplified by PCR, and the amplified‐fragment was inserted into EcoRV restriction enzyme site of pBluescriptSK+ (Stratagene, La Jolla, CA). A kanamicin resistance gene cassette (Km) was inserted into BsmF1 site of the insert DNA for cagE gene. The plasmids (1–2 µg) were used for inactivation of chromosomal genes by natural transformation as previously described. (23) Inactivation of the genes was confirmed by PCR amplification followed by Southern blot hybridization. NF‐κB luciferase reporter gene assay was performed with a Dual Luciferase Assay System (Promega) and luminometer according to the supplier's instructions.
Results
Establishment of MGC1 and MGC2 cells. A gerbil had advanced gastric cancer with strong progression and perineural invasion (Fig. 1a–e). Two weeks after the primary culture, aggregated tumor cell colonies surrounded by fibroblasts were found at the bottom of the culture dishes. After serial passages while decreasing the number of fibroblastic cells, the remaining few fibroblasts were removed with mechanical scraping and the differential attachment selection method. Cells were replaced entirely by the tumor cells, and the obtained cancer cells were designated as MGC cells (Fig. 1f). Next, single‐cell dilution cloning of these MGC cells using a 96‐well culture plate was performed, and finally, cultures of two different types of cell lines forming typical epithelial monolayer were obtained. These two cell colonies, designated MGC1 and MGC2, respectively, have been cultured for more than 12 months with up to 60 passages, without further changes in morphology or growth rate.
Figure 1.
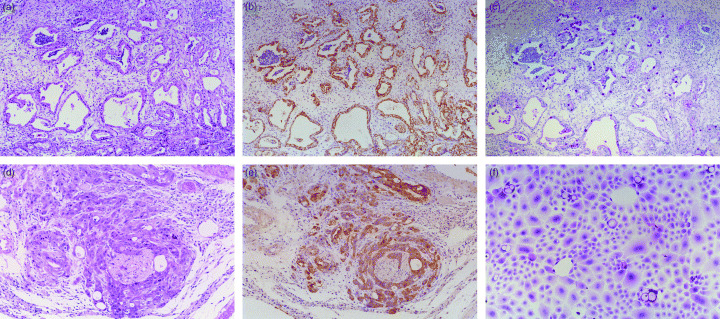
(a) Original gastric cancer tissue occurring in a Mongolian gerbil. The excised cancer tissue was subjected to the following primary culture procedure. (× 10 H&E). (b) Mongolian gerbil gastric cancer tissue comprised of epithelial cells stained with immunohistochemical staining for cytokeratin. (× 10 Cytokeratin). (c) Alcian blue‐periodic acid Schiff staining clearly reveals an aberrant staining pattern in the cancer tissue. (× 10 AB‐PAS). (d) High‐power view of a deep invasive lesion of the gastric cancer. Proliferating and invasive gastric cancer cells surrounding neural tissue show the perineural invasion. (× 40 H&E). (e) Surrounding gastric cancer cells are stained with cytokeratin, in sharp contrast to the neural tissue without such staining. (× 40 Cytokeratin). (f) Cancer cells derived from the Mongolian gerbil gastric cancer tissue, before single colony dilution cloning. (× 80 H&E).
Morphology of MGC cells in culture. The subconfluent culture of MGC1 cells shows a rather large cytoplasm with a monolayer sheet of polygonal‐shaped epithelial cells with a rather high nucleus/cytoplasm ratio. The subconfluent culture of MGC2 cells, on the other hand, reveals a rather small cytoplasm, and much higher nucleus/cytoplasm ratio growing with a cobblestone appearance and rosette formation (Fig. 2a–d). MGC2 cells had 3–4‐fold greater growth activity compared to MGC1 cells.
Figure 2.
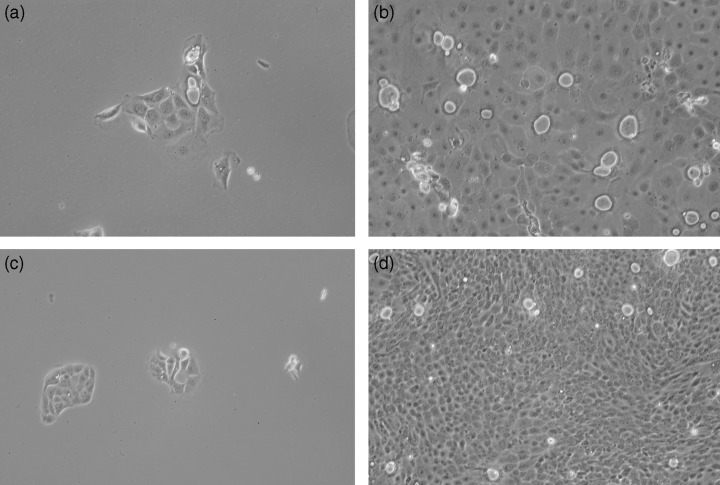
(a) MGC1 cells derived after single‐cell dilution cloning. MGC1 cells show large cytoplasm with a high nucleus/cytoplasm ratio. (× 80). (b) MGC1 cells at confluence show a monolayer sheet of polygonal‐shaped epithelial cells. (× 80). (c) MGC2 cells derived after single‐cell dilution cloning. They reveal rather small cytoplasm and a much higher nucleus/cytoplasm ratio. (× 80). (d) MGC2 cells at confluence with small cytoplasm and higher growth activity. (× 80).
Tumorigenicity in nude mice. Inguinal subcutaneous transplantation of MGC1 and MGC2 cells was attempted using five nude mice. When MGC1 cells were inoculated s.c. into the nude mice, the cells grew slowly to form mainly cystic tumors, which were detected within 4 weeks following inoculation. Four of the five transplanted lesions evidenced stable transplantation, showing cystic lesions consisting of a monolayer of MGC1 cells. Histologically, the tumors were comprised of rather large, pleomorphic, hyperchromatic cells (Fig. 3a–c). Transplanted MGC2 cells show rather high invasiveness of the host tissue, and perineural invasion has been found. Inoculated MGC2 cells grew rapidly to form subcutaneous solid tumors that were detected within 2 weeks after inoculation. In all mice the MGC2 cells showed stable transplantation and glandular‐like configurations. The tumors were composed of rather small, pleomorphic, hyperchromatic cells arranged in a solid pattern (Fig. 3d–f). They grew invasively under the transplanted tissue, evidencing perineural invasion of the muscle layer (Fig. 3f).
Figure 3.
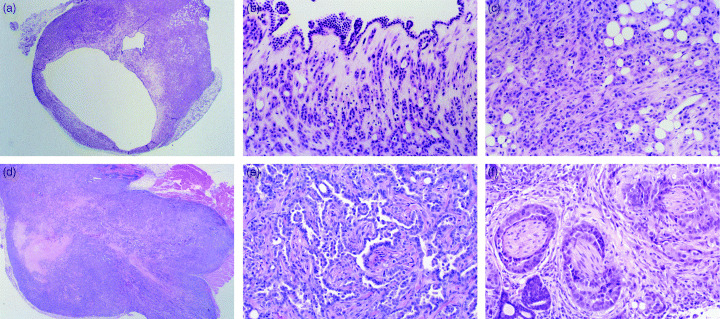
(a) MGC1 cells transplanted subcutaneously into nude mice grew slowly to form cystic tumors, consisting of a monolayer of rather large, pleomorphic, hyperchromatic cells. (× 10 H&E). (b) High‐power view of Fig. 3a. Cystic tumor formation with monolayer of MGC1 cells. (× 40 H&E). (c) High‐power view of Fig. 3a. Tumor formation with MGC1 cells. (× 40 H&E). (d) MGC2 cells transplanted subcutaneously into nude mice grew with stable transplantation and glandular‐like configurations. The tumors were composed of rather small, pleomorphic, hyperchromatic cells arranged in a solid pattern. (× 10 H&E). (e) High‐power view of Fig. 3d. Tumor tended to form glandular‐like constructs with MGC2 cells. (× 40 H&E). (f) Transplanted MGC2 cells with perineural invasion. MGC2 cells grew invasively under the transplanted tissue, with perineural invasion and invasion to the muscle layer. (× 40 H&E).
Stimulation of cells with recombinant IL‐1 beta. To assess the reactivity of MGC cells to pro‐inflammatory cytokines, the phosphorylation status of signaling molecules was evaluated in MGC cells with recombinant human IL‐1β 10 ng/mL during different time periods. IL‐1β stimulation resulted in increased phosphorylation of IκB, Erk1/2 (p44/42 MAPK), and AKT (Fig. 4). Three independent experiments with recombinant human IL‐1β 10 ng/mL activation gave essentially the same results. As shown in Fig. 4, maximal phosphorylation of IκB was seen 5 min after stimulation of IL‐1β, then gradually decreased but was re‐up‐regulated 45 min after stimulation. Maximal phosphorylation of Erk1/2 was observed at 10 and 15 min, and the level gradually decreased with time. By comparison, increased phosphorylation of Akt was seen at 5 and 10 min, and remained elevated at the endpoint.
Figure 4.
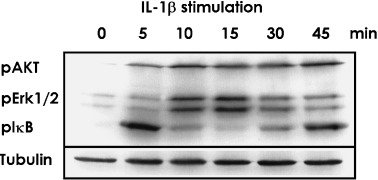
MGC cells were stimulated with recombinant IL‐1β 10 ng/mL. Phosphorylation status of pIκB, pErk1/2 and pAKT was confirmed with western blotting. Anti‐α Tubulin was treated as internal control. Maximal phosphorylation of IκB was seen 5 min after stimulation of IL‐1β, then gradually decreased, but was re‐up‐regulated 45 min after stimulation. Maximal phosphorylation of Erk1/2 was observed at 10 and 15 min, and the level gradually decreased with time. By comparison, the increased phosphorylation of Akt was seen at 5 and 10 min, and was still elevated at the endpoint.
H. pylori attached to MGC cell and induced NFkB activation. To study NF‐κB activation following H. pylori infection on these newly established rodent gastric cancer cells, a luciferase reporter gene under the control of the cis‐acting enhancer element NF‐κB (NF‐κB‐luciferase) was used. Initial experiments were performed in triplicate with the established cells 24 h after cotransfection with 1 µg of pNF‐κB luciferase and 0.1 µg of pRL‐CMV, and activation with culture media with and without the presence of 10 ng/mL lipopolysaccharide. A 1.7‐fold higher transcriptional activation was seen in both MGC1 and MGC2 cells.
The cells cotransfected with pNF‐κB luciferase and pRL‐CMV were challenged to be infected with NCTC11637 or its isogenic cagE knockout mutant (cagE KO) H. pylori for 12 h. Stable attachment on these cells with H. pylori NCTC11637 or its isogenic mutant could be observed with a scanning electron microscope (Fig. 5). Results in Fig. 6 show the representative results of three separate experiments conducted under each condition in triplicate. Elevated transcriptional activation of NF‐κB was induced after infection with NCTC11637, while infection with cagE KO H. pylori did not show such an effect (Fig. 6). The cagE KO H. pylori strain has a breakdown of the Type IV secretion system, and is considered to lack the ability to inject CagA protein into the attached host cells. These results indicate that the results in transcriptional activation of NF‐κB in MGC cells differ in the infected H. pylori‐dependent manner.
Figure 5.
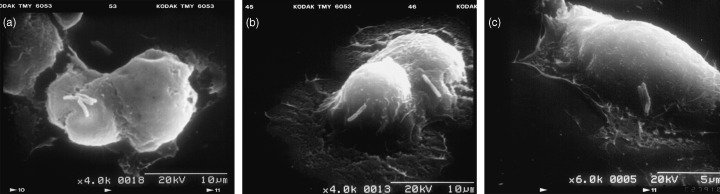
(a) Scanning electron micrograph (SEM) of MGC cell infected with H. pylori. (× 4000). (b) SEM showing MGC cell attached with H. pylori on the cell surface. (× 4000). (c) SEM showing MGC cell attached with H. pylori. (× 6000).
Figure 6.
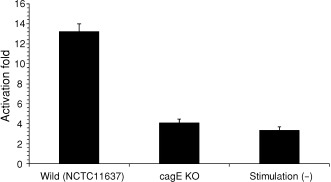
Transcriptional activation of NF‐κB (Luciferase assay on NF‐κB activation after being infected with NCTC11637 or its isogenic cagE knockout mutant (cagE KO) H. pylori). Elevated transcriptional activation of NF‐κB was induced after infection with NCTC11637, while infection with cagE KO H. pylori did not show such an effect. Results shown are the average and standard error of three independent experiments.
Discussion
We succeeded in the establishment and initial characterization of adenocarcinoma cell lines (MGC1 and MGC2) from the gastric cancer tissue of a H. pylori‐infected and MNU‐treated Mongolian gerbil. This is the first report of gastric cancer cells established from a Mongolian gerbil, and the cell lines established may be of potential use in the study of H. pylori‐induced gastric cancers. These cells can easily be cultured, and when the cells were inoculated s.c. into the nude mice, they showed stable transplantation to nude mice and grew firmly, forming solid or cystic tumors. Transplanted MGC2 cells show rather high invasiveness of the host tissue with perineural invasion. Inoculated MGC2 cells grew rapidly to form subcutaneous solid tumors that were detected within 2 weeks after inoculation. Stable attachment on these cells with H. pylori could be observed with scanning electron microscope analysis. The hummingbird phenotype was not clearly observed in the electron microscope analysis; however, this phenotype can differ with the infected H. pylori status or depending on the type of cell line. (21) The cells under study here can also be easily infected with H. pylori ATCC43504 or the Sydney strain of H. pylori. Furthermore, the transcriptional activation through NF‐κB was apparently elevated in the infected H. pylori‐dependent manner.
These established cell lines are unique for the following three reasons. First, they are the first gastric cancer cell lines established in the Mongolian gerbil, a candidate for an ideal H. pylori‐related gastric carcinogenesis model. (24) The derived cells were easily infected with H. pylori and could be used for examinations of the influences after H. pylori infection.
Second, these cell lines can be used to explore the molecular and cellular biological investigations on the basic mechanisms of glandular cell proliferation. The NF‐κB and the signaling pathways that are involved in its activation have great importance for tumor development. (25) In the present study, treatment with recombinant IL‐1β attenuated the phosphorylation status of the IκB/NFκB, MAPK, and the PI3K/Akt pathways. α‐Tubulin was used as an internal control, and the behavior of phosphorylated AKT, ERK1/2, and IκB were clarified. As a result, the activation of pIκB had distinct peaks at 5 and 45 min in the time course. This may indicate a fluctuation resulting from cross‐talk with other signaling pathways such as AKT or ERK, or from the negative feedback of the IκB – NF‐κB signaling cascade. Cross‐talk of these signaling pathways, arising from the cell‐to‐cell interactions between epithelial, mesenchymal, and inflammatory cells, might also be examined from the viewpoint of introducing cancer progression. Furthermore, infection with H. pylori clearly attenuated the transcriptional activation of NF‐κB in these cancer cells, suggesting that the causal effect of H. pylori on the cancer cell progression might be mediated not only by Ras/MAPK but also with the NF‐κB signaling pathway, partly mediated with the epithelial–mesenchymal interactions.
Third, the newly derived two cell lines exhibit markedly different tumorigenesis and invasive activities into the skin, muscle layer, and perineural cavities of the transplanted tissue. They appeared to show a clearly different morphology associated with the invasive activity in the transplanted tissue. One forms a cystic lesion, with lesser invasion, while the other tends to form expansive tumors with diffuse invasion. The two lines were established from the same gastric carcinoma specimen, and are of the same clonal origin. The cells were stable in terms of both morphology and proliferation after being separated into the two lines. Therefore, it may be appropriate to consider the lines to have been derived from heterogenous cell populations contained in the primary cancer; for example, differentiated and undifferentiated components of the adenocarcinoma, rather than the two types differentiating during the process of establishing the two lines. Searching for the factors that determine the differences between the two might be meaningful. Gene expression analysis might provide some information to explain such morphological and biological differences. 26 , 27 , 28 , 29 Since these cell lines are not immortalized cells but derived from actual tumors, they are sure to have properties that reflect the characteristics contained in the original tumor tissue.
The newly established adenocarcinoma cell lines from the in vivo gastric carcinogenesis model showed response to H. pylori infection or IL‐1β stimulation, implying that continuous H. pylori infection can affect the cell proliferation of the induced gastric cancer. In fact, NF‐κB activation was induced with H. pylori infection, and AKT, ERK, and IκB activation were observed as a result of IL‐1β stimulation. Thus, proliferative response of the cell to H. pylori infection should be considered, in terms of the direct actions of H. pylori on cancer cells, and the indirect actions through cytokine stimulation by inflammatory cells.
In conclusion, new adenocarcinoma cell lines were purified from the Mongolian gerbil, the standard H. pylori‐related gastric cancer model. The lines can be used to develop effective therapeutic strategies against gastric cancer, especially in exploring the effect of H. pylori, and thus might contribute greatly to gastric cancer prevention and treatment in humans.
Acknowledgments
This work was supported in part by a Research Grant from the Princess Takamatsu Cancer Research Fund (00‐23207), a Grant‐in‐Aid for the Second‐term Comprehensive 10‐year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare, Japan, and a Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We also thank Ms. Masami Yamamoto and Ms. Hisayo Ban for their skillful assistance.
These authors (Koji Nozaki, Harunari Tanaka) equally contributed to this work.
References
- 1. Warren JR, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active gastritis. Lancet 1983; I: 1273–5. [PubMed] [Google Scholar]
- 2. Marshall BJ, Armstrong JA, Mcgechie DB, Glancy RJ. Attempt to fulfill Koch's postulates for pyloric Campylobacter . Med J Aust 1985; 142: 436–9. [DOI] [PubMed] [Google Scholar]
- 3. Kuipers EJ, Uyterlinde AM, Pena AS, Roosendaal R, Pals G, Nelis GF, Festen HP, Meuwissen SG. Long‐term sequelae of Helicobacter pylori gastritis. Lancet 1995; 345: 1525–8. [DOI] [PubMed] [Google Scholar]
- 4. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127–31. [DOI] [PubMed] [Google Scholar]
- 5. Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez‐Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991; 325: 1132–6. [DOI] [PubMed] [Google Scholar]
- 6. Forman D, Newell DG, Fullerton F, Yarnell JW, Stacey AR, Wald N, Sitas F. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ 1991; 302: 1302–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. The EUROGAST Study Group: An international association between Helicobacter pylori infection and gastric cancer. Lancet 1993; 341: 1359–62. [PubMed] [Google Scholar]
- 8. Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta‐analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998; 114: 1169–79. [DOI] [PubMed] [Google Scholar]
- 9. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784–9. [DOI] [PubMed] [Google Scholar]
- 10. Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Meta‐analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology 2003; 125: 1636–44. [DOI] [PubMed] [Google Scholar]
- 11. Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. SHP‐2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002; 295: 683–6. [DOI] [PubMed] [Google Scholar]
- 12. Tatematsu M, Yamamoto M, Shimizu N, Yoshikawa A, Fukami H, Kaminishi M, Oohara T, Sugiyama A, Ikeno T. Induction of glandular stomach cancers in Helicobacter pylori‐sensitive Mongolian gerbils treated with N‐methyl‐N‐nitrosourea and N‐methyl‐N′‐nitro‐N‐nitrosoguanidine in drinking water. Jpn J Cancer Res 1998; 89: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sugiyama A, Maruta F, Ikeno T, Ishida K, Kawasaki S, Katsuyama T, Shimizu N, Tatematsu M. Helicobacter pylori infection enhances N‐methyl‐N‐nitrosourea‐induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res 1998; 58: 2067–9. [PubMed] [Google Scholar]
- 14. Shimizu N, Ikehara Y, Inada K, Nakanishi H, Tsukamoto T, Nozaki K, Kaminishi M, Kuramoto S, Sugiyama A, Katsuyama T, Tatematsu M. Eradication diminishes enhancing effects of Helicobacter pylori infection on glandular stomach carcinogenesis in Mongolian gerbils. Cancer Res 2000; 60: 1512–4. [PubMed] [Google Scholar]
- 15. Nozaki K, Shimizu N, Ikehara Y, Inoue M, Tsukamoto T, Inada K, Tanaka H, Kumagai T, Kaminishi M, Tatematsu M. Effect of early eradication on Helicobacter pylori‐related gastric carcinogenesis in Mongolian gerbils. Cancer Sci 2003; 94: 235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peek RM Jr, Wirth HP, Moss SF, Yang M, Abdalla AM, Tham KT, Zhang T, Tang LH, Modlin IM, Blaser MJ. Helicobacter pylori alters gastric epithelial cell cycle events and gastrin secretion in Mongolian gerbils. Gastroenterology 2000; 118: 48–59. [DOI] [PubMed] [Google Scholar]
- 17. Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 1998; 115: 642–8. [DOI] [PubMed] [Google Scholar]
- 18. Honda S, Fujioka T, Tokieda M, Satoh R, Nishizono A, Nasu M. Development of Helicobacter pylori‐induced gastric carcinoma in Mongolian gerbils. Cancer Res 1998; 58: 4255–9. [PubMed] [Google Scholar]
- 19. Maeda S, Yoshida H, Ikenoue T, Ogura K, Kanai F, Kato N, Shiratori Y, Omata M. Structure of cag pathogenicity island in Japanese Helicobacter pylori isolates. Gut 1999; 44: 336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, Suzuki T, Sasakawa C. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med 2000; 191: 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirata Y, Maeda S, Mitsuno Y, Tateishi K, Yanai A, Akanuma M, Yoshida H, Kawabe T, Shiratori Y, Omata M. Helicobacter pylori CagA protein activates serum response element‐driven transcription independently of tyrosine phosphorylation. Gastroenterology 2002; 123: 1962–71. [DOI] [PubMed] [Google Scholar]
- 22. Maeda S, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y, Akanuma M, Shiratori Y, Omata M. H. pylori activates NF‐kappaB through a signaling pathway involving IkappaB kinases, NF‐kappaB‐inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 2000; 119: 97–108. [DOI] [PubMed] [Google Scholar]
- 23. Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori . Proc Natl Acad Sci USA 2000; 97: 7533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tatematsu M, Nozaki K, Tsukamoto T. Helicobacter pylori infection and gastric carcinogenesis in animal models. Gastric Cancer 2003; 6: 1–7. [DOI] [PubMed] [Google Scholar]
- 25. Karin M, Cao Y, Greten FR, Li ZW. NF‐kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2002; 2: 301–10. [DOI] [PubMed] [Google Scholar]
- 26. Bebb JR, Letley DP, Thomas RJ, Aviles F, Collins HM, Watson SA, Hand NM, Zaitoun A, Atherton JC. Helicobacter pylori upregulates matrilysin (MMP‐7) in epithelial cells in vivo and in vitro in a Cag dependent manner. Gut 2003; 52: 1408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaneda A, Kaminishi M, Yanagihara K, Sugimura T, Ushijima T. Identification of silencing of nine genes in human gastric cancers. Cancer Res 2002; 62: 6645–50. [PubMed] [Google Scholar]
- 28. Cao X, Tsukamoto T, Nozaki K, Mizoshita T, Ogasawara N, Tanaka H, Takenaka Y, Kaminishi M, Tatematsu M. Beta‐catenin gene alteration in glandular stomach adenocarcinomas in N‐methyl‐N‐nitrosourea‐treated and Helicobacter pylori‐infected Mongolian gerbils. Cancer Sci 2004; 95: 487–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sakakura C, Hagiwara A, Miyagawa K, Nakashima S, Yoshikawa T, Kin S, Nakase Y, Ito K, Yamagishi H, Yazumi S, Chiba T, Ito Y. Frequent downregulation of the runt domain transcription factors RUNX1, RUNX3 and their cofactor CBFB in gastric cancer. Int J Cancer 2005; 113: 221–8. [DOI] [PubMed] [Google Scholar]