

Abstract
Arsenic trioxide (As2O3) is a potent antitumor agent used to treat acute promyelocytic leukemia (APL) and, more recently, solid tumors. However, the dose of As2O3 required to suppress human xenographs in mice is markedly higher than that used to treat APL in humans. Paradoxically, low doses of As2O3 stimulate angiogenesis, which might be expected to promote tumor growth. Clearly, appropriate dosages of As2O3 are required to treat human patients to avoid toxicity and undesirable side effects. In the present study, we investigated As2O3 with respect to its toxicity and effects on tumor growth, angiogenesis and cell apoptosis using H22 hepatocellular carcinoma (HCC) cells in a mouse model of HCC. As2O3 inhibited tumor growth and angiogenesis, and enhanced tumor cell apoptosis at doses greater than 1 mg/kg, but mice lost weight and failed to thrive at doses of 4 mg/kg and greater. In contrast, low doses (<1 mg/kg) of As2O3 promoted tumor growth, upregulated the expression of vascular endothelial growth factor and tumor angiogenesis, and had no effect on tumor cell apoptosis. In vitro studies demonstrated that As2O3 inhibited the proliferation of H22 tumor cells and bovine aortic endothelial cells, and induced their apoptosis in a dose‐ and time‐dependent fashion, suggesting that the mechanism of As2O3‐mediated inhibition of tumor growth is due to direct effects of the drug on both tumor cells and endothelia. In summary, different doses of As2O3 have opposing effects on tumor growth and angiogenesis. The results demonstrate that As2O3 has a narrow window of therapeutic opportunity with respect to dosage, and that low doses of the drug as used in metronomic therapy should be used with extreme caution. (Cancer Sci 2006; 97: 675–681)
Abbreviations:
- AI
apoptosis index
- As2O3
arsenic trioxide
- APL
acute promyelocytic leukemia
- BAEC
bovine aortic endothelial cells
- HCC
hepatocellular carcinoma
- TUNEL
terminal deoxynucleotidyl transferase biotin‐dUTP nick‐end labeling
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide
- PI
proliferation index
- VEGF
vascular endothelial growth factor.
Arsenic trioxide has been used as an anticancer agent in traditional Chinese medicine for several thousand years. It was originally shown to induce complete clinical remission in patients with APL at Harbin Medical University in China in the 1970s.( 1 ) It has subsequently been widely employed to treat APL, including the treatment of patients that are resistant to conventional chemotherapies.( 2 )
The therapeutic potential and antitumor activity of As2O3 extends to non‐APL leukemia, and to a variety of solid tumors and tumor cell lines including neuroblastoma,( 3 ) head and neck,( 4 ) gastric,( 5 ) transitional,( 6 ) renal,( 7 ) esophageal,( 8 ) prostate,( 9 ) colorectal( 10 ) and hepatocellular( 11 ) cancers or cell lines. However, the activity of As2O3 against solid tumors has not been as effective as against APL. As2O3 induces the apoptosis of various cancer cell lines( 3 , 4 , 5 , 6 , 7 ) and inhibits the growth of tumors,( 8 , 9 , 12 ) but the dosages of As2O3 required to exert these effects( 9 , 12 , 13 , 14 ) are much higher than those required to inhibit hematologic malignancies.( 15 , 16 ) Such high doses are not clinically achievable without the risk of severe side effects due to toxicity. A review of recently published reports in which As2O3 was used to treat solid tumors in mice reveals that effective dosages of As2O3 range from 2 to 6.5 mg/kg in treatments that lasted for 1–6 weeks.( 9 , 12 , 13 , 14 ) Such dosages are considerably higher (approximately 12–40‐fold) than the standard dosage of 0.16 mg/kg used to treat human patients with APL over a period of 6 weeks.( 15 , 16 ) Few studies in mice have reported data about the toxicity of As2O3.
The anticancer activity of As2O3 is in part related to its effects on mitochondria,( 17 ) and the generation of nitric oxide and reactive oxygen species.( 18 ) Accordingly, As2O3 exerts antitumor effects as a result of its highly cytotoxic nature. Perhaps not surprisingly therefore, systemically administered As2O3 has undesirable side effects. As2O3 has been reported to exert cardiac toxicity in human patients even at the low dose of 0.16 mg/kg,( 19 , 20 ) and repeated injection of As2O3 produces cardiac toxicities in mice.( 21 ) Intraperitoneally administered As2O3 has binding affinity for liver and kidneys, and causes damage to both organs.( 22 ) In one recent report, oral administration of As2O3 to mice at doses of 3 or 6 mg/kg for 30 days resulted in significant reductions in bodyweight, protein and glycogen, as well as reduction in the ascorbic acid content of both livers and kidneys.( 23 ) The data suggest that As2O3 is highly toxic when systemically administered to mice at the doses required to treat solid tumors. In contrast, low doses of As2O3 were shown to stimulate angiogenesis and tumorigenesis.( 24 ) The latter results suggest that the therapeutic dosage of As2O3 should be carefully investigated before it is applied to treat solid tumors in patients.
In the present study, we investigated As2O3 with respect to its toxicity, and effects on tumor growth, angiogenesis and cell apoptosis in a mouse model of HCC.
Materials and Methods
Mice, cell lines and As2O3
Male BALB/c mice (H‐2b), 8 weeks old, were obtained from the Animal Research Center, Harbin Medical University, China. The syngeneic H22 HCC cells and BAEC were supplied by Dianzang Cell Bank of Wuhan University, Wuhan, China. An As2O3 solution was purchased from Yida Pharmaceutical Co. Ltd, Harbin Medical University.
Animal experimental design
All procedures administered to animals were in accordance with institutional animal ethics approvals and guidelines. All experiments included 10 mice per treatment group.
Assessment of toxicity Mice were treated by intraperitoneal injection of As2O3 at different doses every 2 days. Control mice were injected with an equivalent volume of PBS. The mice were carefully monitored for symptoms of toxicity, and weighed once each week. The mean weight of each group of mice was calculated and used as a parameter of toxicity, as described previously.( 23 )
Tumor implantation and treatment H22 tumor cells were maintained by successive transplantation into the abdominal cavity of mice. The cancerous ascites were collected, stained with Trypan blue to record viability, and counted by microscopy. Tumors were established by subcutaneous injection of 2 × 105 H22 cells into a site on the left flank of mice, as described previously.( 25 ) Animals were randomly assigned to treatment when tumors reached around 0.2 cm in diameter approximately 7 days after injection of tumor cells. As2O3 was administered intraperitoneally at different doses every alternate day. Control mice were given an equivalent volume of PBS. The growth of tumors was monitored and tumor size determined by measuring two perpendicular diameters.
Assessment of tumor vascularity
The methodology to determine tumor vascularity has been described previously.( 25 ) Briefly, 10 µm frozen tumor sections prepared from tumors 4 weeks after treatment were fixed with acetone, rinsed with PBS, blocked with 2% bovine serum albumin for 2 h, and incubated overnight with an anti‐CD31 antibody (MEC13.3, Pharmingen, San Diego, CA, USA). They were subsequently incubated for 30 min with secondary antibodies using the SABC kit (Boster Biological Technology, Wuhan, China), developed with Sigma FAST DAB (3,3′‐diaminobenzidine tetrahydrochloride) and CoCl2 enhancer tablets (Sigma, St Louis, MO, USA) and counterstained with hematoxylin. Stained blood vessels were counted in five blindly chosen random fields (0.155 mm2) at 400× magnification, and the mean of the highest three counts was calculated.
In situ detection of apoptotic cells
Serial sections of 6 µm thickness were prepared from tumors 4 weeks after treatment, stained with the TUNEL agent (Boehringer Mannheim, Mannheim, Germany), and examined by fluorescence microscopy. Adjacent sections were counterstained with hematoxylin and eosin. The total number of apoptotic cells in 10 randomly selected fields was counted. The AI was calculated as the percentage of positive‐staining cells, namely:
| AI = number of apoptotic cells × 100/total number of nucleated cells. |
Proliferation, viability and apoptosis of cells in vitro
The cells (5 × 103 H22 or 1 × 104 BAEC) were seeded in 96‐well plates in 200 µL of RPMI‐1640 media (Life Technologies, Beijing, China) supplemented with 50 U/mL penicillin/streptomycin. They were cultured at 37°C for 24 h, and the medium was replaced with fresh RPMI‐1640 or the same media containing different concentrations of As2O3. After a further 12, 24, 36 or 48 h incubation, 20 µL of MTT was added to each well followed by incubation for 4 h. The medium was discarded and 150 µL of dimethylsulfoxide was added into each well and incubated for 20 min. The OD570nm was measured and the PI calculated according to the formula:
| experimental OD value/control OD value × 100%. |
To measure cell apoptosis, the cells harvested after As2O3 treatment as above were fixed with 4% paraformaldehyde solution, and permeabilized with a solution of 0.1% Triton‐X100 and 0.1% sodium citrate. They were incubated with TUNEL reagent for 60 min at 37°C, examined by fluorescence microscopy, and the AI was calculated as above. The experiments were repeated three times.
Expression of VEGF by western blot analysis
Tumors excised from mice 4 weeks after treatment were minced, and homogenized in protein lysate buffer. Debris was removed by centrifugation, and the lysates were resolved on 10% polyacrylamide sodium dodecylsulfate gels, and electrophoretically transferred to nitrocellulose Hybond C extra membranes (Amersham Life Sciences, Buckinghamshire, UK). The membranes were incubated with an anti‐VEGF monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by a horseradish peroxidase‐conjugated secondary antibody, and developed by enhanced chemiluminescence (Amersham) followed by exposure to X‐ray film. The density of each band was evaluated using a densitometric analysis program (FR200; Furi, Shanghai, China). Blots were stained with an antitubulin antibody to confirm that each lane contained similar amounts of tumor homogenate. The relative levels of VEGF were normalized with respect to tubulin band density.
H22 cells were cultured in 2 mL of RPMI‐1640 or the same media containing different concentrations of As2O3 for 36 h, and the levels of VEGF in cell lysates were measured as above.
Statistical analysis
Results were expressed as mean values ± SD, and a Student's t‐test was used for evaluating statistical significance. A value of less than 0.05 (P < 0.05) was used for statistical significance.
Results
Toxicity of As2O3 in mice
Different doses of As2O3 (0.5, 1, 2, 4 and 8 mg/kg) were administered intraperitoneally every 2 days for 6 weeks into mice weighing on average 24.8 ± 1.2 g. Systemic administration of As2O3 at a dose of either 4 or 8 mg/kg caused the coats of mice to become dull and lose hair, pigmented spots appeared on the skin, and hyperkeratinosis was evident. The feet of mice showed signs of swelling, and mice became lethargic. No mice died as a consequence of As2O3 administration. Weight loss was used as a parameter to quantify toxicity. The results demonstrated that As2O3 had no significant effect on the weight of mice when administered at a dose of 0.5 or 1 mg/kg (Fig. 1). There was no significant difference in bodyweight between mice treated with PBS and As2O3 at a dose of 2 mg/kg until the sixth week when a significant difference (8% reduction, P < 0.05) in bodyweight was observed, suggesting that the effects of As2O3 are cumulative (Fig. 1). However, when As2O3 was administered at doses of 4 and 8 mg/kg, the difference in weight became significant (11 and 16% reduction, both P < 0.05) and highly significant (19 and 28% reduction, both P < 0.01) after the fourth and sixth weeks, respectively. The results suggest that for long‐term treatment, As2O3 should not be administered to mice at a dosage over 2 mg/kg in order to avoid severe side effects.
Figure 1.
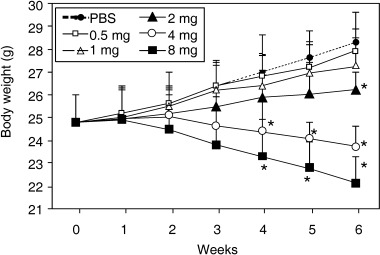
Effects of As2O3 on the body weight of mice. Different doses of As2O3 (0.5, 1, 2, 4, and 8 mg/kg) were administered i.p. to 8‐week‐old male BABL/c mice every 2 days. Control mice were injected with phosphate‐buffered saline (PBS). *P < 0.05, significantly different from control mice.
Effects of As2O3 on tumor growth
Tumors were established by subcutaneous injection of 2 × 105 H22 cells into the flanks of mice. Animals were randomly assigned to treatment when tumors reached around 0.2 cm in diameter after approximately 7 days. As2O3 was administered inrtaperitoneally at doses of 0.2, 0.5, 1, 2 and 4 mg/kg every 2 days for 30 days. Tumor growth became significantly inhibited (by 17, 35 and 53%, respectively, all P < 0.05) by the 20th day of treatment with doses of 1, 2 and 4 mg/kg of As2O3, and by the 15th day in the case of the highest dose of 4 mg/kg (46% reduction, P < 0.05). In marked contrast, the two lowest doses of 0.2 and 0.5 mg/kg had significantly promoted the growth of tumors (19 and 15% increase, respectively, both P < 0.05) by the 30th day of treatment compared to the growth of tumors of control mice (Fig. 2).
Figure 2.
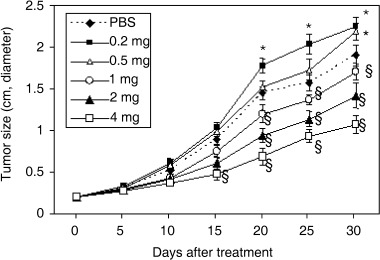
Effects of As2O3 on tumor growth. Different doses of As2O3 (0.2, 0.5, 1, 2, and 4 mg/kg) were administered i.p. into BABL/c mice bearing tumors of approximately 0.2 cm in diameter, established by subcutaneous injection of H22 cells 7 days earlier. Control mice were injected with phosphate‐buffered saline (PBS). The size of the tumors is plotted against days after treatment. *A significant (P < 0.05) increase in tumor size in response to As2O3 versus PBS. §A significant (P < 0.05) reduction in tumor size in response to As2O3 versus PBS.
Effects of As2O3 on tumor angiogenesis
The vascularity of tumors of mice 4 weeks after treatment was examined by enumeration of vessels in tumor sections stained with an anti‐CD31 monoclonal antibody. Low doses of As2O3 surprisingly promoted tumor angiogenesis as evidenced by increased blood vessel density following treatment (Fig. 3a,b). There was a significant increase (37%, P < 0.01) in tumor blood vessel counts of mice treated with 0.2 mg/kg As2O3 compared to mice treated with PBS. In contrast, As2O3 at a dose of 0.5 mg/kg only slightly increased vessel counts such that there was no significant difference compared to control mice. High doses of As2O3 had an opposing effect in that tumor blood vessel density was significantly decreased (by 25–42%, P < 0.05) in a dose‐dependent fashion when the dosage of As2O3 was raised to either 1, 2 or 4 mg/kg (Fig. 3a,b).
Figure 3.

Effects of As2O3 on tumor vascularity and expression of vascular endothelial growth factor (VEGF). (a) Representative sections from tumors of mice treated by i.p. administration of phosphate‐buffered saline (PBS), 0.2 mg/kg of As2O3, or 4 mg/kg of As2O3 for 4 weeks. Sections were stained with an anti‐CD31 mAb to visualize blood vessels. (b) The blood vessels were counted to record mean vessel density. *A significant (P < 0.05) increase in mean vessel counts of tumors treated with As2O3 versus PBS (control). §A significant reduction in mean vessel counts of tumors treated with As2O3 versus PBS (control). (c) Western blot analysis of VEGF expression in homogenates of tumors from mice treated with PBS (control, lane 1) or As2O3 (lane 2: 0.2 mg/kg; lane 3: 0.5 mg/kg; lane 4: 1 mg/kg; lane 5: 2 mg/kg; lane 6: 4 mg/kg). (d) Western blot analysis of VEGF expression in lysates of H22 cells incubated with PBS (control, lane 1) or As2O3 (lane 2: 0.1 µg/mL; lane 3: 0.2 µg/mL; lane 4: 0.4 µg/mL; lane 5: 0.8 µg/mL). The density of each band was measured and compared to that of the internal control, tubulin. *A significant difference at P < 0.05 in the band intensities of VEGF between control and As2O3‐treated groups.
Effects of As2O3 on expression of VEGF in vivo and in vitro
Given that As2O3 affected the vascularity of tumors, we investigated whether it affected the expression of VEGF, a key promoter of angiogenesis commonly expressed in tumors. Western blot analysis of tumor homogenates revealed, surprisingly, that As2O3 upregulated the expression of VEGF (by 89–140%, P < 0.01) at all of the doses of As2O3 used in this study compared with PBS‐treated control tumors. Quantification of the densities of bands on the blot indicated that there was no significant difference in the levels of VEGF induced in response to different doses of As2O3, implying that low and high doses of As2O3 induce VEGF expression to similar extents (Fig. 3c). Similarly, western blot analysis of cell lysates also showed that As2O3 upregulated the expression of VEGF at all of the concentrations of As2O3 used in this study compared with the control. Quantification of the densities of bands on the blot indicated that there was no significant difference in the levels of VEGF induced in response to different concentrations of As2O3, in accordance with the in vivo studies (Fig. 3d).
As2O3 induces cell apoptosis in situ
We examined whether tumors underwent programmed cell death after administration of As2O3, as measured by in situ labeling of fragmented DNA using the TUNEL method. Adjacent sections were stained with hematoxylin and eosin, and the AI was calculated. The AI for tumors treated with As2O3 at doses of 1, 2 and 4 mg/kg was significantly higher (by 39, 51 and 69%, respectively, all P < 0.01) than that for PBS‐treated tumors. However, As2O3 treatment at the lowest dose of 0.2 mg/kg did not increase the AI of tumors. The dose of 0.5 mg/kg of As2O3 slightly increased the AI compared to that of control tumors, but the difference was not significant (Fig. 4a,b). The results indicate that As2O3 induces tumor cell apoptosis in a dose‐dependent manner.
Figure 4.
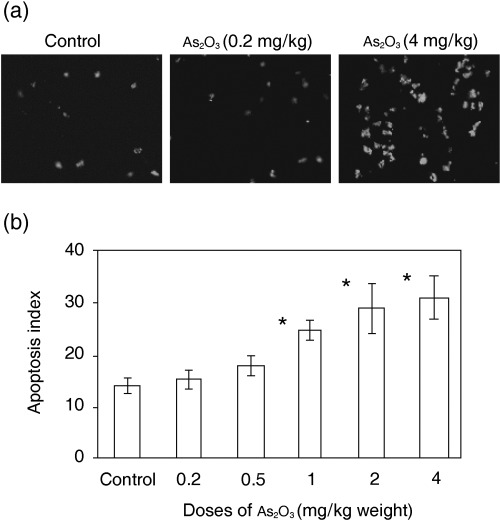
As2O3 induces tumor cell apoptosis in situ. (a) Illustrated are representative terminal deoxynucleotidyl transferase biotin‐dUTP nick‐end labeling (TUNEL)‐stained sections from the tumors of mice treated by i.p. administration of either phosphate‐buffered saline (PBS), 0.2 mg/kg of As2O3, or 4 mg/kg of As2O3 for 4 weeks. (b) TUNEL‐positive cells in tumor sections from mice treated by i.p. administration of either PBS, or 0.2, 0.5, 1, 2 or 4 mg/kg of As2O3 for 4 weeks, were counted to record the apoptosis index. *A significant difference at P < 0.01 in tumor cell apoptosis between the tumors of PBS (control) and As2O3‐treated mice.
As2O3 inhibits the proliferation and viability of BAEC and H22 cells and induces their apoptosis in vitro
Angiogenesis is a complex process that encompasses activation, migration and proliferation of endothelial cells. We investigated the effects of As2O3 in vitro on BAEC to determine whether As2O3 has direct effects on vascular endothelial cells. As2O3 inhibited the proliferation and viability of BAEC and induced their apoptosis in a dose‐ and time‐dependent manner. Differences in both the PI and AI of the As2O3‐ and PBS‐treated groups became significant at concentrations of 0.2 µg/mL or greater, whereas concentrations less than 0.2 µg/mL had no significant effect (Fig. 5a,b). As2O3 also inhibited the proliferation and viability of H22 cells and induced their apoptosis in a dose‐ and time‐dependent manner (Fig. 5c,d). Furthermore, at the same concentration, As2O3 was shown to have stronger inhibitory effects on H22 tumor cells than on BAEC.
Figure 5.

As2O3 inhibits the proliferation of H22 cells and BAEC and induces their apoptosis in vitro. H22 cells and BAEC were incubated in the absence or presence of As2O3 at different concentrations, and harvested at different time‐points for analysis. The proliferation/viability of H22 cells (a) and BAEC (b) was assessed using the MTT method and the proliferation index was calculated. The apoptosis of H22 cells (c) and BAEC (d) was assessed using terminal deoxynucleotidyl transferase biotin‐dUTP nick‐end labeling (TUNEL) analysis and the apoptosis index was calculated. *A significant difference at P < 0.05; **a highly significant difference at P < 0.001, from control at the indicated time‐point.
Discussion
The present study has for the first time simultaneously investigated the toxicity of systemically delivered As2O3, and its effects on tumor growth, angiogenesis and apoptosis in a mouse model of HCC. The data indicate that different doses of As2O3 have opposing effects on HCC tumor growth and angiogenesis. Dosages ranging from 1 to 2 mg/kg are appropriate for treating HCC tumors in mice, as tumor growth is suppressed and the side effects are endurable. It is not known whether the results are translatable to the affects of As2O3 in human cancer patients, but the fact that the optimum dosage of cancer drugs can vary markedly between different species warrants follow‐up studies in humans.
Tumors must establish an adequate vascular network to facilitate metastasis and acquire the nutrition necessary for growth. The dependency of tumors on angiogenesis provides the rationale for antiangiogenesis therapy of cancers aimed at shutting down the tumor blood supply. The frequent or continuous administration of low doses of chemotherapeutic agents, denoted metronomic chemotherapy, exerts a marked antiangiogenic effect, resulting in a superior antitumor effect compared with conventional scheduling.( 26 ) These findings herald the start of a new era in chemotherapy, as metronomic treatment targeting genomically stable endothelial cells may reduce the risk of development of drug resistance while reducing the toxic side effects of drugs. A number of chemotherapeutic drugs have been shown to inhibit the tumor angiogenesis of several different types of cancers when administered at low doses (metronomic).( 26 ) However, the metronomic administration of As2O3 might not be desirable to treat solid tumors as it stimulates tumor angiogenesis when administered at low doses.( 24 , 27 ) Low doses of As2O3 have been demonstrated to increase endothelial cell growth and tube formation in three dimensions( 28 ) and induce the expression of VEGF in endothelial and cervical cancer cells,( 28 , 29 , 30 ) but are unable to induce apoptosis of endothelial cells.( 31 ) In the studies in which As2O3 was shown to inhibit angiogenesis, it was used at a very high dose.( 32 , 33 ) In the present study we demonstrated that As2O3 induced the expression of VEGF at both low and high doses, but the apoptosis of vascular endothelial cells was induced only at high concentrations. The validity and generality of augmented synthesis of VEGF by As2O3 treatment has already been established by several prior studies investigating the effects of As2O3 or arsenite on B16 melanoma,( 24 ) ovarian cancer cells,( 29 , 34 ) and human umbilical vein endothelial cells,( 28 ) yet Roboz et al. reported that As2O3 inhibited the expression of VEGF in a leukemic cell line HEL.( 32 ) The opposing effects of As2O3 on VEGF expression are probably cell‐specific and dose‐specific. Notably, the dose of As2O3 used by Roboz et al. was higher than the highest dose used in the present study. We hypothesize that low doses of As2O3 promote tumor angiogenesis because they upregulate the expression of VEGF, but are unable to induce the apoptosis of endothelial cells. In contrast, high doses of As2O3 inhibit tumor angiogenesis because the apoptosis of vascular endothelial cells by As2O3 overweighs upregulation of VEGF. In accord, low concentrations of As2O3 significantly increased vascularity and blood vessel density in a chorioallantoic membrane assay of angiogenesis, whereas high concentrations of As2O3 generated pools of blood instead of vessels.( 30 ) As2O3 induces apoptosis in several cancer cell lines through either activation of caspase‐3, caspase‐7 and caspase–9,( 9 , 35 , 36 ) downregulation of the anti‐apoptotic protein Bcl‐2,( 35 ) production of free radical species,( 36 , 37 ) activation of c‐Jun NH2‐terminal kinase and p38,( 9 , 31 ) or induction of p53 protein.( 31 )
Emerging evidence suggests that angiogenesis plays a seminal role in the proliferation of liquid tumors, where endothelial cells release cytokines that stimulate the growth of leukemic cells, and leukemic cells in turn release endothelial growth factors such as VEGF.( 32 ) As2O3 interrupts this reciprocal stimulatory loop between leukemic cells and endothelial cells by causing apoptosis of both cell types and by inhibiting VEGF production by leukemic cells.( 32 ) The potent antileukemic effect of As2O3 is attributed, in part, to the ‘fluid’ feature of leukemia, which allows high local concentrations of As2O3 and close contact with cancer cells. The results presented herein indicate that caution should be taken in the systemic administration of As2O3 to treat HCC, and possibly other solid tumors. As2O3 is effective against solid tumors, but there is a narrow window of therapeutic opportunity, given that low doses of As2O3 stimulate tumor growth and high doses are toxic. In an alternative approach, localized delivery of As2O3 to treat solid tumors has been used to achieve therapeutic efficacy and reduce toxicity. Delivery of As2O3 directly to the tumor improves the tumor/normal tissue uptake ratios, and permits higher and more frequent doses of the anticancer drug, and at the same time avoids toxicity by reducing uptake of the drug by other organs.( 8 , 38 , 39 ) Alternatively, lower doses of As2O3 could be effectively used if As2O3 was to be combined with other therapeutic modalities such as B7H3‐mediated immunotherapy,( 25 ) non‐calcemic vitamin D analogs,( 40 ) l‐buthionine‐sulfoximine( 41 ) or docosahexaenoic acid.( 42 )
Acknowledgments
This work was financially supported by the National Natural Scientific Foundation of China (30471681, 30571808), and the Collaborative Fund for Overseas Scholars from the Scientific and Technological Bureau of Heilongjiang Province, China (WH05C02).
References
- 1. Soignet SL. Clinical experience of arsenic trioxide in relapsed acute promyelocytic leukemia. Oncologist 2001; 6: 11–16. [DOI] [PubMed] [Google Scholar]
- 2. Shen Y, Shen ZX, Yan H et al. Studies on the clinical efficacy and pharmacokinetics of low‐dose arsenic trioxide in the treatment of relapsed acute promyelocytic leukemia: a comparison with conventional dosage. Leukemia 2001; 15: 735–41. [DOI] [PubMed] [Google Scholar]
- 3. Akao Y, Nakagawa Y, Akiyama K. Arsenic trioxide induces apoptosis in neuroblastoma cell lines through the activation of caspase3 in vitro . FEBS Lett 1999; 455: 59–62. [DOI] [PubMed] [Google Scholar]
- 4. Seol JG, Park WH, Kim ES et al. Effect of arsenic trioxide of cell cycle arrest in head and neck cancer cell line PCI‐1. Biochem Biophys Res Commun 1999; 265: 400–4. [DOI] [PubMed] [Google Scholar]
- 5. Zhang TC, Cao EH, Li JF, Ma W, Qin JF. Induction of apoptosis and inhibition of human gastric cancer MCG‐803 cell growth by arsenic trioxide. Eur J Cancer 1999; 35: 1258–63. [DOI] [PubMed] [Google Scholar]
- 6. Pu YS, Hour TC, Chen J, Huang CY, Guan JY, Lu SH. Arsenic trioxide as a novel anticancer agent against human transitional carcinoma − characterizing its apoptotic pathway. Anticancer Drugs 2002; 13: 293–300. [DOI] [PubMed] [Google Scholar]
- 7. Hyun PW, Hee CY, Won JC et al. Arsenic trioxide inhibits the growth of A498 renal cell carcinoma cells via cell cycle arrest or apoptosis. Biochem Biophys Res Commun 2003; 300: 230–5. [DOI] [PubMed] [Google Scholar]
- 8. Shen ZY, Zhang Y, Chen JY et al. Intratumoral injection of arsenic to enhance antitumor efficacy in human esophageal carcinoma cell xenografts. Oncol Rep 2004; 11: 155–9. [PubMed] [Google Scholar]
- 9. Maeda H, Hori S, Nishitoh H et al. Tumor growth inhibition by arsenic trioxide in the orthotopic metastasis model of androgen‐independent prostate cancer. Cancer Res 2001; 61: 5432–40. [PubMed] [Google Scholar]
- 10. Nakagawa Y, Akao Y, Morikawa H et al. Arsenic trioxide‐induced apoptosis through oxidative stress in cells of colon cancer cell lines. Life Sci 2002; 70: 2253–62. [DOI] [PubMed] [Google Scholar]
- 11. Kito M, Akao Y, Ohishi N, Yagi K, Nozawa Y. Arsenic trioxide‐induced apoptosis and its enhancement by buthionine sulfoximine in hepatocellular carcinoma cell lines. Biochem Biophys Res Commun 2002; 291: 861–7. [DOI] [PubMed] [Google Scholar]
- 12. Monzen H, Griffin RJ, Williams BW, Amano M, Ando S, Hasegawa T. Study of arsenic trioxide‐induced vascular shutdown and enhancement with radiation in solid tumor. Radia Med 2004; 22: 205–11. [PubMed] [Google Scholar]
- 13. Kito M, Matsumoto K, Wada N et al. Antitumor effect of arsenic trioxide in murine xenograft model. Cancer Sci 2003; 94: 1010–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu HY, Yang YL, Liu SM, Bi L, Chen SX. Effect of arsenic trioxide on human hepatocarcinoma in nude mice. World J Gastroenterol 2004; 10: 3677–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shen ZX, Shi ZZ, Fang J et al. All‐trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Nat Acad Sci USA 2004; 101: 5328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niu C, Yan H, Yu T et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow‐up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 1999; 94: 3315–24. [PubMed] [Google Scholar]
- 17. Shen ZY, Shen J, Cai WJ, Hong C, Zheng MH. The alteration of mitochondria is an early event of arsenic trioxide induced apoptosis in esophageal carcinoma cell. Int J Mol Med 2000; 5: 155–8. [DOI] [PubMed] [Google Scholar]
- 18. Woo SH, Park IC, Park MJ et al. Arsenic trioxide induces apoptosis through a reactive oxygen species‐dependent pathway and loss of mitochondria membrane potential in HeLa cells. Int J Oncol 2002; 21: 57–63. [PubMed] [Google Scholar]
- 19. Takeshita A, Uehara A, Shinjo K et al. Impairment of heart rate variability control during arsenic trioxide treatment for acute promyelocytic leukemia. Leukemia 2004; 18: 647–8. [DOI] [PubMed] [Google Scholar]
- 20. Unnikrishnan D, Dutcher JP, Varshneya N et al. Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood 2001; 97: 1514–16. [DOI] [PubMed] [Google Scholar]
- 21. Li Y, Sun X, Wang L, Zhou Z, Kang YJ. Myocardial toxicity of arsenic trioxide in a mouse model. Cardiovasc Toxicol 2002; 2: 63–73. [DOI] [PubMed] [Google Scholar]
- 22. Shen J, Wu MH, Chen MH, Li QS, Shen ZY. The effects of arsenic trioxide on the transplanted hepatic carcinoma in mice. Chin J Cancer Biother 2002; 9: 450–4. (In Chinese.) [Google Scholar]
- 23. Verma RJ, Vasu A, Sayyed AA. Arsenic toxicity in mice and its possible amelioration. J Environ Sci (China) 2004; 16: 447–53. [PubMed] [Google Scholar]
- 24. Soucy NV, Ihnat MA, Kamat CD et al. Arsenic stimulates angiogenesis and tumorigenesis in vivo . Toxicol Sci 2003; 76: 271–9. [DOI] [PubMed] [Google Scholar]
- 25. Luo L, Qiao H, Meng F et al. Arsenic trioxide synergizes with B7H3‐mediated immunotherapy to eradicate hepatocellular carcinomas. Int J Cancer 2006; 118: 1823–30. [DOI] [PubMed] [Google Scholar]
- 26. Browder T, Butterfield CE, Kraling BM et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug‐resistant cancer. Cancer Res 2000; 60: 1878–86. [PubMed] [Google Scholar]
- 27. Kamat CD, Green DE, Curilla S et al. Role of HIF signalling on tumorigenesis in response to chronic low‐dose arsenic administration. Toxicol Sci 2005; 86: 248–57. [DOI] [PubMed] [Google Scholar]
- 28. Kao Y, Yu CL, Chang LW, Yu HS. Low concentrations of arsenic induce vascular endothelial growth factor and nitric oxide release and stimulate angiogenesis in vitro . Chem Res Toxicol 2003; 16: 460–8. [DOI] [PubMed] [Google Scholar]
- 29. Duyndam MC, Hulscher TM, Fontijn D, Pinedo HM, Boven E. Induction of vascular endothelial growth factor expression and hypoxia‐inducible factor 1α protein by the oxidative stressor arsenite. J Biol Chem 2001; 276: 48 066–76. [DOI] [PubMed] [Google Scholar]
- 30. Soucy NV, Mayka D, Klei LR, Nemec AA, Bauer JA, Barchowsky A. Neovascularization and angiogenic gene expression following chronic arsenic exposure in mice. Cardiovasc Toxicol 2005; 5: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu M, Xia L, Luo D, Waxman S, Jing Y. Dual effects of glutathione‐S‐transferase, π on As2O3 action in prostate cancer cells: enhancement of growth inhibition and inhibition of apoptosis. Oncogene 2004; 23: 3945–52. [DOI] [PubMed] [Google Scholar]
- 32. Roboz GJ, Dias S, Lam G et al. Arsenic trioxide induces dose‐ and time‐dependent apoptosis of endothelium and may exert an antileukemic effect via inhibition of angiogenesis. Blood 2000; 96: 1525–30. [PubMed] [Google Scholar]
- 33. Lew YS, Brown SL, Griffin RJ, Song CW, Kim JH. Arsenic trioxide causes selective necrosis in solid murine tumors by vascular shutdown. Cancer Res 1999; 59: 6033–7. [PubMed] [Google Scholar]
- 34. Duyndam MC, Hulscher STM, Van Der Wall E, Pinedo HM, Boven E. Evidence for a role of p38 kinase in hypoxia‐inducible factor 1‐independent induction of vascular endothelial growth factor expression by sodium arsenite. J Biol Chem 2003; 278: 6885–95. [DOI] [PubMed] [Google Scholar]
- 35. Li X, Ding X, Adrian TE, Path FRC. Arsenic trioxide causes redistribution of cell cycle, caspase activation, and GADD expression in human colonic, breast, and pancreatic cancer cells. Cancer Invest 2004; 22: 389–400. [DOI] [PubMed] [Google Scholar]
- 36. Chen YC, Lin‐Shiau SY, Lin JK. Involvement of reactive oxygen species and caspase 3 activation in arsenite‐induced apoptosis. J Cell Physiol 1998; 177: 324–33. [DOI] [PubMed] [Google Scholar]
- 37. Jing Y, Dai J, Chalmers‐Redman RM, Tatton WG, Waxman S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide‐dependent pathway. Blood 1999; 94: 2102–11. [PubMed] [Google Scholar]
- 38. Zhu AL, Liu LX, Piao DX, Lin YX, Zhao JP, Jiang HC. Liver regional continuous chemotherapy: use of femoral or subclavian artery for percutaneous implantation of catheter‐port systems. World J Gastroenterol 2004; 10: 1659–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gochi A, Orita K, Fuchimoto S, Tanaka N, Ogawa N. The prognosis advantage of preoperative intratumoral injection of OK‐432 for gastric cancer patients. Br J Cancer 2001; 84: 443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumagai T, Shih LY, Hughes SV et al. 19‐Nor‐1,25(OH) 2D2 (a novel, noncalcemic vitamin D analogue), combined with arsenic trioxide, has potent antitumor activity against myeloid leukemia. Cancer Res 2005; 65: 2488–97. [DOI] [PubMed] [Google Scholar]
- 41. Maeda H, Hori S, Ohizumi H et al. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and 1‐buthionine‐sulfoximine. Cell Death Differ 2004; 11: 737–46. [DOI] [PubMed] [Google Scholar]
- 42. Baumgartner M, Sturlan S, Roth E, Wessner B, Bachleitner‐Hofmann T. Enhancement of arsenic trioxide‐mediated apoptosis using docosahexaenoic acid in arsenic trioxide‐resistant solid tumor cells. Int J Cancer 2004; 112: 707–12. [DOI] [PubMed] [Google Scholar]