

Abstract
Melanogenesis substrate, N‐propionyl‐4‐S‐cysteaminylphenol (NPrCAP) is specifically taken up by melanoma cells and inhibits their growth by producing cytotxic free radicals. By taking advantage of this unique chemical agent, we have established melanoma‐targeting intracellular hyperthermia by conjugating NPrCAP with magnetite nanoparticles (NPrCAP/M) upon exposure to an alternating magnetic field (AMF). This treatment causes cytotoxic reaction as well as heat shock responses, leading to elicitation of antitumor immune response, which was proved by tumor rechallenge test and CTL induction. We found the level of heat shock protein 72 (Hsp72) to be increased in the cell lysate and culture supernatant after intracellular hyperthermia. Melanoma‐specific CD8+ T‐cell response to dendritic cells loaded with hyperthermia‐treated tumor lysate was enhanced when compared with non‐treated tumor lysate. When heat shock protein, particularly Hsp72, was immuno‐depleted from hyperthermia‐treated tumor cell lysate, specific CD8+ T‐cell response was abolished. Thus, it is suggested that antitumor immune response induced by hyperthermia using NPrCAP/M is derived from the release of HSP‐peptide complex from degraded tumor cells. Therefore, this chemo‐thermo‐immuno (CTI)‐therapy might be effective not only for primary melanoma but also for distant metastasis because of induction of systemic antimelanoma immune responses. (Cancer Sci 2010)
Melanoma has been increasing in incidence leading to a rise in morbidity and mortality in recent decades. Metastatic melanoma is extremely difficult to cure and continues to have a poor prognosis. Only 12% with metastatic melanoma survive for 5 years.( 1 ) The reason for this poor prognosis is the lack of effective conventional therapies. Various types of therapies such as immunotherapy, chemotherapy, and biologic therapy have been studied in melanoma management. However, a very modest effect was recorded in advanced malignant melanoma. Therefore, there is an emerging need for innovative therapies for the control of advanced melanoma.
It has been reported that the intracellular hyperthermia using magnetic nanoparticles is effective for treating certain types of cancer in not only primary but also metastatic lesions.( 2 , 3 , 4 , 5 , 6 , 7 , 8 ) Incorporated magnetic nanoparticles generate heat within the cells after exposure to the alternating magnetic field (AMF) due to hysteresis loss or relaxational loss.( 9 , 10 ) One of us has shown that hyperthermic treatment using magnetite cationic liposomes (MCLs), which are cationic liposomes containing 10‐nm magnetite nanoparticles, induces antitumor immunity by enhancement of heat shock protein (HSP) expression.( 3 , 11 , 12 , 13 ) We previously proposed that cross‐presentation of extracellular HSP‐peptide complex released from hyperthermia‐induced necrotic tumor cells is the mechanism for inducing antitumor immunity.( 7 ) In this paper, we present evidence that tumor‐derived HSP‐peptide complex is responsible for the hyperthermia‐mediated antitumor immunity.
In addition, exploitation of biological properties unique to melanoma cells may provide a novel approach to improve the effect of hyperthermic treatment. We have previously shown that the sulfur‐amine analog of tyrosine, 4‐S‐cysteaminyl phenol (4‐S‐CAP), and its N‐acetyl or propionyl derivatives (NAcCAP or NPrCAP) are good substrates for melanoma‐specific targeting and therapy. They have been shown to cause selective cytotoxicity against melanocytes and melanoma cells after selective uptake. Therefore, they can be good candidates for developing antimelanoma chemotherapy because melanogenesis is inherently toxic and expressed uniquely in melanocytic cells.( 14 , 15 , 16 , 17 ) Recently, we synthesized new magnetite nanoparticles, NPrCAP/M, on which NPrCAP was directly conjugated on the surface of magnetite nanoparticles.( 18 , 19 ) We have shown that NPrCAP/M is specifically targeted to melanoma cells, and internalized and aggregated within their cell cytoplasm. In addition, we have observed that B16 melanoma cells, which were subjected to intracellular hyperthermia using NPrCAP/M with AMF exposure, were brought to necrotic cell death, resulting in tumor growth retardation.( 18 ) Here we show that the intracellular hyperthermic treatment using NPrCAP/M with AMF exposure induces tumor‐specific immune responses and therefore we call this antimelanoma therapy “chemo‐thermo‐immuno (CTI)” therapy. We demonstrate that CTI therapy‐induced antimelanoma immunity is mediated through cross‐presentation of up‐regulated intracellular and extracellular HSPs‐peptide complex derived from melanoma cells.
Materials and Methods
Mice and cells. Female C57BL/6 mice were obtained from Hokudo (Sapporo, Japan) and used at 4–6 weeks of age. B16‐OVA is a B16F1 melanoma cells stably transfected with chicken ovalbumin (OVA) cDNA (kindly provided by Dr Y. Nishimura, Kumamoto University, Kumamoto, Japan). B16‐OVA was cultured in DMEM supplemented 10% FCS and 250 μg/mL of hygromycin B. B3Z is a CD8+ T‐cell hybridoma that expresses LacZ in response to activation of T‐cell receptors specific for the SIINFEKL peptide (SL8; OVA‐immunodominant peptide) in the context of H‐2Kb (kindly provided by Dr N. Shastri, University of California, Berkeley, CA, USA). B16F1 melanoma cells, CT26 colon carcinoma cells, and LLC lung carcinoma cells were cultured in DMEM supplemented with 10% FCS. EL4 lymphoma cells, YAC‐1 cells, and B3Z cells were cultured in complete RPMI supplemented with 10% FCS. Bone marrow‐derived dendritic cells (DCs) were generated from the femurs and tibiae of C57BL/6 mice. The bone marrow was flushed out, and the leukocytes were obtained and cultured in complete RPMI‐1640 with 10% FCS and 20 ng/mL GM‐CSF (Endogen, Woburn, MA, USA) for 5 days. On day 3, fresh medium with GM‐CSF was added to the plates for the day 5 cultures.
Preparation of NPrCAP/M. Magnetite nanoparticles (Fe3O4; average particle size, 10 nm) were kindly provided by Toda Kogyo (Hiroshima, Japan). The details of the preparation of NPrCAP/M are described elsewhere.( 18 ) Briefly, magnetite nanoparticles were coated with aminosilane and conjugated with NPrCAP via maleimide cross‐linkers. The resultant NPrCAP/M was suspended in 10 mL of H2O. The degree of incorporation of NPrCAP to magnetite was 61.0 nmol/mg magnetite.
Antibodies. For depletion of HSPs from cell lysate, anti‐Hsp72/Hsc73 mAb, anti‐Hsp90α polyclonal antibody, anti‐Hsp90 mAb, and anti‐lysine‐aspartic acid‐glutamic acid‐leucine (KDEL) mAb were used. Anti‐Hsp72/Hsc73 mAb and anti‐Hsp90mAb were used for western blotting. These antibodies were obtained from StressGen Biotechnologies Victoria, BC, Canada). Mouse IgG and Rabbit IgG were purchased from IBL (Takasaki, Japan).
Measurement of iron concentration in the NPrCAP/M‐exposed cells. Subconfluently growing melanoma and non‐melanoma cells (8 × 104/cm2) in a 25‐cm2 flask were re‐fed with the medium containing 5.94 mg of NPrCAP/M or magnetite (84 mg/mL). To discriminate between incorporation of NPrCAP/M and magnetite by direct attachment to cells and that by diffusion from the medium, culture flasks were fixed on a slanted disc and rotated slowly for 30 min. After the cells were washed with PBS twice and collected, they were dissolved completely in 200 μL of concentrated HCl and incubated at 43°C for 30 min. Then, 10 mL of H2O2 and 4 mL of 1% potassium thiocyanate were added in sequence to the cell solution. The concentration of NPrCAP/M used in these experiments was 24.4 μM as NPrCAP.
Transplantation of tumor cells and intracellular hyper‐thermia. All of the animal experiments were conducted with the approval of the Animal Experiment Ethics Committee of Sapporo Medical University. B16‐OVA cells (1 × 106) were subcutaneously transplanted into the right flank of C57BL/6 mice on day 0. NPrCAP/M nanoparticles (24.4 μM as NPrCAP, 100 μL) were injected subcutaneously into the tumor on days 7, 9, 11, and 13. A magnetic field was created using a horizontal coil (inner diameter, 7 cm; length, 7 cm) with a transistor inverter (LTG‐100‐05; Dai‐ichi High Frequency, Tokyo, Japan).( 4 ) The magnetic field frequency and intensity were 118 KHz and 30.6 KA/m (386 Oe), respectively. Twenty‐four hours after injection, mice were subjected to AMF exposure to heat the tumor at 43°C for 30 min. The heated field was three dimensions, which was created using a horizontal coil with a transistor inverter. The mice whose tumors were injected with NPrCAP/M were put in a horizontal coil and exposed to AMF. Moreover, we monitored the temperature of the tumor surface as well as the rectal temperature using an optical fiber probe. The temperature of the tumor surface was maintained at 43°C and rectal temperature was about 38°C. Tumor growth was recorded once every 2 days. The cured mice were then rechallenged with a subcutaneous injection of B16‐OVA cells (1 × 106) or irrelevant 3LL lung carcinoma cells (1 × 106) on the left flank. Tumor size was determined by the following formula: tumor volume = 0.5 × (length × width2), where length and width were measured in millimeters.
Histopathology of tumor sections. Twenty‐one days after tumor challenge, subcutaneous B16‐OVA tumors were harvested and fixed in 10% formalin in PBS, then paraffin embedded and sectioned. Hematoxylin–eosin (H&E)‐stained sections were prepared for analysis of therapeutic effect and gross infiltrate. For immunohistochemical analysis, the frozen tissues were stained with an antimouse CD4 mAb (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or an antimouse CD8 mAb (Chemicon International, Temecula, CA, USA) and then incubated with HRP‐conjugated goat antirat Ig (Dako, Tokyo, Japan), followed by hematoxylin counterstaining.
In vitro cytotoxicity assay. After mice were treated by intracellular hyperthermia as described above, spleens were harvested on day 28, then 5 × 106 spleen cells were restimulated in vitro with irradiated B16‐OVA cells in 2 mL of RPMI‐1640 supplemented with 50 μM of β‐mercaptethanol (Invitrogen, Carlsbad, CA, USA) and 10% FCS for 5 days. Cytotoxic activity of the effector cells against target cells (B16‐OVA, EL4, EL4 loaded with SL8 peptide and YAC‐1) was determined by standard 51Cr release assay.
Quantification of HSPs. Cultured B16‐OVA cells were exposed to NPrCAP/M for 30 min and irradiated by AMF to heat them at 43°C. After NPrCAP/M exposure with or without AMF irradiation, cells (1 × 106) were cultured in 1 mL of 10% RPMI for 72 h. Culture supernatant was collected at 12, 24, and 48 h, or cells escaping cell death were lysed at 72 h after intracellular hyperthermia by freezing and thawing and centrifugation at 2380 g for 5 min. The expression of Hsp72/Hsc73 and Hsp90 was determined by western blotting with an anti‐Hsp72/Hsc73 mAb or anti‐Hsp90 mAb. Heat shock protein (HSP) in the lysate or culture supernatant was quantified by the Hsp90α ELISA and Hsp70 ELISA kits (StressGen), which can detect and quantify Hsp90α and inducible Hsp72, respectively.
Cross‐presentation of antigen derived from hyperthermia‐treated tumor cell lysate by DCs. Cultured B16‐OVA cells were exposed to NPrCAP/M for 30 min and irradiated by AMF to heat them at 43°C. After NPrCAP/M exposure with or without AMF irradiation, cells were cultured for 72 h and 1 × 107 of cells in 1 mL of 10% RPMI medium were lysed by three cycles of freezing and thawing and centrifugation at 2380 g for 5 min. Dendritic cells DCs (1 × 105) derived from bone marrow of C57BL/6 mice were pulsed with the cell lysate (100 μL) and incubated with 1 × 105 B3Z T cell hybridoma. After overnight incubation, LacZ activity was measured by addition of 100 μL of chlorophenol red‐β‐d‐galactopyranoside (CPRG, Roche, Basel, Switzerland) solution. The absorbance was measured at 595 nm after 4‐h incubation at 37°C.
Immunodepletion of HSP. Cultured B16‐OVA cells were treated and lysed as described above. The cell lysate (100 μL) was incubated with antibodies (5 μg each) against Hsp90, Hsp72/Hsc73, KDEL, or all of them. The mixture was added to 10 μL of protein A‐Sepharose beads (50% slurry; GE Healthcare Japan, Tokyo, Japan), and the suspension was rotated at RT for 1 h. Then, the suspension was spun at 14 000 g for 1 min. After removal of the beads, the supernatant was used for cross‐presentation assay using B3Z as described above. Mouse IgG (15 μg) was used as an experimental control. Depletion was assessed by immunoblotting with anti‐Hsp90, Hsp72/Hsc73, or KDEL antibodies.
In vivo cross‐presentation of antigen derived from melanoma cells after CTI therapy. B16‐OVA cells (1 × 106) were transplanted into the footpads of C57BL/6 mice on day 0. NPrCAP/M nanoparticles (24.4 μM as NPrCAP, 100 μL) were injected into the tumor on days 7, 9, 11, and 13. Twenty‐four hours after injection, mice were subjected to AMF exposure to heat the tumor at 43°C for 30 min. Control mice were injected with PBS or NPrCAP/M nanoparticles alone without hyperthermia. After 5 h of the last CTI therapy against B16‐OVA, popliteal nodes were removed and DCs were isolated using CD11c MACS beads (Miltenyi Biotec).Then, B3Z cells (1 × 105) were added to the DC culture (1 × 105) in 96‐well flat‐bottom plates and incubated at 37°C. Twenty‐four hours after incubation, absorbance at 595 nm was measured.
Statistical analyses. All experiments were independently performed three times in triplicate. Comparisons between two groups were performed using Student’s t‐test. In the tumor transplantation assay, we determined statistical significance using Kruskal–Wallis one‐way analysis. In all experiments, differences were considered statistically significant at P < 0.05.
Results
N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanoparticles (NPrCAP/M) was preferentially incorporated into melanoma cells. To examine whether NPrCAP/M could be incorporated into melanoma cells more preferentially than magnetite alone, we compared amounts of iron molecules in cells after culture in the NPrCAP/M‐ or magnetite‐containing media. As shown in Figure 1, B16F1, B16‐OVA melanoma cells incorporated large amounts of iron derived from NPrCAP/M compared to that from magnetite alone. Non‐melanoma CT26 colon carcinoma and LLC lung carcinoma cells captured a small amount of NPrCAP/M; however, the amount was not significantly different from that with magnetite alone or was almost the same as for magnetite. These data suggested that NPrCAP/M nanoparticle was an ideal agent for specific targeting to melanoma cells.
Figure 1.
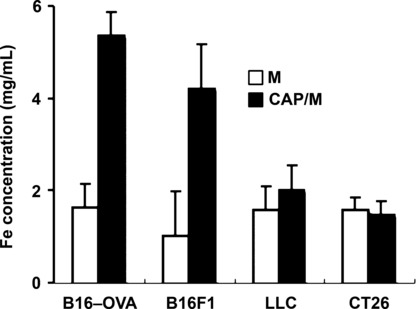
N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanopar‐ticles (NPrCAP/M) was preferentially incorporated into melanoma cells. Subconfluently growing melanoma and non‐melanoma cells (8 × 104/cm2) in a 25 cm2 flask were fed with medium containing 5.94 mg of NPrCAP/M or magnetite (84 mg/mL) for 30 min. Iron concentration of magnetite nanoparticles was measured using the potassium thiocyanate method.
Antitumor effect of intracellular hyperthermia using NPrCAP/M with AMF exposure and its ability to induce antitumor immunity. We have recently reported the efficacy of melanoma growth inhibition by combination therapy of NPrCAP/M administration with AMF exposure.( 19 ) B16F1 melanoma bearing mice treated with NPrCAP/M injection followed by hyperthermia showed apparent tumor growth retardation. In addition, we have observed that treatment with NPrCAP/M alone (without hyperthermia) also showed the growth suppression of B16F1 melanoma, indicating that NPrCAP/M has a chemotherapeutic effect. In this paper, we used B16‐OVA, which was B16 melanoma transfected with OVA as a surrogate tumor antigen, to examine the antigen‐specific antitumor immune response. Mice were transplanted with B16‐OVA melanoma cells and treated with NPrCAP/M injection followed by hyperthermia or NPrCAP/M injection alone as described in the Materials and Methods. Histopathological examination of the day 21 tumors without treatment showed that inflammatory infiltrates were poorly detected including CD8+ T cells and CD4+ T cells (Fig. 2a‐i, ‐ii, ‐iii, ‐iv). In contrast, treatment with combination of NPrCAP/M injection and hyperthermia induced apparent tumor destruction and necrosis with deposit of NPrCAP/M particles (Fig. 2a‐v, ‐vi). In addition, a dense inflammatory infiltrate including neutrophiles, macrophages, plasma cells, and lymphocytes was observed around the residual tumor cells (Fig. 2a‐vii, ‐viii). Furthermore, we have observed that this infiltrate included both CD8+ T cells and CD4+ T cells (Fig. 2a‐ix, ‐x). These T cells were hardly seen around the tumor without treatment or with NPrCAP/M without AMF exposure (data not shown). Tumor volume in the group treated by combination of NPrCAP/M injection and hyperthermia was significantly reduced compared with the non‐treated control group (P = 0.0025) and the group of NPrCAP alone (P = 0.023) (Fig. 2b). Six out of 10 mice in the group treated by NPrCAP/M injection and hyperthermia were cured. In contrast, all mice died of tumor burden in the treatment with NPrCAP/M without AMF and control groups. Interestingly, both NPrCAP/M with and without AMF exposure resulted in a significant and equal reduction of melanoma tumor volume by 17 days after tumor challenge. However, the tumors of mice treated with NPrCAP/M without AMF exposure grew rapidly after day 17. This suggested that hyperthermia might be required for the complete elimination of melanoma tumors. To examine whether cured mice developed antitumor immune responses, these mice were rechallenged with live B16‐OVA melanoma cells or irrelevant mouse lung carcinoma LLC 4 weeks after NPrCAP/M and hyperthermic treatment. As a result, all cured mice rejected a rechallenge of live B16‐OVA melanoma cells, but not LLC lung carcinoma cells (Fig. 2c). These data indicated that intracellular hyperthermia using NPrCAP/M with AMF exposure induced specific antitumor immunity.
Figure 2.
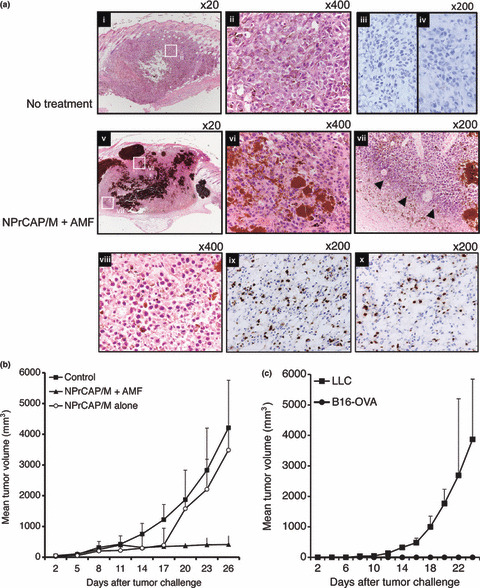
Antitumor effect and tumor‐specific immunity induced by intracellular hyperthermia using N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanoparticles (NPrCAP/M) and alternating magnetic field (AMF) exposure. (a) Pre‐established subcutaneous B16‐OVA tumors without treatment (i,ii,iii,iv) or intracellular hyperthermia using NPrCAP/M with AMF exposure (v,vi,vii,viii,ix,x) were harvested and analyzed histologically using H&E‐stained sections. Intracellular hyperthermia using NPrCAP with AMF exposure induced tumor destruction and inflammatory infiltrate (arrow head). The frozen tissues excised from mice untreated or treated with NPrCAP/M with AMF exposure were stained with an antimouse CD8 mAb (iii,ix) or CD4 mAb (iv,x). (b) Tumor growth of B16‐OVA melanoma cells in non‐treated control mice (n = 10), mice treated with NPrCAP/M with AMF exposure (n = 10), or mice treated with NPrCAP/M alone (n = 10). Points, mean tumor volume (mm3); bars, SD. (c) The development of tumor specific immunity by intracellular hyperthermia. Mice cured by intracellular hyperthermia were rechallenged with B16‐OVA melanoma cells (n = 3) or lung carcinoma LLC (n = 3). Mice were transplanted with 1 × 106 B16‐OVA cells and then the tumor growth rates of each group were compared using the average tumor size. Points, mean tumor volume (mm3); bars, SD. Results shown are representative of three different experiments.
Induction of tumor‐specific CTL by intracellular hyper‐thermia. To analyze the mechanism for the generation of antitumor immunity by NPrCAP/M and hyperthermia, we examined CTL induction in mice after intracellular hyperthermia. Spleen cells of mice after hyperthermia showed high cytotoxicity against B16‐OVA melanoma cells compared to EL4 lymphoma and YAC‐1 cells. In addition, spleen cells also showed high cytotoxicity against EL4 pulsed with SL8 peptide derived from OVA protein (Fig. 3). These results suggest that intracellular hyperthermia using NPrCAP/M can elicit specific tumor immunity by inducing CTL against B16‐OVA melanoma cells.
Figure 3.
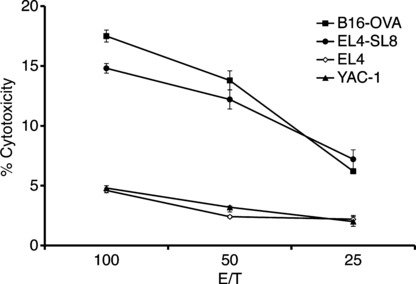
Induction of tumor‐specific CTL by intracellular hyperthermia. Analysis of spelenocytes cytotoxic activity against B16‐OVA melanoma cells, EL4, EL4 pulsed with SL8 peptide, and YAC‐1 in 51Cr release assay. Mice were treated by N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanoparticles (NPrCAP/M) injection followed by alternating magnetic field (AMF) exposure. Four weeks after treatment, spleen cells were removed and stimulated with irradiated B16‐OVA cells. Cytotoxic activity of spleen cells against B16‐OVA cells, EL4 cells, EL4 cells pulsed with SL8 peptide, or YAC‐1 cells was determined by standard 51Cr‐release assay. Bars, SD.
Enhanced expression of Hsp72 in B16‐OVA melanoma cell after intracellular hyperthermia. We examined the expression of HSPs in tumor cells treated with NPrCAP with AMF exposure in vitro by western blotting and ELISA. The protein level of Hsp72 but not Hsc73 or Hsp90 was increased at 48 h after hyperthermia (Fig. 4a). Similarly, the concentration of Hsp72 in cell lysate resulted in a three‐fold increase at 72 h after hyperthermia, compared to cells without treatment (Fig. 4b). However, the concentration of Hsp90 did not change (Fig. 4c). Treatment with NPrCAP alone decreased the level of intracellular Hsp72 and Hsp90. We are currently investigating the underlying mechanism. From the results obtained, we hypothesized that hyperthermia using NPrCAP/M might induce tumor immunity through up‐regulation of Hsp72.
Figure 4.
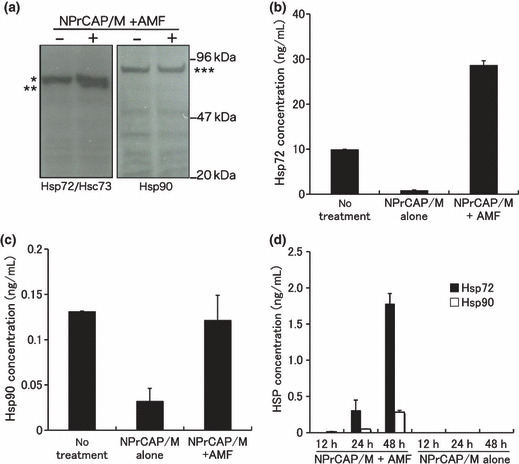
Heat shock protein (HSP) expression in tumor cells after intracellular hyperthermia. (a–c) B16‐OVA cells were subjected to hyperthermia using N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanoparticles (NPrCAP/M) with alternating mag‐netic field (AMF) exposure in vitro. Seventy‐two hours after intracellular hyperthermia (+) or left untreated (–), the expression of Hsp72 and Hsp90 was determined by western blotting with an anti‐Hsp72/Hsc73 mAb or anti‐Hsp90 mAb (a). *Hsc73, **Hsp72, ***Hsp90. The expression of Hsp72 was markedly enhanced by intracellular hyperthermia. Heat shock protein (HSP) in the lysate was quantified by the Hsp72 ELISA (b) and Hsp90 ELISA kits (c). (d) Hsp72 and Hsp90 in the 12‐, 24‐ and 48‐h culture supernatant after intracellular hyperthermia were measured using ELISA. Bars, SD. Results shown are representative of three different experiments.
Intracellular hyperthermia using NPrCAP/M with AMF exposure results in the release of HSPs into the extracellular milieu. It has been demonstrated that once cancer cells become necrotic, several HSPs, above all, Hsp72 and Hsp90, are released from cells and might act as a danger signal, subsequently eliciting cell‐specific immune responses. We therefore examined whether Hsp72 and Hsp90 would be released from necrotic melanoma cells after intracellular hyperthermia in vitro. Culture supernatants from B16‐OVA were collected at 12, 24, and 48 h after intracellular hyperthermia and the quantity of Hsp72 and Hsp90 was evaluated using ELISA. Although Hsp72 and Hsp90 were detected at 48 h after hyperthermia, concentration of extracellular Hsp72 was a 4.5‐fold higher than that of Hsp90 (Fig. 4d). These in vitro results suggested that treatment of B16‐OVA melanoma with intracellular hyperthermia would release HSPs, in particular Hsp72, into extracellular milieu in vivo and these extracellular HSPs might play an important role in inducing antitumor immunity.
CD8+ T‐cell response against DCs loaded with B16‐OVA melanoma cell lysate after intracellular hyperthermia. To analyze the mechanism of tumor specific CTL induction, we examined B3Z CD8+ T‐cell response against DCs loaded with supernatant from B16‐OVA culture after intracellular hyperthermia. However, only a very modest response was observed (data not shown). One of the reasons for this modest response may be the degradation of peptide chaperoned by HSPs by protease in the culture medium. We therefore decided to use melanoma cell lysate after hyperthermia. NPrCAP/M loaded B16‐OVA melanoma cells were subjected to AMF irradiation and cultured for 72 h. Melanoma cells were lysed by three cycles of freezing and thawing. Dendritic cells (DCs) derived from mouse bone marrow were loaded with the lysate for 2 h and then cultured with B3Z CD8+ T‐cell hybridoma. B3Z response against DC loaded with B16‐OVA melanoma cell lysate increased after intracellular hyperthermia using NPrCAP/M, compared with non‐heated cells and cells loaded NPrCAP/M without AMF exposure (Fig. 5a). These data demonstrated that loading DCs with lysate derived from melanoma cells treated with hyperthermia enhanced the cross‐presentation of B16‐OVA‐specific antigen peptide.
Figure 5.
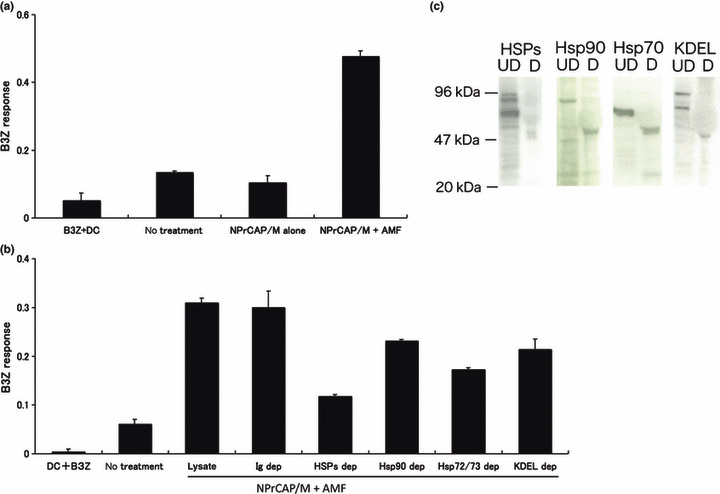
CD8+ T‐cell response against dendritic cells (DCs) pulsed with B16‐OVA melanoma cell lysate after intracellular hyperthermia and effect of heat shock protein (HSP) depletion. (a) B16‐OVA cells were subjected to N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanoparticles (NPrCAP/M) exposure and alternating magnetic field (AMF) irradiation, then lysed by freezing and thawing. Dendritic cells (DCs) were pulsed with the cell lysate, and cultured overnight with B3Z and the absorbance of β‐galactosidase activity was measured at 595 nm. Bars, SD. (b) Lysates of the cells were immunoprecipitated with anti‐Hsp72/Hsc73 antibody, anti‐Hsp90 antibody and anti‐KDEL antibody. Immunoprecipitates were removed from lysate. After being pulsed with the HSP‐depleted lysate, DCs were incubated with B3Z T cell hybridoma and the absorbance of β ‐galactosidase activity was measured at 595 nm. Bars, SD. (c) Cell lysates were depleted of HSPs (above lanes) with antibodies specific for each HSP. Immunoblots were of depleted (D) or undepleted (UD) lysates. Results shown are representative of five different experiments.
Effects of immunodepletion of HSPs on CD8+ T‐cell response. Next, we investigated the underlying mechanism responsible for the enhancement of cross‐presentation by intracellular hyperthermia. Hyperthermia has long been shown to induce the expression of HSPs within tumor cells, which have been shown to chaperone tumor‐associated antigen peptides. To investigate the role of HSPs in intracellular hyperthermia‐induced CD8+ T‐cell response, we depleted HSPs from lysate using anti‐HSP antibody, and measured CD8+ T‐cell response against DCs loaded with the HSP‐depleted lysate. Depletion of major HSPs (Hsp72/Hsc73, Hsp90, and ER‐resident HSPs) from NPrCAP/M and hyperthermic treated B16‐OVA cell lysate caused a loss of 59% of initial B3Z response (P = 0.0001 vs depletion with control Ig) (Fig. 5b), whereas depletion with control Ig did not show any effect. Importantly, depletion of Hsp72/Hsc73 exhibited a 44% reduction of activity and it was best decreased in response in the HSP depletion assay. The inhibition rate was statistically significant compared with the depletion with control Ig (P = 0.001). Depletion of Hsp90 or ER resident HSPs caused a loss of 25% (P = 0.0857) or 31% (P = 0.0034) of the initial activity, respectively. Immunoblots showed that depletion of each HSP was complete (Fig. 5c). These results suggested that Hsp72/Hsc73, Hsp90, and ER‐resident HSPs were involved in the induction of CTL response at various extents. In addition, our data demonstrated that these HSPs chaperoned antigenic peptides and extracellular HSP‐peptide complexes were cross‐presented by DCs, followed by specific CTL activation. Notably, Hsp72/Hsc73 was largely responsible for the observed T‐cell response. As shown in Figure 4, these data were consistent with the enhanced expression of Hsp72 within the melanoma cells. Thus, DCs loaded with intracellular hyperthermia‐treated melanoma cell lysate are more efficient than DCs loaded with untreated melanoma cell lysate in cross‐presentation to CTLs.
Dendritic cells (DCs) derived from tumor‐draining lymph nodes cross‐present melanoma‐associated antigen after CTI therapy. We examined whether melanoma‐associated antigen was indeed cross‐presented by DCs within tumor‐draining lymph nodes after CTI therapy. To test this, we transplanted B16 melanoma cells into the footpads of mice. After three rounds of CTI therapy, tumor‐draining popliteal nodes were removed and DCs were isolated and cultured with B3Z. B3Z response against regional lymph node‐derived DCs of CTI‐treated mice was evident when compared to PBS control or NPrCAP/M injection without hyperthermia (Fig. 6). We therefore conclude that intracellular hyperthermia using NPrCAP/M with AMF exposure promotes OVA‐derived peptide presentation on DCs both in vitro and in vivo.
Figure 6.
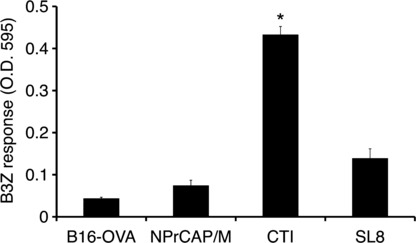
Dendritic cells (DCs) derived from tumor‐draining lymph nodes cross‐present melanoma‐associated antigen after CTI therapy. B16‐OVA cells (1 × 106) were transplanted into the footpads of C57BL/6 mice on day 0. N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanoparticles (NPrCAP/M) nanoparticles (24.4 mM as NPrCAP, 100 mL) were injected into the tumor on days 7, 9, 11, and 13. Twenty‐four hours after injection, mice were subjected to alternating magnetic field (AMF) exposure to heat the tumor at 43°C for 30 min. Control mice were injected with PBS or NPrCAP/M nanoparticles alone without hyperthermia. After 5 h of the last CTI therapy against B16‐OVA, tumor‐draining popliteal nodes were removed and DCs were isolated, then cultured with B3Z cells. Twenty‐four hours after incubation, absorbance at 595 nm was measured. Bars, SD. *P < 0.01; paired Student’s t‐test. Data are representative of three independent experiments.
Discussion
In this study, we showed that intracellular hyperthermia of melanoma cells using NPrCAP/M with AMF exposure elicited antitumor immune responses via cross‐presentation of HSP‐chaperoned antigen. Moreover, we demonstrated that DCs derived from tumor‐draining lymph nodes indeed cross‐presented melanoma‐associated antigen after CTI therapy. It has been believed that enhanced expression of intracellular HSPs by hyperthermia plays an important role in the induction of antitumor immunity.( 11 , 20 ) Moreover, it has been demonstrated that overexpression of HSPs, particularly Hsp72, causes increased tumor immunogenicity due to augmentation of the chaperoning ability of antigenic peptide, thereby augmenting the presentation of antigenic peptide in the context of MHC class I molecules.( 21 , 22 ) However, in order to prime tumor‐specific immunity, it is necessary to present tumor antigen in the context of MHC class I in conjunction with the co‐stimulation signal through co‐stimulatory molecules such as B7.1 and B7.2 by professional antigen‐presenting cells such as DCs. Dendritic cells (DCs) have the unique capacity to take up, process, and present exogenous antigens in association with MHC class I molecules. This process is termed cross‐presentation and the resulting CD8+ T‐cell priming is referred to as cross‐priming. It has been demonstrated that some exogenous antigens such as HSPs( 23 , 24 , 25 , 26 , 27 ) and particulate protein antigens( 28 ) gain access to the MHC class I processing pathway and initiate CTL responses. This exogenous pathway is important for the development of CD8+ CTL responses against tumors and infectious pathogens that do not have access to the classical MHC class I pathway.
Here, we showed that the HSPs‐antigen peptide complex released from melanoma cells treated with intracellular hyperthermia is taken up by DCs and cross‐presented HSP‐chaperoned peptide in the context of MHC class I molecules (Fig. 7). Our CTI therapy induced NPrCAP‐ as well as heat‐mediated melanoma cell necrosis to NPrCAP/M incorporated cells. We have reported that repeated hyperthermia (three cycles of NPrCAP/M injection and AMF irradiation) was required to induce the maximal antitumor immune responses.( 19 ) If melanoma cells escape from necrotic cell death, repeated hyperthermia should produce necrotic cell death of previously heat shocked‐melanoma cells in which HSPs were induced. In fact, our data suggested that Hsp72/Hsc73, Hsp90, and ER‐resident HSPs participated in the induction of CD8+ T‐cell response. In particular, among HSPs, Hsp72 was largely responsible for the augmented antigen presentation to CD8+ T cells. As Hsp72 is known to up‐regulate in response to hyperthermia or heat shock treatment,( 11 ) newly synthesized Hsp72 has a chance to bind to the heat‐denatured melanoma‐associated antigen.
Figure 7.
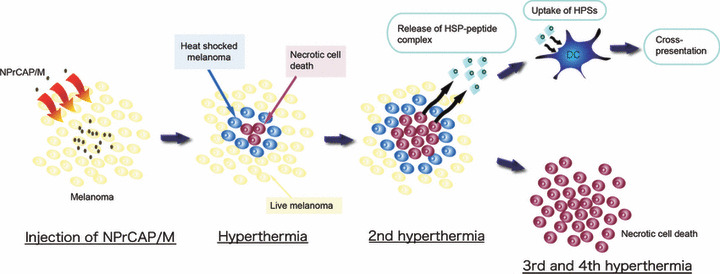
Schema of intracellular hyperthermia using N‐propionyl‐4‐S‐cysteaminylphenol with magnetite nanoparticles (NPrCAP/M) with alternating magnetic field (AMF) exposure. Injected NPrCAP/M nanoparticles are specifically incorporated in melanoma cells. Intracellular hyperthermia can induce necrotic cell death and adjacent live melanoma cells suffer heat shock, resulting in increased level of intracellular heat shock protein (HSP)‐peptide complexes. Repeated hyperthermia turns heat‐shocked cells to necrotic cells, leading to the release of their intracellular contents, including HSPs‐peptide complexes, into extracellular milieu. The released HSPs‐peptide complexes are taken up by dendritic cells (DCs). Then, DCs migrate into regional lymph nodes and cross‐present HSP‐chaperoned antigenic peptides to CD8+ T cells in the context of MHC class I molecules, thereby inducing antimelanoma CTLs. Finally, the remaining melanoma cells are killed by repeated hyperthermia or by the melanoma‐specific CTLs.
Taken together, intracellular hyperthermia using NPrCAP/M is a promising treatment for improvement of clinical effects, especially for patients with advanced metastatic melanomas, and even for prevention of recurrence and/or metastasis for early melanomas because of induction of systemic antimelanoma immunity.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
We thank Dr Shastri for providing B3Z T cell hybridoma, Dr Nishimura for providing B16‐OVA, and Toda Kogyo Co. for providing the magnetite. This work was supported by a Health and Labor Sciences Research Grant‐in‐Aid for Research on Advanced Medical Technology from the Ministry of Health, Labor and Welfare of Japan.
References
- 1. Balch CM, Buzaid AC, Soong SJ et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol 2001; 19: 3635–48. [DOI] [PubMed] [Google Scholar]
- 2. Yanase M, Shinkai M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T. Intracellular hyperthermia for cancer using magnetite cationic liposomes: an in vivo study. Jpn J Cancer Res 1998; 89: 463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kawai N, Ito A, Nakahara Y et al. Anticancer effect of hyperthermia on prostate cancer mediated by magnetite cationic liposomes and immune‐response induction in transplanted syngeneic rats. Prostate 2005; 64: 373–81. [DOI] [PubMed] [Google Scholar]
- 4. Ito A, Shinkai M, Honda H, Kobayashi T. Medical application of functionalized magnetic nanoparticles. J Biosci Bioeng 2005; 100: 1–11. [DOI] [PubMed] [Google Scholar]
- 5. Yanase M, Shinkai M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T. Antitumor immunity induction by intracellular hyperthermia using magnetite cationic liposomes. Jpn J Cancer Res 1998; 89: 775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ito A, Shinkai M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T. Augmentation of MHC class I antigen presentation via heat shock protein expression by hyperthermia. Cancer Immunol Immunother 2001; 50: 515–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ito A, Kobayashi T, Honda H. A mechanism of antitumor immunity induced by hyperthermia. Jpn J Hyperthermic Oncol 2005; 21: 1–19. [Google Scholar]
- 8. Ito A, Takeshi K. Intracellular hyperthermia using magnetite nanoparticles: a novel method for hyperthermia clinical applications. Thermal Medicine 2008; 24: 113–29. [Google Scholar]
- 9. Shinkai M, Yanase M, Honda H, Wakabayashi T, Yoshida J, Kobayashi T. Intracellular hyperthermia for cancer using magnetite cationic liposomes: in vitro study. Jpn J Cancer Res 1996; 87: 1179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hergt R, Dutz S, Mueller R, Zeisberger M. Magnetite particle hyperthermia: nanoparticle magnetism and materials development for cancer therapy. J Phys Condens Matter 2006; 18: S2919–34. [Google Scholar]
- 11. Ito A, Shinkai M, Honda H et al. Heat shock protein 70 expression induces antitumor immunity during intracellular hyperthermia using magnetite nanoparticles. Cancer Immunol Immunother 2003; 52: 80–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ito A, Matsuoka F, Honda H, Kobayashi T. Antitumor effects of combined therapy of recombinant heat shock protein 70 and hyperthermia using magnetic nanoparticles in an experimental subcutaneous murine melanoma. Cancer Immunol Immunother 2004; 53: 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ito A, Honda H, Kobayashi T. Cancer immunotherapy based on intracellular hyperthermia using magnetite nanoparticles: a novel concept of “heat‐controlled necrosis” with heat shock protein expression. Cancer Immunol Immunother 2006; 55: 320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito Y, Jimbow K. Selective cytotoxicity of 4‐S‐cysteaminylphenol on follicular melanocytes of the black mouse: rational basis for its application to melanoma chemotherapy. Cancer Res 1987; 47: 3278–84. [PubMed] [Google Scholar]
- 15. Miura S, Ueda T, Jimbow K, Ito S, Fujita K. Synthesis of cysteinylphenol, cysteaminylphenol, and related compounds, and in vivo evaluation of antimelanoma effect. Arch Dermatol Res 1987; 279: 219–25. [DOI] [PubMed] [Google Scholar]
- 16. Miura T, Jimbow K, Ito S. The in vivo antimelanoma effect of 4‐S‐cysteaminylphenol and its n‐acetyl derivative. Int J Cancer 1990; 46: 931–4. [DOI] [PubMed] [Google Scholar]
- 17. Thomas PD, Kishi H, Cao H et al. Selective incorporation and specific cytocidal effect as the cellular basis for the antimelanoma action of sulphur containing tyrosine analogs. J Invest Dermatol 1999; 113: 928–34. [DOI] [PubMed] [Google Scholar]
- 18. Sato M, Yamashita T, Ohkura M et al. N‐propionyl‐ cysteaminylphenol ‐magnetite conjugate (NPrCAP/M) is a nanoparticle for the targeted growth suppression of melanoma cells. J Invest Dermatol 2009; 129: 2233–41. [DOI] [PubMed] [Google Scholar]
- 19. Takada T, Yamashita T, Sato M et al. Growth inhibition of re‐challenge B16 melanoma transplant by conjugates of melanogenesis substrate and magnetite nanoparticles as the basis for developing melanoma‐targeted chemo‐thermo‐immunotherapy. J Biomed Biotechnol 2009; 2009: 457936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mise K, Kan N, Okino T et al. Effect of heat treatment on tumor cells and antitumor effector cells. Cancer Res 1990; 50: 6199–202. [PubMed] [Google Scholar]
- 21. Wells AD, Rai SK, Salvato MS, Band H, Malkovsky M. Hsp72‐mediated augmentation of MHC class I surface expression and endogenous antigen presentation. Int Immunol 1998; 10: 609–17. [DOI] [PubMed] [Google Scholar]
- 22. Ito A, Matsuoka F, Honda H, Kobayashi T. Heat shock protein 70 gene therapy combined with hyperthermia using magnetic nanoparticles. Cancer Gene Ther 2003; 10: 918–25. [DOI] [PubMed] [Google Scholar]
- 23. Udono H, Srivastava PK. Comparison of tumor‐specific immunogenicities of stress‐induced proteins gp96, hsp90, and hsp70. J Immunol 1994; 152: 5398–403. [PubMed] [Google Scholar]
- 24. Tamura Y, Peng P, Liu K, Daou M, Srivastava PK. Immunotherapy of tumors with autologous tumor‐derived heat shock protein preparations. Science 1997; 278: 117–20. [DOI] [PubMed] [Google Scholar]
- 25. Moroi Y, Mayhew M, Trcka J et al. Induction of cellular immunity by immunization with novel hybrid peptides complexed to heat shock protein 70. Proc Natl Acad Sci USA 2000; 97: 3485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kurotaki T, Tamura Y, Ueda G et al. Efficient Cross‐Presentation by Heat Shock Protein 90‐Peptide Complex‐Loaded Dendritic Cells via an Endosomal Pathway. J Immunol 2007; 179: 1803–13. [DOI] [PubMed] [Google Scholar]
- 27. Kutomi G, Tamura Y, Okuya K et al. Targeting to static endosome is required for efficient cross‐presentation of endoplasmic reticulum‐resident oxygen‐regulated protein 150‐peptide complexes. J Immunol 2009; 183: 5861–9. [DOI] [PubMed] [Google Scholar]
- 28. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP‐independent MHC class I crosspresentation in vivo. Immunity 2004; 21: 155–65. [DOI] [PubMed] [Google Scholar]