

Abstract
Almost all cancer cells have multiple epigenetic abnormalities, which combine with genetic changes to affect many cellular processes, including cell proliferation and invasion, by silencing tumor‐suppressor genes. In this review, we focus on the epigenetic mechanisms of DNA hypomethylation and CpG island hypermethylation in gliomas. Aberrant hypermethylation in promoter CpG islands has been recognized as a key mechanism involved in the silencing of cancer‐associated genes and occurs at genes with diverse functions related to tumorigenesis and tumor progression. Such promoter hypermethylation can modulate the sensitivity of glioblastomas to drugs and radiotherapy. As an example, the methylation of the O6‐methylguanine DNA methyltransferase (MGMT) promoter is a specific predictive biomarker of tumor responsiveness to chemotherapy with alkylating agents. Further, we reviewed reports on pyrosequencing – a simple technique for the accurate and quantitative analysis of DNA methylation. We believe that the quantification of MGMT methylation by pyrosequencing might enable the selection of patients who are most likely to benefit from chemotherapy. Finally, we also evaluated the potential of de novo NY‐ESO‐1, the most immunogenic cancer/testis antigen (CTA) discovered thus far, as an immunotherapy target. The use of potent epigenetics‐based therapy for cancer cells might restore the abnormally regulated epigenomes to a more normal state through epigenetic reprogramming. Thus, epigenetic therapy may be a promising and potent treatment for human neoplasia. (Cancer Sci 2010)
Epigenetic abnormalities in human neoplasia
Aberrant epigenetic mechanisms, such as promoter hypermethylation, histone modifications, or non‐coding RNA expression, are recognized as being important in tumor formation.( 1 ) These epigenetic mechanisms comprise the “third pathway” in Knudson’s model of tumor‐suppressor gene inactivation and can affect gene expression without effecting genetic changes.( 2 , 3 ) It is now known that almost all types of cancer cells have multiple epigenetic abnormalities, which combine with genetic changes to affect many cellular processes, including cell proliferation and invasion, by silencing tumor‐suppressor genes.( 4 ) Therefore, research on epigenetic dysregulation is now as commonplace as that on the genetic etiology of human cancers. The fundamental components of cancer epigenetics are the DNA methylation pattern; nucleosome remodeling; and a series of acetylation, methylation, and other modifications at key amino acid residues in histones. Overall, these epigenetic alterations disturb the chromatin structure, leading to abnormal gene expression and tumor formation.
During the early development of mammals, DNA methylation occurs at the cytosine residues in cytosine‐guanine sequences (CpGs) in the DNA; this methylation pattern is heritable.( 5 ) In humans, approximately 70% CpG dinucleotides, which are generally located in repetitive DNA sequences, are methylated. Other than the methylated CpGs, clusters of unmethylated CpGs are present in the genome, and these clusters are called CpG islands.( 6 , 7 ) Around 60% of genes have CpG islands in their 5′‐promoter regions, and DNA methylation of these islands results in irreversible inhibition of gene expression. A recent genome‐wide analysis revealed that CpG islands are also found in non‐promoter regions: around 50% of the CpG islands are not present in association with annotated promoters. In addition, epigenetic abnormalities causing the loss of gene function are more frequent than genetic abnormalities in cancer cells.( 4 , 8 , 9 )
Thus, cellular epigenetic inheritance mediated by aberrant DNA methylation that leads to gene silencing, gene imprinting, and/or activation of cancer‐associated genes is now accepted as an important factor defining the transformed phenotype.
DNA hypomethylation and CpG island hypermethylation in gliomas
Hypomethylation has been reported to occur in repetitive elements localized in satellite sequences or pericentromeric regions, thus resulting in genomic instability in several cancers including glioblastoma.( 10 , 11 ) Genome‐wide hypomethylation occurs at a high frequency (∼80%) in primary glioblastomas.( 11 , 12 , 13 ) The level of hypomethylation ranges from nearly normal to approximately 50% of the normal level in approximately 20% cases; the latter reflects the massive demethylation of approximately 10 million CpG sites per tumor cell.
In glioblastomas, both repetitive sequences and single‐copy loci can be hypomethylated. Glioblastomas with global hypomethylation show remarkable hypomethylation (22–55% of normal brain) of the tandem repeat satellite (Sat2) at the pericentromeric regions of chromosomes 1, 9, and 16, and moderate hypomethylation (71–82% of normal brain) of D4Z4 at the subtelomeric regions of chromosomes 4q35 and 10q26.( 11 , 13 ) Although it is not as frequent as regional hypermethylation in promoter CpG islands, regional hypomethylation at single‐copy loci in glioblastomas is associated with the activation of some cancer‐associated genes, such as the melanoma antigen gene (MAGEA1).( 14 ) Activation of the cancer‐testis antigen genes by the epigenetic regulation is described in greater detail in the following section. Another example associated with hypomethylation in glioma is the loss of imprinting of the insulin‐like growth factor 2 (IGF‐2) gene,( 15 ) and it has been known to induce tumor development.( 16 )
Locus‐specific hypermethylation, which mostly occurs at promoter CpG islands, is also frequently observed in gliomas. In gliomas as well as other malignancies, CpG island hypermethylation in promoters occurs in genes with diverse functions related to tumorigenesis and tumor progression, DNA repair, apoptosis, angiogenesis, invasion, and drug resistance. For example, promoter hypermethylation in the genes CDKN/p16, RB, PTEN, TP53, and p14 ARF affects the RB, PI3K, and p53 pathways.( 17 , 18 , 19 , 20 , 21 ) In glioma cells, CpG island hypermethylation may occur at genes that are not expressed in the brain; this suggests that not all instances of CpG island methylation are functionally important for tumorigenesis. Loss of heterozygosity in chromosome 19q that is frequently observed in gliomas has suggested the presence of a tumor suppressor gene.( 22 , 23 , 24 ) Screens for promoter hypermethylation in glioma discovered a candidate tumor suppressor, epithelial membrane protein 3 (EMP3).( 25 ) EMP3 is a myelin‐related gene involved in cell proliferation and cell–cell interaction.
Methylation of the MGMT promoter and the resultant response to drugs and radiotherapy
Promoter hypermethylation can modulate the sensitivity of glioblastomas to drugs and radiotherapy. This area has been intensively investigated. One example is suppressor of cytokine signaling 1 (SOCS1), which is silenced by hypermethylation. Re‐activation of SOCS1 in glioma sensitized to radiation via inactivation of the MAPK pathway.( 26 ) Epigenetic profiling might shed light on the catalog of glioma and patient‐specific therapy. The best known example is MGMT promoter methylation and the resultant response to DNA alkylating agents. An alkylating agent kills cells by forming cross‐links between adjacent strands of DNA, thus inhibiting DNA replication. However, the effectiveness of such alkylating agents is frequently hampered by inherent or acquired resistance. The main determinant of the resistance to alkylating agents is the activity of MGMT, which directly and specifically removes the cytotoxic alkyl adducts formed at the O6 position of guanine by these alkylating agents.( 27 ) Because the MGMT gene is not usually mutated or deleted, DNA methylation – the main epigenetic modification seen in cancer cell lines and primary tumors – may cause a reduction in the MGMT levels.( 28 ) These observations highlight the importance of MGMT methylation as a specific predictive biomarker for the responsiveness to chemotherapy with alkylating agents. Indeed, MGMT methylation is associated with significantly longer survival in glioblastoma and other gliomas treated with radiation and alkylating agents, such as temozolomide (TMZ).( 29 , 30 ) Although it is unclear if this is directly due to reduced MGMT expression, the predictive value of MGMT methylation is largely confirmed in a number of prospective and retrospective clinical investigations. The clinical outcome after TMZ therapy depends on the methylation status of the MGMT promoter; MGMT modification is one of the key factors that could enhance the clinical benefits of this treatment. In a previous in vitro study on human glioma cells, we found that β‐interferon markedly enhanced chemosensitivity to TMZ;( 31 ) this finding suggested that one of the major mechanisms by which β‐interferon enhances chemosensitivity is the down‐regulation of MGMT transcription via p53 induction. This effect was also observed in an experimental animal model.( 32 ) The results of these two studies suggested that compared to chemotherapy with TMZ alone and concomitant radiotherapy, chemotherapy with β‐interferon and temozolomide with concomitant radiotherapy might further improve the clinical outcome of malignant gliomas.
Pyrosequencing for the quantification of DNA methylation
MGMT levels vary widely according to the type of tumor and also among tumors of the same type.( 33 ) For example, approximately 30% gliomas have reduced levels of MGMT because of methylation of the corresponding gene, whereas MGMT methylation is rare or absent in other tumor types.( 34 , 35 , 36 ) Some reports describe the detection of MGMT methylation based on a modified methylation‐specific polymerase chain reaction (MSP).( 37 , 38 ) This method enables cost‐efficient analysis of MGMT promoter methylation. However, it is not quantitative and poses the risk of false‐positive or false‐negative results, especially when the DNA quality and/or quantity are low; this is often the case in clinical settings wherein samples are typically obtained from formalin‐fixed paraffin‐embedded specimens. Further technical studies are necessary for the development of an MGMT methylation assay. Mikeska et al. compared and optimized three quantitative techniques of MGMT methylation, that is combined bisulfite restriction analysis (COBRA), SNuPE ion pair‐reverse phase high‐performance liquid chromatography, and pyrosequencing.( 39 ) They concluded that pyrosequencing assay provides the most accurate and most robust MGMT methylation marker.
Pyrosequencing technology is based on the sequencing‐by‐synthesis principle, and it involves a simple technique for accurate and quantitative analysis of DNA sequences.( 40 , 41 , 42 ) In this method, a sequencing primer is hybridized to a single‐stranded, PCR‐amplified DNA template and incubated with a DNA polymerase. Each event of incorporation of a deoxynucleotide triphosphate (dNTP) is accompanied by the release of pyrophosphate (PPi) in a quantity equimolar to the amount of nucleotide incorporated. This PPi is quantitatively converted to ATP by ATP sulfurylase in the presence of adenosine 5′‐phosphosulfate. The ATP thus obtained drives the luciferase‐mediated conversion of luciferin to oxyluciferin, thus generating visible light in amounts proportional to the amount of ATP generated. The light produced in this luciferase‐catalyzed reaction is detected by a charge‐coupled device (CCD) camera and seen as a peak in a pyrogram. Apyrase – a nucleotide‐degrading enzyme – continuously degrades unincorporated dNTPs and excess ATP, during synthesis of the complementary DNA, and the nucleotide sequence is determined from the signal peak during pyrogram tracing( 43 , 44 , 45 ) (Fig. 1). We have shown that the methylation value obtained using pyrosequencing correlates well with the methylation status and MGMT expression in glioma cell lines (Fig. 2). However, in contrast to cell lines which comprise relatively homogenous cells, bulk tumors comprise histologically heterogenous cells. Pyrosequencing revealed that one part of the tumor exhibited high MGMT expression with 18% methylation in the nucleus, whereas a different lesion in the same tumor exhibited low MGMT expression with 53% methylation (Fig. 3). However, with MSP, both these areas were found to be unmethylated; that is, MSP failed to detect the difference between them. The quantification of MGMT methylation by pyrosequencing might enable the selection of patients who are most likely to benefit from chemotherapy.
Figure 1.
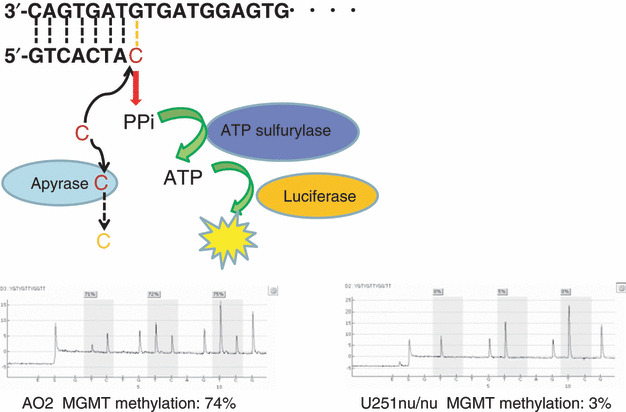
Pyrosequencing technology. A sequencing primer is hybridized to a single‐stranded, PCR‐amplified DNA template and incubated with a DNA polymerase. Each event of incorporation of a dNTP is accompanied by the release of pyrophosphate (PPi) in a quantity equimolar to the amount of nucleotide incorporated. This PPi is quantitatively converted to ATP by ATP sulfurylase in the presence of adenosine 5′‐phosphosulfate. The ATP thus obtained drives the luciferase‐mediated conversion of luciferin to oxyluciferin, thus generating visible light in amounts proportional to the amount of ATP generated. The light produced in this luciferase‐catalyzed reaction is detected by a charge‐coupled device (CCD) camera and seen as a peak in a pyrogram. Apyrase – a nucleotide‐degrading enzyme – continuously degrades unincorporated dNTPs and excess ATP, during synthesis of the complementary DNA. Gray columns indicate quantified CpG sites.
Figure 2.
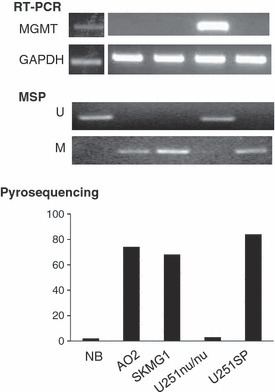
Methylation status and mRNA expression in glioma cell lines and normal brain cells. Normal brain (NB) cells and the U251nu/nu glioma cells expressed MGMT, as determined by RT‐PCR (upper panel), and contained an unmethylated MGMT promoter, as determined by methylation‐specific PCR (MSP) (middle panel). Pyrosequencing revealed that the methylation levels in NB and U251nu/nu cells were 2% and 3%, respectively (lower panel). MGMT was not expressed in three other glioma cell lines (i.e. AO2, SKMG1, and U251SP), but the level of promoter methylation was very high in these cells (70–85%).
Figure 3.
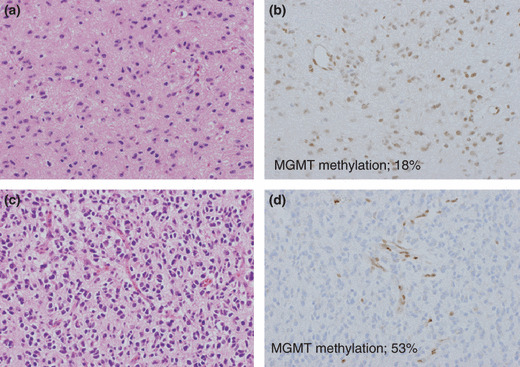
Intratumoral heterogeneity of MGMT expression and pyrosequencing. One part of the tumor showed high MGMT expression with 18% methylation in the nucleus (a,b), as determined by pyrosequencing, whereas a different lesion in the same tumor showed low MGMT expression with 53% methylation (c,d). However, with methylation‐specific PCR (MSP), both these areas were found to be unmethylated. (a,c) Hematoxylin & Eosin stained sections of two different lesions in a single tumor. (b,d) MGMT immunohistochemical stained sections.
Epigenetic therapy for human glioblastoma multiformes (GBMs)
Malignant glioma represents about 20% of all intracranial tumors. Despite advances in radiation therapy and chemotherapy administered after the surgical resection, the prognosis of malignant glioma remains poor with a median survival of less than10 months.( 46 ) In contrast to genetic alterations, epigenetic modifications such as promoter hypermethylation and histone acetylation are theoretically reversible by drug treatment. In the context of epigenetic therapy for cancer, while it is necessary to understand the precise contribution of epigenetic abnormalities to the development of glioblastomas, treatment directed toward restoring the function of genes that have been silenced by epigenetic changes in glioma cells has the potential of “normalizing” cancer cells. Therefore, DNA methylation has now become a therapeutic target for human cancers.( 47 , 48 ) DNA methylation is strongly inhibited by the cytosine analogs 5‐azacytidine and 5‐aza‐2′‐deoxycytidine (DAC); these agents are incorporated into DNA during cell division, following which they trap DNA methyltransferases and degrade them.( 48 , 49 , 50 ) Low‐dose exposure to these agents induces cell differentiation and represses cell growth through demethylation. The use of these demethylating agents in the treatment of myelodysplastic syndrome has received FDA approval, and clinical trials in this regard are currently underway in Japan. But DAC has never been tested in clinical trials for gliomas.
One of the most promising treatments for solid tumors by epigenetic drugs may be the combination of DAC and immunotherapy. Under normal conditions, a subgroup of tumor‐specific antigens called the cancer/testis antigens (CTAs) is expressed only in the tissues of the testis, ovary, and placenta; however, these antigens are also expressed in various types of human tumors.( 51 , 52 ) Since normal CTA‐expressing tissues do not express major histocompatibility complex (MHC) class I molecules, CD8 T cells cannot recognize the CTAs expressed on them; this suggests that CTAs are ideal targets for tumor immunotherapy. The expression of CTAs in tumors results in the recognition of tumor‐specific antigens on the cell surface by cytotoxic T lymphocytes (CTLs). Previously, we demonstrated that the expression of CTA genes such as LAGE‐1, CT7, SCP‐1, SSX‐1, SSX‐2, SSX‐4, and NY‐ESO‐1 was nearly imperceptible in 30 glioma tissues.( 53 ) Among these, NY‐ESO‐1 is the most immunogenic CTA discovered thus far and is considered to be a very promising immunotherapy target.( 54 ) It has been shown that NY‐ESO‐1 is expressed in cancerous cells, probably because of the loss of epigenetic regulation that is observed when methylated chromatin regions are demethylated or deacetylated histones are acetylated.( 55 ) We demonstrated that even though NY‐ESO‐1 is not expressed in human glioma cells, the prototypic inhibitor of DNA methylation – DAC – markedly reactivates NY‐ESO‐1 expression in glioma cells but not in normal human cells. In addition, glioma cells induced to express NY‐ESO‐1 exhibit in vitro and in vivo sensitivity to antigen‐specific CTLs.( 56 )
It is widely accepted that histone modification and DNA methylation are intricately interrelated; these mechanisms act together to regulate gene expression. The synergistic effect of DNA demethylation and histone deacetylase inhibition has been investigated in detail.( 57 , 58 , 59 ) DNA folds around a core of eight histones to form nucleosomes, which constitute the smallest structural unit of chromatin. The basic amino‐terminal tails of histones protrude out of the nucleosomes and undergo posttranslational modifications, including histone acetylation and methylation. The acetylation status of the lysine residues in histones H3 and H4 is controlled by the balanced action of histone acetyltransferases and HDACs. Acetylated histones have often been associated with transcriptionally active or open chromatin. In contrast, the deacetylation induced by HDACs results in chromatin compaction and gene inactivation. Histone methylation, mediated by histone methyltransferases, exerts different effects on gene expression, depending on the target residue. In fact, while the methylation of histone H3 lysine 9 (H3‐K9) is a marker of transcriptionally inactive chromatin, the methylation of H3‐K4 is associated with transcriptionally active chromatin.( 60 ) We demonstrated that DAC and the HDAC inhibitor valproic acid synergistically induced the de novo expression of NY‐ESO‐1.( 61 ) This synergistic combination decreased the levels of methylated H3‐K9 (H3‐K9‐diMe) and increased those of acetylated H3‐K9 (H3‐K9‐Ac), while causing DNA demethylation in the NY‐ESO‐1 promoter region (Fig. 4). These data are consistent with the results of many previous intensive studies. We also examined the expression of H3‐K4‐diMe – the marker of active chromatin – in the same region. However, we did not find any significant changes in the methylation status of H3‐K4; this finding suggested that compared to H3‐K4‐diMe, DNA methylation and H3‐K9 modification are more dominantly associated with the expression of NY‐ESO‐1. A reason for the absence of a close correlation between NY‐ESO‐1 expression and H3‐K4 modification might be that H3K4‐diMe is associated with “permissive” chromatin that is either active or potentially active.( 62 ) Nevertheless, our data are quite consistent with the findings that DAC and valproic acid (VPA) synergistically reactivate NY‐ESO‐1. Thus, our results not only identify a potential epigenetic immunotherapy but also suggest that the silencing mechanism of NY‐ESO‐1 in gliomas is mediated by both DNA methylation and histone modification.
Figure 4.

A schematic representation of the proposed mechanism underlying histone modifications in the NY‐ESO‐1 promoter. (a) Methylation of lysine 9, moderate methylation of lysine 4, and hypermethylation of the CpG islands caused the formation of a folded chromatin structure, leading to NY‐ESO‐1 gene silencing. (b) 5‐aza‐2′‐deoxycytidine (DAC) dramatically decreases lysine 9 methylation, demethylates the CpG sites, and unfolds the chromatin, thus reactivating gene expression. (c) VPA increases lysine 9 acetylation but has no effect on the methylation of lysine 9 or lysine 4 and does not reverse transcriptional silencing. (d) The combination of DAC and VPA decreases lysine 9 methylation and increases lysine 9 acetylation, but does not affect lysine 4 methylation. This combination unfolds the chromatin to a considerable extent and reactivates gene expression with high efficiency.
Future perspectives
Beyond the issues raised by the clinical trial on the use of demethylating agents for the treatment of MDS and the extension of the trial findings to other human malignancies, epigenetic therapy may be a promising and potent treatment for human neoplasia. However, a large amount of research is still required for understanding the role of other components of the epigenetic machinery in human neoplasia. For example, key amino acid residues in histones are also targets of aberrant epigenetic processes such as a series of acetylation, methylation, and other modifications.( 1 ) Polycomb group proteins can regulate gene expression independent of DNA methylation via H3‐K27 trimethylation, and early data suggest that this pathway is disrupted in cancer cells; thus, this pathway could represent a promising target for cancer treatment.( 63 , 64 )
Overall, it is important to elucidate the precise role of epigenetic abnormalities in human neoplasia. The use of potent epigenetics‐based therapy for cancer cells might restore the abnormally regulated epigenomes to a more normal state through epigenetic reprogramming. DNA methylation inhibitors and/or a combination of agents targeting other epigenetic modifications can facilitate this process in vitro,( 65 ) and the use of such agents might be a promising treatment for cancer.
References
- 1. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128: 683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999; 21: 163–7. [DOI] [PubMed] [Google Scholar]
- 3. Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev 2004; 23: 29–39. [DOI] [PubMed] [Google Scholar]
- 4. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 5. Turker MS. The establishment and maintenance of DNA methylation patterns in mouse somatic cells. Semin Cancer Biol 1999; 9: 329–37. [DOI] [PubMed] [Google Scholar]
- 6. Bestor TH, Gundersen G, Kolsto AB, Prydz H. CpG islands in mammalian gene promoters are inherently resistant to de novo methylation. Genet Anal Tech Appl 1992; 9: 48–53. [DOI] [PubMed] [Google Scholar]
- 7. Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev 1995; 5: 309–14. [DOI] [PubMed] [Google Scholar]
- 8. Wood LD, Parsons DW, Jones S et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318: 1108–13. [DOI] [PubMed] [Google Scholar]
- 9. Schuebel KE, Chen W, Cope L et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 2007; 3: 1709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300: 455. [DOI] [PubMed] [Google Scholar]
- 11. Cadieux B, Ching TT, VandenBerg SR, Costello JF. Genome‐wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res 2006; 66: 8469–76. [DOI] [PubMed] [Google Scholar]
- 12. Gama‐Sosa MA, Slagel VA, Trewyn RW et al. The 5‐methylcytosine content of DNA from human tumors. Nucleic Acids Res 1983; 11: 6883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fanelli M, Caprodossi S, Ricci‐Vitiani L et al. Loss of pericentromeric DNA methylation pattern in human glioblastoma is associated with altered DNA methyltransferases expression and involves the stem cell compartment. Oncogene 2008; 27: 358–65. [DOI] [PubMed] [Google Scholar]
- 14. Yu J, Zhang H, Gu J et al. Methylation profiles of thirty‐four promoter‐CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer 2004; 4: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Uyeno S, Aoki Y, Nata M et al. IGF2 but not H19 shows loss of imprinting in human glioma. Cancer Res 1996; 56: 5356–9. [PubMed] [Google Scholar]
- 16. Sakatani T, Kaneda A, Iacobuzio‐Donahue CA et al. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science 2005; 307: 1976–8. [DOI] [PubMed] [Google Scholar]
- 17. Costello JF, Berger MS, Huang HS, Cavenee WK. Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Res 1996; 56: 2405–10. [PubMed] [Google Scholar]
- 18. Baeza N, Weller M, Yonekawa Y, Kleihues P, Ohgaki H. PTEN methylation and expression in glioblastomas. Acta Neuropathol 2003; 106: 479–85. [DOI] [PubMed] [Google Scholar]
- 19. Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest 2001; 81: 77–82. [DOI] [PubMed] [Google Scholar]
- 20. Amatya VJ, Naumann U, Weller M, Ohgaki H. TP53 promoter methylation in human gliomas. Acta Neuropathol 2005; 110: 178–84. [DOI] [PubMed] [Google Scholar]
- 21. Bello MJ, Rey JA. The p53/Mdm2/p14ARF cell cycle control pathway genes may be inactivated by genetic and epigenetic mechanisms in gliomas. Cancer Genet Cytogenet 2006; 164: 172–3. [DOI] [PubMed] [Google Scholar]
- 22. Cairncross G, Berkey B, Shaw E et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 2006; 24: 2707–14. [DOI] [PubMed] [Google Scholar]
- 23. Cairncross JG, Macdonald DR. Successful chemotherapy for recurrent malignant oligodendroglioma. Ann Neurol 1988; 23: 360–4. [DOI] [PubMed] [Google Scholar]
- 24. Van Den Bent MJ, Carpentier AF, Brandes AA et al. Adjuvant procarbazine, lomustine, and vincristine improves progression‐free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligo‐astrocytomas: a randomized European Organisation for Research and Treat‐ment of Cancer phase III trial. J Clin Oncol 2006; 24: 2715–22. [DOI] [PubMed] [Google Scholar]
- 25. Alaminos M, Davalos V, Ropero S et al. EMP3, a myelin‐related gene located in the critical 19q13.3 region, is epigenetically silenced and exhibits features of a candidate tumor suppressor in glioma and neuroblastoma. Cancer Res 2005; 65: 2565–71. [DOI] [PubMed] [Google Scholar]
- 26. Zhou H, Miki R, Eeva M et al. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res 2007; 13: 2344–53. [DOI] [PubMed] [Google Scholar]
- 27. Day RS III, Ziolkowski CH, Scudiero DA et al. Defective repair of alkylated DNA by human tumour and SV40‐transformed human cell strains. Nature 1980; 288: 724–7. [DOI] [PubMed] [Google Scholar]
- 28. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 2000; 16: 168–74. [DOI] [PubMed] [Google Scholar]
- 29. Hegi ME, Diserens AC, Gorlia T et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Eng J Med 2005; 352: 997–1003. [DOI] [PubMed] [Google Scholar]
- 30. Everhard S, Kaloshi G, Criniere E et al. MGMT methylation: a marker of response to temozolomide in low‐grade gliomas. Ann Neurol 2006; 60: 740–3. [DOI] [PubMed] [Google Scholar]
- 31. Natsume A, Ishii D, Wakabayashi T et al. IFN‐beta down‐regulates the expression of DNA repair gene MGMT and sensitizes resistant glioma cells to temozolomide. Cancer Res 2005; 65: 7573–9. [DOI] [PubMed] [Google Scholar]
- 32. Natsume A, Wakabayashi T, Ishii D et al. A combination of IFN‐beta and temozolomide in human glioma xenograft models: implication of p53‐mediated MGMT downregulation. Cancer Chemother Pharmacol 2008; 61: 653–9. [DOI] [PubMed] [Google Scholar]
- 33. Margison GP, Povey AC, Kaina B, Santibanez Koref MF. Variability and regulation of O6‐alkylguanine‐DNA alkyltransferase. Carcinogenesis 2003; 24: 625–35. [DOI] [PubMed] [Google Scholar]
- 34. Silber JR, Mueller BA, Ewers TG, Berger MS. Comparison of O6‐methylguanine‐DNA methyltransferase activity in brain tumors and adjacent normal brain. Cancer Res 1993; 53: 3416–20. [PubMed] [Google Scholar]
- 35. Silber JR, Bobola MS, Ghatan S, Blank A, Kolstoe DD, Berger MS. O6‐methylguanine‐DNA methyltransferase activity in adult gliomas: relation to patient and tumor characteristics. Cancer Res 1998; 58: 1068–73. [PubMed] [Google Scholar]
- 36. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999; 59: 793–7. [PubMed] [Google Scholar]
- 37. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Derks S, Lentjes MH, Hellebrekers DM, De Bruine AP, Herman JG, Van Engeland M. Methylation‐specific PCR unraveled. Cell Oncol 2004; 26: 291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mikeska T, Bock C, El‐Maarri O et al. Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. J Mol Diagn 2007; 9: 368–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. BioTechniques 2003; 35: 146–50. [DOI] [PubMed] [Google Scholar]
- 41. Tost J, Dunker J, Gut IG. Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. BioTechniques 2003; 35: 152–6. [DOI] [PubMed] [Google Scholar]
- 42. Dupont JM, Tost J, Jammes H, Gut IG. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem 2004; 333: 119–27. [DOI] [PubMed] [Google Scholar]
- 43. Ronaghi M, Uhlen M, Nyren P. A sequencing method based on real‐time pyrophosphate. Science (New York, NY) 1998; 281: 363–5. [DOI] [PubMed] [Google Scholar]
- 44. Tost J, El abdalaoui H, Gut IG Serial pyrosequencing for quantitative DNA methylation analysis. BioTechniques. 2006; 40: 721–2, 4, 6. [DOI] [PubMed] [Google Scholar]
- 45. Uhlmann K, Brinckmann A, Toliat MR, Ritter H, Nurnberg P. Evaluation of a potential epigenetic biomarker by quantitative methyl‐single nucleotide polymorphism analysis. Electrophoresis 2002; 23: 4072–9. [DOI] [PubMed] [Google Scholar]
- 46. Fine HA, Dear KB, Loeffler JS, Black PM, Canellos GP. Meta‐analysis of radiation therapy with and without adjuvant chemotherapy for malignant gliomas in adults. Cancer 1993; 71: 2585–97. [DOI] [PubMed] [Google Scholar]
- 47. Issa JP. Decitabine. Curr Opin Oncol 2003; 15: 446–51. [DOI] [PubMed] [Google Scholar]
- 48. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006; 5: 37–50. [DOI] [PubMed] [Google Scholar]
- 49. Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5‐azacytidine. Cell 1979; 17: 771–9. [DOI] [PubMed] [Google Scholar]
- 50. Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res 2009; 15: 3938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen YT, Scanlan MJ, Sahin U et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA 1997; 94: 1914–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boon T, Cerottini JC, Van den Eynde B, Van Der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol 1994; 12: 337–65. [DOI] [PubMed] [Google Scholar]
- 53. Natsume A, Wakabayashi T, Tsujimura K et al. The DNA demethylating agent 5‐aza‐2′‐deoxycytidine activates NY‐ESO‐1 antigenicity in orthotopic human glioma. Int J Cancer 2008; 122: 2542–53. [DOI] [PubMed] [Google Scholar]
- 54. Jager E, Nagata Y, Gnjatic S et al. Monitoring CD8 T cell responses to NY‐ESO‐1: correlation of humoral and cellular immune responses. Proc Natl Acad Sci USA 2000; 97: 4760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dos Santos NR, Torensma R, De Vries TJ et al. Heterogeneous expression of the SSX cancer/testis antigens in human melanoma lesions and cell lines. Cancer Res 2000; 60: 1654–62. [PubMed] [Google Scholar]
- 56. Natsume A, Wakabayashi T, Tsujimura K et al. The DNA demethylating agent 5‐aza‐2′‐deoxycytidine activates NY‐ESO‐1 antigenicity in orthotopic human glioma. Int J Cancer 2008; 122: 2542–53. [DOI] [PubMed] [Google Scholar]
- 57. Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re‐expression of genes silenced in cancer. Nat Genet 1999; 21: 103–7. [DOI] [PubMed] [Google Scholar]
- 58. Nguyen CT, Weisenberger DJ, Velicescu M et al. Histone H3‐lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5‐aza‐2′‐deoxycytidine. Cancer Res 2002; 62: 6456–61. [PubMed] [Google Scholar]
- 59. Suzuki H, Gabrielson E, Chen W et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 2002; 31: 141–9. [DOI] [PubMed] [Google Scholar]
- 60. Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants. Mol endocrinol (Baltimore, MD) 2005; 19: 563–73. [DOI] [PubMed] [Google Scholar]
- 61. Oi S, Natsume A, Ito M et al. Synergistic induction of NY‐ESO‐1 antigen expression by a novel histone deacetylase inhibitor, valproic acid, with 5‐aza‐2′‐deoxycytidine in glioma cells. J Neurooncol 2009; 92: 15–22. [DOI] [PubMed] [Google Scholar]
- 62. Santos‐Rosa H, Schneider R, Bannister AJ et al. Active genes are tri‐methylated at K4 of histone H3. Nature 2002; 419: 407–11. [DOI] [PubMed] [Google Scholar]
- 63. Kirmizis A, Bartley SM, Kuzmichev A et al. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev 2004; 18: 1592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kondo Y, Shen L, Cheng AS et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 2008; 40: 741–50. [DOI] [PubMed] [Google Scholar]
- 65. Mikkelsen TS, Hanna J, Zhang X et al. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008; 454: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]