

Abstract
Malignant pleural mesothelioma (MPM), an aggressive and refractory tumor type, is increasing in frequency throughout the world. Peroxisome proliferator activated receptor‐γ (PPAR‐γ) agonists have anticancer activity against several cancer cell lines in vitro and in vivo. However, there have been no reports that PPAR‐γ agonists induce growth inhibition of MPM cell lines. In this study, we investigated the inhibitory effect of a PPAR‐γ agonist in combination with an anticancer agent on MPM cell growth in vitro and in vivo. We examined the therapeutic efficacy of the PPAR‐γ agonist troglitazone (TGZ) in combination with cisplatin against a human MPM cell line, both in vitro and orthotopically inoculated into severe combined immunodeficient (SCID) mice. Troglitazone (TGZ) alone inhibited MPM cell growth in vitro in a dose‐dependent manner via induction of G1 cell cycle arrest and apoptosis. The combination of TGZ and cisplatin showed an additive inhibitory effect on MPM cell growth compared to treatment with either individual drug. Treatment with 500 mg/kg or 1000 mg/kg TGZ effectively inhibited the production of thoracic tumors and pleural effusion in EHMES‐10 cell‐bearing SCID mice. Moreover, treatment with 500 mg/kg TGZ in combination with 3 mg/kg cisplatin more effectively prolonged survival compared to treatment with either individual drug. These results suggest that TGZ in combination with cisplatin may become a novel therapy for MPM. (Cancer Sci 2010)
Malignant pleural mesothelioma (MPM), an aggressive tumor arising in the mesothelium, in general tends to occur between 30 and 40 years after initial asbestos exposure.( 1 ) Malignant pleural mesothelioma (MPM) remains a serious problem, as the worldwide incidence of this disease continues to increase. In Japan, the peak incidence is predicted to occur in 2025, and 103 000 deaths are anticipated over the next 40 years. Malignant pleural mesothelioma (MPM) is an extremely difficult disease to treat, with the median overall survival ranging between 9 and 17 months, regardless of disease stage( 2 ).
The combination of cisplatin and pemetrexed was established as a standard of care front‐line regimen. The regimen had a 41.3% response rate, a median time to progression of 5.7 months, and an overall median survival of 12.1 months.( 1 , 2 ) As observed in a meta‐analysis of phase II studies,( 3 ) other cisplatin‐based combinations produced a response rate of approximately 25–30%, as follows: cisplatin plus etoposide; cisplatin plus doxorubicin; cisplatin plus gemcitabine; cisplatin plus interferon; and oxaliplatin plus raltitrexed (or gemcitabine or vinorelbine) and methotrexate. However, the efficacies of these regimens remain insufficient. Therefore, novel therapeutic strategies are needed to improve the disease prognosis.
Peroxisome proliferator activated receptor‐γ (PPAR‐γ) is a member of the nuclear hormone receptor superfamily of ligand activated transcription factors.( 4 ) Peroxisome proliferator activated receptor‐γ (PPAR‐γ) forms a heterodimeric complex with the retinoid X receptor which then binds to a PPAR response element. This interaction is responsible for the regulation of cellular events ranging from glucose and lipid homeostasis to cell differentiation and apoptosis.( 5 ) In addition, PPAR‐γ has been shown to regulate growth, differentiation, and gene expression in a number of cancer cell lines.( 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ) However, a role for PPAR‐γ in MPM regulation is unknown.
In the present study, we examined whether PPAR‐γ agonists induce an inhibition of MPM cell line growth in vitro and in vivo. Furthermore, we investigated the possibility of a synergistic effect between a PPAR‐γ agonist and cisplatin for MPM treatment.
Materials and Methods
Human MPM cell lines and culture conditions. EHMES‐10 was established from the pleural effusion of a patient with MPM in our institution.( 14 , 15 ) MSTO‐211H was purchased from the American Type Culture Collection (Manassas, VA, USA). Malignant pleural mesothelioma (MPM) cells were cultured in RPMI‐1640 medium (Nikken Bio Medical Laboratories, Kyoto, Japan) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), penicillin (100 U/mL) and streptomycin (50 mg/mL) at 37°C in a 5% CO2 humidified atmosphere.
Reagents. We used the PPAR‐γ agonists troglitazone (TGZ) (kindly provided by Daiichi‐Sankyo, Tokyo, Japan), ciglitazone (CGZ) (Calbiochem, Darmstadt, Germany), rosiglitazone (RGZ) (Alexis Biochemicals, Lausen, Switzerland), and 15‐deoxy‐Δ12,14‐prostaglandin J2 (15d‐PGJ2) (Calbiochem). For in vitro experiments, these agents at various concentrations were dissolved in dimethylsulfoxide (DMSO) (Sigma‐Aldrich, St. Louis, MO, USA), and were added to cells in medium with a final DMSO concentration of 1.0%. For the in vivo study, TGZ was prepared as a suspension in a vehicle consisting of 0.5% carboxymethyl cellulose (CMC) (Wako Pure Chemical Industries, Osaka, Japan) in sterile water. Cisplatin was kindly provided by Nippon Kayaku (Tokyo, Japan). Rabbit polyclonal antibodies against p27kip1 and cyclin D1 were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit monoclonal antibodies against p21waf1/cip1 and mouse monoclonal antibodies against Ki‐67 and PPAR‐γ were purchased from Cell Signaling Technology, Dako Japan (Kyoto, Japan), and Santa Cruz Biotechnology (Santa Cruz, CA, USA), respectively. Horseradish peroxidase conjugated goat antirabbit IgG and horse antimouse IgG were purchased from Cell Signaling Technology.
Cell proliferation assay (WST‐1 assay). The effect of individual test agents on cell viability was assessed using cell proliferation reagent WST‐1 (4‐[3‐(4‐lodophenyl)‐2‐(4‐nitrophenyl)‐2H‐5‐tetrazolio]‐1,3‐benzene disulfonate) (Roche Diagnostics, Mannheim, Germany) assay in 8 to 16 replicates. Malignant pleural mesothelioma (MPM) cells (1–2 × 104 cells/well) were plated in 96‐well plates (Nunc, Roskilde, Denmark) and were exposed to various concentrations of test agents dissolved in DMSO. The final concentration of DMSO in the culture medium was 1.0%, which did not affect MPM cell proliferation (data not shown). Controls received DMSO vehicle at a concentration equal to that of drug‐treated cells. After drug treatment, 10 μL of WST‐1 reagent were added to each well, and the culture plates were incubated for 1 h at 37°C. Absorbance was measured at 450 nm with a reference wavelength at 690 nm by an E max precision microplate reader (Molecular Devices, Tokyo, Japan). The cell viability percentage was determined as the ratio of the absorbance of the sample versus the control. Eight replicate wells were used for each drug concentration, and the assay was carried out independently three times.
Western blot analysis. Total protein was extracted from MPM cells. The cells were lysed in a lysis buffer (25 mM Tris‐HCl [pH 7.5], 150 mM NaCl, 1% Triton X, 50 mM NaF, 1 mM Na3VO4) containing a complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) on ice. The cell lysates were centrifuged at 267 g for 5 min. Protein concentrations were measured using a Bio‐Rad Protein Assay Reagent (Bio‐Rad Lab., Hercules, CA, USA). Twenty micrograms of protein were separated on a 12.5% Tris‐Glycine gel (Bio‐Rad Lab.). After electrophoresis, the protein was transferred to a nitrocellulose membrane using an iBlotTM Dry Blotting System (Invitrogen, Carlsbad, CA, USA), and blocked in Tris‐buffered saline containing 0.1% Tween‐20 (TBS‐T) with 10% skim milk at 4°C for 1 h. The membrane was incubated with the appropriate primary antibody in TBS‐T containing 1% skim milk at 4°C overnight. All primary antibodies were diluted 1:1000 in TBS‐T containing 1% skim milk. After treatment with a primary antibody, the membrane was washed three times with TBS‐T and incubated with the horseradish peroxidase‐conjugated secondary antibody (diluted 1:2000) for 1 h at room temperature. Immune complexes were visualized using ECL detection reagents (Amersham, Buckinghamshire, UK).
Cell cycle analysis. Malignant pleural mesothelioma (MPM) cells, treated with or without test agents over time periods from 8 to 48 h, were collected after a brief trypsinization using 0.25% Trypsin‐EDTA (Invitrogen), washed with phosphate‐buffered saline (PBS) (Wako Pure Chemical Industries), and the cell nuclei were stained using the CycleTEST PLUS DNA Reagent Kit (Becton Dickinson, San Jose, CA, USA). Cells were sorted by FACScan analysis, and cell cycle profiles were determined using ModFitLT software (Becton Dickinson, San Diego, CA, USA).
Detection of apoptosis. Cell apoptosis was detected using an Annexin V‐fluorescein isothiocyanate (FITC)/7‐amino‐actinomycin D (7‐AAD) kit (Beckman Coulter, Marseille, France). Malignant pleural mesothelioma (MPM) cells treated with or without test agents were collected after a brief trypsinization, washed with PBS, and stained by 10 μL of Annexin V‐FITC solution and 20 μL of 7‐AAD. Cells were sorted by FACScan analysis, and apoptosis profiles were determined using ModFitLT software.
Animals. Male severe combined immunodeficient (SCID) mice (6–8 weeks old) were obtained from Clea Japan (Osaka, Japan) and maintained under specific pathogen‐free conditions throughout the study. Experiments were carried out in accordance with the guidelines established by the Ehime University Committee on Animal Care and Use.
Orthotopic implantation model. Cultured EHMES‐10 cells were harvested, washed twice, and re‐suspended in PBS. Subsequently, the tumor cells (3 × 106) were injected into the thoracic cavity of the SCID mice as previously described.( 15 ) Treatments were started 1 week after orthotopic implantation because this time period was necessary for the EHMES‐10 cells to adhere to the pleural surface and begin proliferating. To evaluate the effect of treatments on tumor weight, pleural effusion, and survival time, mice were divided into seven experimental groups (n = 7 in each group) that included: vehicle alone (0.5% CMC in sterile water per day, by gavage), TGZ alone (250, 500, and 1000 mg/kg per day, by gavage), cisplatin alone (3 and 6 mg/kg per week, by intraperitoneal injection), and a combination of TGZ (500 mg/kg per day, by gavage) and cisplatin (3 mg/kg per week, by intraperitoneal injection). Four weeks after tumor cell inoculation, the mice were dissected. The thoracic tumors were carefully removed and weighed, the pleural effusion was harvested using a 1‐mL syringe, and the volume of the pleural effusion was measured.
In vivo analyses for apoptosis, proliferation, and the cell cycle regulator p21waf1/cip1. Two hours after TGZ gavage and/or cisplatin injection, the tumors were excised from the tumor‐bearing SCID mice, and formalin‐fixed, paraffin‐embedded tissue sections were subsequently prepared for immunohistochemistry with the anti ki‐67 monoclonal antibody and the anti p21waf1/cip1 monoclonal antibody, and for in situ apoptosis detection by terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL) with the In Situ Apoptosis Detection Kit (Takara Biomedicals, Shiga, Japan). The p21waf1/cip1 and Ki‐67 immunoreactivities were assessed with visualization under a light microscope. In areas with the highest nuclear labeling density and the lowest amount of inflammatory cell infiltrate, the percentage of tumor nuclei expressing p21waf1/cip1 and Ki‐67 was determined by counting 1000 cells per slide. TUNEL‐positive cells were assessed in the same way.
Statistical analysis. In vitro data are expressed as mean ± SD. For comparisons between groups in the proliferation assay, we used one‐way anova with Dunnett’s post test. Survival was compared using the Kaplan–Meier method, and statistical analysis was performed using the log‐rank test. For between‐group comparisons in p21waf1/cip1, Ki‐67, and TUNEL‐positive cells in vivo, we used one‐way anova followed by a Scheffe post‐hoc test. P‐values below 0.05 were considered statistically significant. Drug interactions were analyzed using CalcuSyn software (version 2.0; Biosoft, Cambridge, UK), which is based on the median effect model of Chou and Talaly.( 16 ) The combination index (CI) for each fraction affected was simulated, and for the final evaluation, the averaged CI at 0.5, 0.75, 0.90, and 0.95 fraction affected was determined. A CI < 0.3, 0.3–0.7, 0.7–0.9, 0.9–1.1, 1.1–1.45, 1.45–3.3, and >3.3 indicates highly synergistic, synergistic, moderate to slight synergistic, nearly additive, slight to moderate antagonistic, antagonistic, and strong antagonistic, respectively.
Results
Effects of PPAR‐γ agonists on MPM cell growth. To investigate the effect of PPAR‐γ agonists on the proliferation of MPM cells, cells were treated with TGZ, CGZ, RGZ, and 15d‐PGJ2 (0–200 μM). Peroxisome proliferator activated receptor‐γ (PPAR‐γ) agonists inhibited EHMES‐10 and MSTO‐211H cell growth in a dose‐dependent manner (Fig. 1a,b). We chose TGZ from the four PPAR‐γ agonists because TGZ had the most potent inhibitory effect on cell proliferation. In addition, a large amount of TGZ was available for in vivo study.
Figure 1.
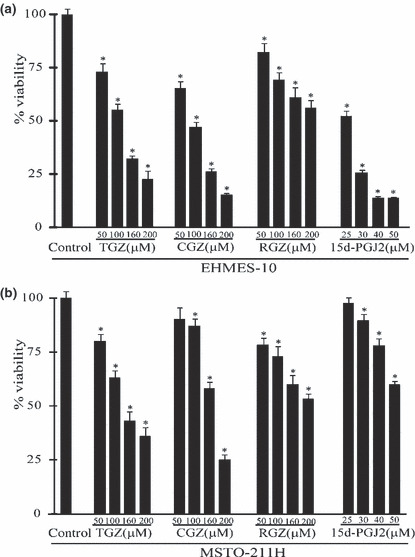
Effect of peroxisome proliferator activated receptor‐γ (PPAR‐γ) agonists on proliferation of malignant pleural mesothelioma (MPM) cells. (a) EHMES‐10 cells were treated with troglitazone (TGZ), ciglitazone (CGZ), rosiglitazone (RGZ), or 15‐deoxy‐Δ12,14‐prostaglandin J2 (15d‐PGJ2) for 3 days. (b) MSTO‐211H cells were treated with TGZ, CGZ, RGZ, or 15d‐PGJ2 for 3 days. Data represent mean ± SD. *P < 0.01 (one‐way anova with Dunnett’s post test).
Induction of G1 arrest after TGZ treatment in MPM cells. To investigate the mechanism of growth inhibition by TGZ, cell cycle progression in EHMES‐10 cells and MSTO‐211H cells following treatment with TGZ was evaluated by FACS analysis. Of the EHMES‐10 cells that were treated with vehicle (DMSO) for 48 h, 58.03% were in the G1 phase and 27.37% were in the S phase. Treatment with TGZ for 48 h significantly increased the percentage of EHMES‐10 cells in the G1 phase (83.24%). Much higher concentrations of TGZ induced the subG1 phase, representing apoptosis (data not shown). Similar results were obtained from experiments using MSTO‐211H cells (Fig. 2a). The assay was carried out independently three times.
Figure 2.
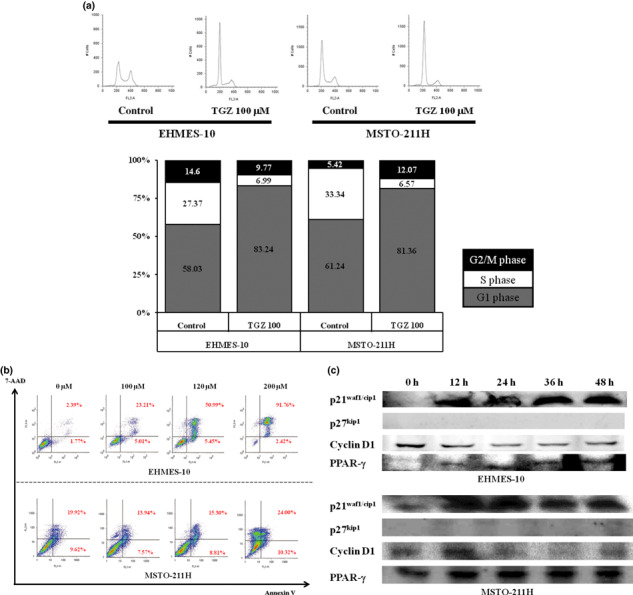
The mechanism of troglitazone (TGZ)‐induced growth inhibition in vitro. (a) Effects of peroxisome proliferator activated receptor‐γ (PPAR‐γ) agonists on the cell cycle profile. After treatment with 100 μM TGZ or DMSO (control) for 48 h, EHMES‐10 cells and MSTO‐211H cells were collected, fixed, stained with propidium iodide (PI), and analyzed by flow cytometry. The values represent the number of cells in a cell cycle phase as a percentage (%) of total cells. (b) Induction of apoptosis by TGZ in EHMES‐10 cells and MSTO‐211H cells. EHMES‐10 cells and MSTO‐211H cells were treated with different concentrations of TGZ for 48 h, and apoptosis was analyzed by flow cytometry staining with Annexin V‐FITC and 7‐amino‐actinomycin D (7‐AAD). (c) Effects of TGZ on the expression of p21waf1/cip1, p27kip1, cyclin D1, and PPAR‐γ protein. EHMES‐10 cells and MSTO‐211H cells were treated with 100 μM TGZ for the indicated time. Whole cell lysates were prepared and used for detection of the expression of each protein with antibodies against p21waf1/cip1, p27kip1, cyclin D1, and PPAR‐γ by western blot analysis.
Induction of apoptosis after TGZ treatment. EHMES‐10 cells were treated with different concentrations of TGZ for 48 h. High doses of TGZ induced apoptosis of EHMES‐10 cells in a dose‐dependent manner. Similar results were obtained from experiments using MSTO‐211H cells (Fig. 2b). The assay was carried out independently three times.
Expression of cell cycle regulatory proteins and PPAR‐γ. We analyzed the protein expression of p21waf1/cip1, p27kip1, cyclin D1, and PPAR‐γ in EHMES‐10 cells after treatment with 100 μM TGZ. Troglitazone (TGZ) increased the expression of p21waf1/cip1 and PPAR‐γ proteins, whereas the expression of cyclin D1 protein was decreased. EHMES‐10 cells did not express the p27kip1 protein. Similar results were obtained from experiments with MSTO‐211H cells (Fig. 2c). The assay was carried out independently three times.
Effect of cisplatin on MPM cell growth. EHMES‐10 cells and MSTO‐211H cells were treated with cisplatin (0–80 μM) to investigate the effect of cisplatin on cell viability. Cisplatin inhibited EHMES‐10 and MSTO‐211H cell growth in a dose‐dependent manner (Fig. 3a). To investigate the mechanism of growth inhibition by cisplatin, cell cycle progression and apoptotic cells following treatment of both cell lines with cisplatin were evaluated by FACS analysis. A low concentration (1.25 μM) of cisplatin tended to increase the G2/M phase population, but a high concentration (5 μM) of cisplatin did not have this effect in either cell line (Fig. 3b,c). A much higher concentration (20 μM) of cisplatin increased the number of apoptotic cells in a time‐dependent manner in both cell lines (Fig. 3d). The assay was carried out independently three times.
Figure 3.
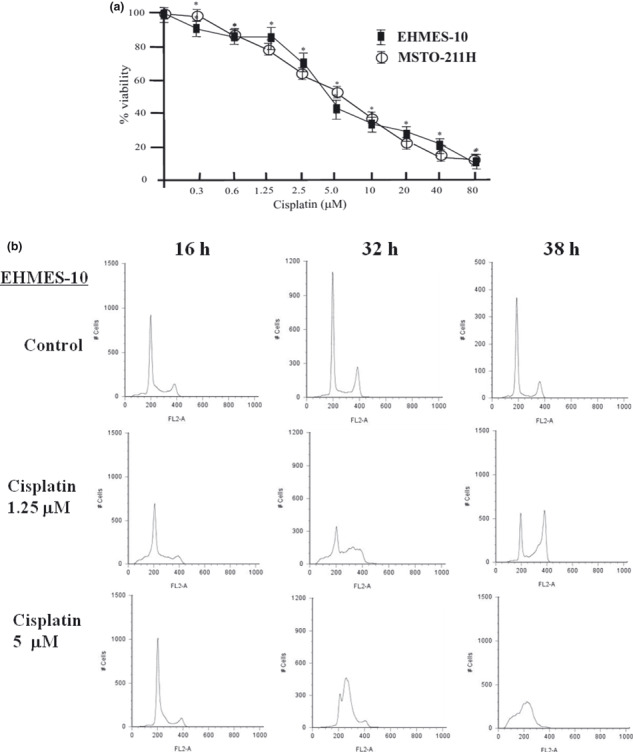
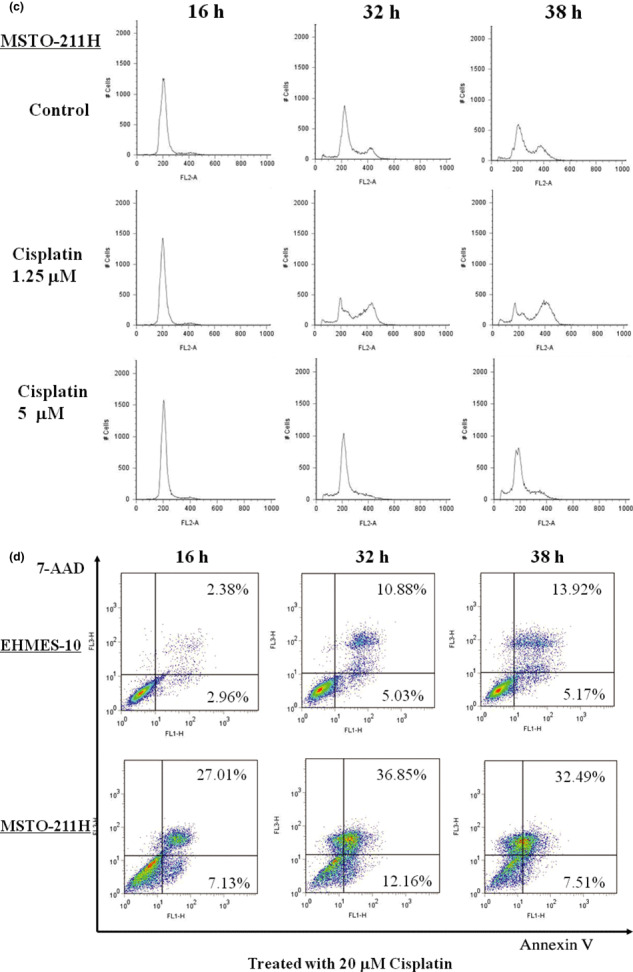
Effect of cisplatin on malignant pleural mesothelioma (MPM) cell growth. (a) Effect of cisplatin on proliferation of EHMES‐10 cells and MSTO‐211H cells. EHMES‐10 cells and MSTO‐211H cells were treated with cisplatin at concentrations of 0.3 μM, 0.6 μM, 1.25 μM, 2.5 μM, 5.0 μM, 10 μM 20 μM, 40 μM, or 80 μM for 16 h followed by a 56‐h drug‐free period. After 72 h, cell viability was assessed with the WST‐1 assay. Data represent mean ± SD. *P < 0.01 (one‐way anova with Dunnett’s post test). (b) Effects of cisplatin on the cell cycle profile. After treatment with 1.25 μM and 5 μM cisplatin for 16 h, 32 h, and 38 h, EHMES‐10 cells and MSTO‐211H cells were collected, fixed, stained with propidium iodide (PI), and analyzed by flow cytometry. (c) Induction of apoptosis by cisplatin in EHMES‐10 cells and MSTO‐211H cells. EHMES‐10 cells and MSTO‐211H cells were treated with 20 μM cisplatin for 16 h, 32 h, and 38 h, and cell apoptosis was analyzed by flow cytometry staining with Annexin V‐FITC and 7‐amino‐actinomycin D (7‐AAD).
Effects of combination treatment with TGZ and cisplatin. Since cisplatin is inactivated by DMSO, combination treatment with TGZ and cisplatin was tested by a sequential treatment protocol as previously described.( 17 ) EHMES‐10 cells were treated with various concentrations of cisplatin for 16 h followed by an 8‐h drug‐free washout and then treatment with various concentrations of TGZ for 48 h. After 72 h, cell viability was assessed with the WST‐1 assay. We observed that EHMES‐10 cells treated with cisplatin followed by TGZ demonstrated maximum growth inhibition as compared with cisplatin alone (Fig. 4).
Figure 4.
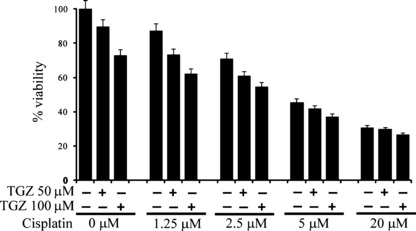
Effect of troglitazone (TGZ) in combination with cisplatin on EHMES‐10 cell growth in vitro. EHMES‐10 cells were treated with various concentrations of cisplatin and with TGZ at concentrations of 50 μM or 100 μM.
We used the CalcuSyn software to determine synergistic, additive, or antagonistic effects of the drug combinations. In EHMES‐10 cells, the IC50 values of TGZ and cisplatin were 124.81 μM and 6.19 μM, respectively (Fig. 5a,b). Because the ratio of IC50 values for TGZ and cisplatin against EHMES‐10 cells was 20:1, EHMES‐10 cells were exposed to varying concentrations of TGZ and cisplatin sequentially at a fixed ratio of 20:1 (TGZ/cisplatin), and then cell viability was assessed with the WST‐1 assay. The averaged CI was 0.924, which indicates a nearly additive inhibitory effect for TGZ and cisplatin (Fig. 5c). To investigate the mechanism of the additive inhibitory effect of TGZ and cisplatin, apoptotic cells were evaluated by FACS analysis following treatment of EHMES‐10 cells with TGZ and cisplatin. A larger number of apoptotic cells was observed after combination treatment than after single‐agent treatment. The assay was carried out independently three times (Fig. 6).
Figure 5.
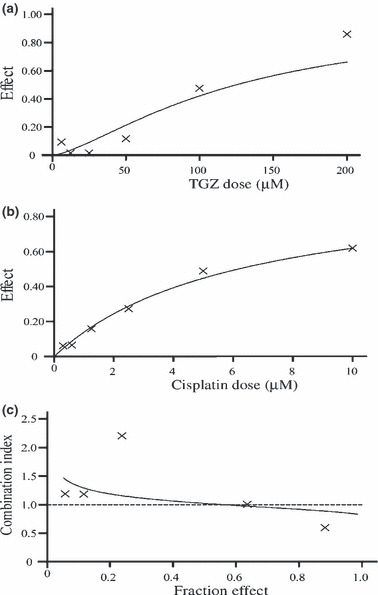
Analysis of the combination of troglitazone (TGZ) and cisplatin in EHMES‐10 cells. (a) Dose‐effect curves for TGZ. (b) Dose‐effect curves for cisplatin. (c) The fraction‐effect versus combination index (Fa‐CI) curve calculated with the CalcuSyn software.
Figure 6.
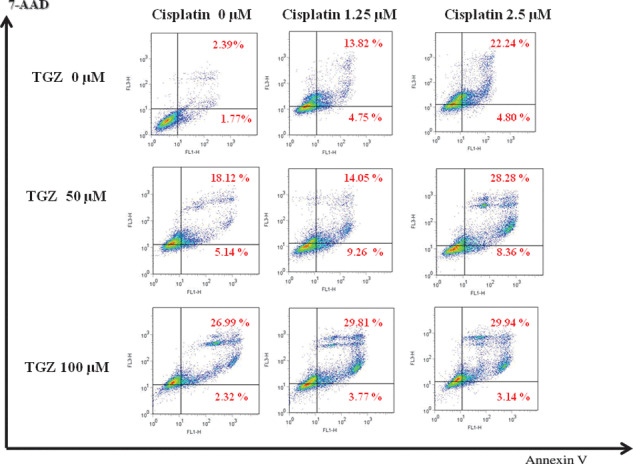
Effect of the combination of troglitazone (TGZ) and cisplatin in EHMES‐10 cells in vitro. Induction of apoptosis by the combination of TGZ and cisplatin in EHMES‐10 cells. EHMES‐10 cells were treated with cisplatin (1.25 and 2.5 μM) for 16 h, followed by an 8‐h drug‐free washout and TGZ (50 and 100 μM) for 48 h. After 72 h, cell apoptosis was analyzed by flow cytometry staining with Annexin V‐FITC and 7‐amino‐actinomycin D (7‐AAD).
Effects of several treatments in the orthotopic implantation model with EHMES‐10 cells. We examined the therapeutic effects of TGZ, cisplatin, and the combination of TGZ and cisplatin on tumors and pleural effusion caused by EHMES‐10 cells. Table 1 shows that tumor weight and pleural effusion tended to be lower in the 500 mg/kg TGZ treatment group compared to the control group. In the 1000 mg/kg TGZ treatment group, the cisplatin treatment groups, and the TGZ/cisplatin combination group, tumor weight and pleural effusion were significantly reduced. With respect to the survival of the tested mice, administration of 500 mg/kg or 1000 mg/kg TGZ and 3 mg/kg cisplatin significantly prolonged the survival time of the EHMES‐10 cell‐bearing SCID mice compared with control (Fig. 7a,b). The combination treatment with TGZ and cisplatin significantly prolonged the survival time of EHMES‐10 cell‐bearing SCID mice compared with either individual drug treatment (Fig. 7b).
Table 1.
Effect of TGZ and in combination with cisplatin on thoracic tumor and pleural effusion produced by MPM cells in SCID mice
| Treatment | Thoracic tumor | Pleural effusion | ||
|---|---|---|---|---|
| Incidence | Weight (mg) | Incidence | Volume (μL) | |
| Control | 5/5 | 390 (246–416) | 5/5 | 450 (250–500) |
| 250 mg/kg TGZ | 5/5 | 277 (174–398) | 5/5 | 540 (250–610) |
| 500 mg/kg TGZ | 5/5 | 313 (21–384) | 2/5 | 0 (0–650) |
| 1000 mg/kg TGZ | 5/5 | 153* (94–334) | 3/5 | 40 (0–680) |
| 3 mg/kg Cisplatin | 5/5 | 89** (57–139) | 0/5 | 0** |
| 6 mg/kg Cisplatin | 5/5 | 16** (11–18) | 0/5 | 0** |
| 3 mg/kg Cisplatin+500 mg/kg TGZ | 5/5 | 82** (22–180) | 0/5 | 0** |
EHMES‐10 cells were inoculated into the thoracic cavity of SCID mice on day 0. Treatments were started 1 week after orthotopic implantation. Experimental groups included: control (0.5% carboxymethyl cellulose [CMC] in sterile water per day, by gavage), troglitazone (TGZ) alone (250, 500, and 1000 mg/kg per day, by gavage), cisplatin alone (3 and 6 mg/kg per week, by intraperitoneal injection), and a combination of TGZ (500 mg/kg per day, by gavage) and cisplatin (3 mg/kg per week, by intraperitoneal injection). Four weeks after tumor cell inoculation, the mice were dissected and thoracic tumors and pleural effusions were evaluated as described in the Materials and Methods. Data are expressed as median with ranges in parentheses. *P < 0.05 compared with control; **P < 0.01 compared with control. MPM, malignant pleural mesothelioma, SCID, severe combined immunodeficient.
Figure 7.
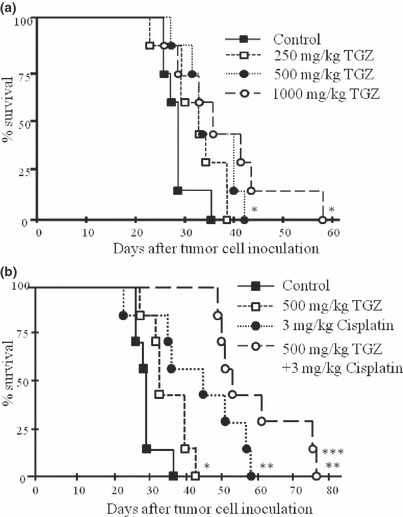
Survival of severe combined immunodeficiency (SCID) mice inoculated with EHMES‐10 cells. EHMES‐10 cells (3 × 106) were inoculated into the thoracic cavity of SCID mice. When the mice became moribund, they were sacrificed. Survival was therefore determined up to the day the mice were sacrificed. (a) Survival time of EHMES‐10 cell‐bearing SCID mice treated with troglitazone (TGZ). (b) Survival time of EHMES‐10 cell‐bearing SCID mice treated with TGZ, cisplatin, and the combination of TGZ and cisplatin. *P < 0.05 versus control; **P < 0.01 versus control; ***P < 0.05 versus the individual drug.
Previous reports demonstrated that TGZ increased liver toxicity.( 18 , 19 , 20 ) Therefore, we measured the blood concentration of TGZ and evaluated liver and renal function. The mean blood concentrations of TGZ in the 500 mg/kg TGZ, 1000 mg/kg TGZ, and combination treatment groups were 0.546 (n = 4), 0.685 (n = 3), and 0.566 (n = 5) μg/mL, respectively. In previous clinical data, the maximum blood concentration of TGZ was 0.9 μg/mL. No abnormal findings in liver and renal function tests were observed in our experiments.
Immunohistochemical analyses of the antitumor mechanisms of TGZ, cisplatin, and combination treatment. Numbers of p21waf1/cip1‐positive cells tended to increase in the TGZ treatment group compared with the control and other treatment groups. Numbers of Ki‐67‐positive cells decreased significantly in the cisplatin treatment group and tended to decrease in the TGZ and combination treatment groups compared with the control group. Numbers of TUNEL‐positive cells increased significantly in the cisplatin and combination treatment groups and tended to increase in the TGZ treatment group compared with the control group (Fig. 8a,b).
Figure 8.
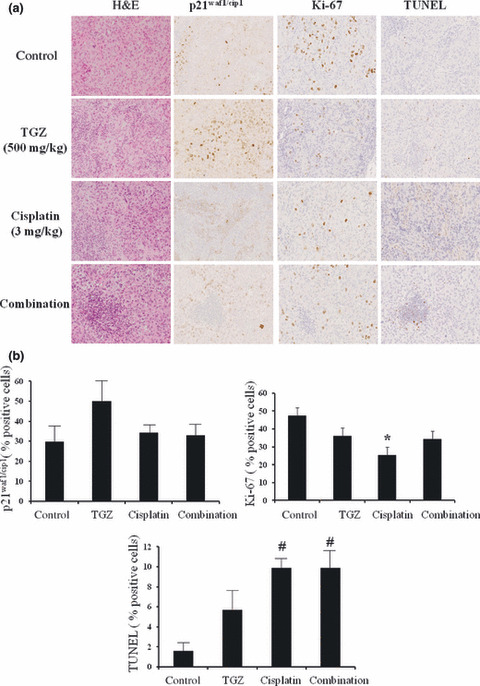
Histologic analyses of thoracic tumors produced by EHMES‐10 cells. EHMES‐10 cell‐bearing SCID mice were treated with 500 mg/kg troglitazone (TGZ), 3 mg/kg cisplatin, and the combination of TGZ and cisplatin. Four weeks after tumor cell inoculation, the mice were killed (day 28). (a) The thoracic tumors were analyzed by H&E and immunohistochemistry. Magnification, ×400. (b) Quantitative immunohistochemical analysis. Columns, mean number from five independent areas; bars, SD. *P < 0.01 versus control; #P < 0.01 versus control.
Discussion
The present study demonstrates that TGZ inhibits MPM cell growth via cell cycle arrest and apoptosis in vitro and in vivo. In addition, administration of TGZ prolonged the survival time of EHMES‐10 cell‐bearing SCID mice. The combination treatment with TGZ and cisplatin inhibits tumor cell proliferation and prolonged the survival time of EHMES‐10 cell‐bearing SCID mice as compared to either individual drug.
Treatment with PPAR‐γ agonists has been shown to inhibit the growth of many types of cancer cells via induction of cell cycle arrest and/or apoptosis.( 21 ) It has been reported that overexpression of p21waf1/cip1 and p27kip1 inhibits the activities of cyclin–CDK complexes (cyclin–cyclin‐dependent kinase complexes), which regulate cell cycle progression in mammalian cells.( 22 , 23 ) This study is the first to show that TGZ increases the level of PPAR‐γ expression and induces cell growth inhibition via induction of cell cycle arrest and apoptosis in MPM. These results indicate that PPAR‐γ acts as a suppressor gene. Functional impairment of the cell cycle is reconstituted with expression of PPAR‐γ. In the present study, we found that TGZ induced an increased expression of p21waf1/cip1 and a decreased expression of cyclin D1, which resulted in an increase in the G1 phase population of MPM cells. Furthermore, we found that a high dose of TGZ induces apoptosis in a dose‐dependent manner. The mechanisms by which TGZ induced p21waf1/cip1 expression, but not p27kip1 expression, were not clarified. Previous reports indicated that TGZ up‐regulates the p21waf1/cip1 protein in hepatocellular carcinoma cell lines and p27kip1 expression in pancreatic tumors.( 21 ) p21waf1/cip1, but not p27kip1, induced by TGZ might mediate G1 cell cycle arrest induced by TGZ in EHMES‐10 and MSTO‐211H cells.
After observing substantial suppression of MPM cell growth by TGZ treatment in vitro, we conducted experiments designed to test the potential of TGZ to exert an inhibitory effect against MPM in vivo. The previously reported animal model, which included EHMES‐10 cells being orthotopically inoculated into the thoracic cavity of SCID mice to reproducibly develop thoracic tumors and bloody pleural effusions, has shown a human patient‐like progression of MPM.( 15 , 24 ) A high dose of TGZ reduced tumor weight and pleural effusion in the EHMES‐10 cell‐bearing SCID mice in addition to prolonging their survival. To the best of our best knowledge, this is the first preclinical report that clearly shows the therapeutic efficacy of TGZ against the progression of MPM cells.
A high dose of TGZ was effective in the treatment of EHMES‐10 cell‐bearing SCID mice, but its clinical application is limited by increased liver toxicity( 18 , 19 , 20 ) and an unsatisfactory control of pleural effusion. Cisplatin, a key drug used for MPM, reduced the number of pleural effusions and the tumor weight in mice. The combination treatment of TGZ and cisplatin significantly extended the survival period for mice compared with either individual drug. These findings have important implications for development of new therapeutic strategies. Why did the combination of TGZ and cisplatin show an additive inhibitory effect? Troglitazone (TGZ) and cisplatin used different mechanisms to inhibit cell growth. Our in vitro study showed that TGZ increased the G1 phase cell population, whereas a low concentration (1.25 μM), but not a higher concentration (5 μM), of cisplatin tended to increase the G2/M phase cell population. Much higher concentrations of TGZ and cisplatin (20 μM) induced apoptosis. It was suggested that the duration and magnitude of the cell cycle arrest are dependent on the length of treatment and the concentrations of the drugs. Troglitazone (TGZ) and cisplatin are known to induce G1 phase arrest and G2‐M phase arrest, respectively, followed by apoptosis in vitro.( 21 , 25 , 26 , 27 , 28 ) Our in vivo study revealed that TGZ increased the numbers of p21waf1/cip1 and TUNEL‐positive cells and that cisplatin increased the number of TUNEL‐positive cells.
Troglitazone (TGZ) monotreatment has been evaluated in phase II clinical trials for metastatic colorectal and refractory breast cancers and has demonstrated little clinical benefit.( 29 , 30 ) Four recent studies have reported that a combination of PPAR‐γ agonists and chemotherapeutic agents showed antitumor activity. Rosiglitazone (RGZ)‐mediated down‐regulation of metallothioneins, which have been shown to be involved in resistance to platinum‐based therapy, demonstrated a striking synergistic effect between RGZ and platinum‐based drugs in non‐small‐cell lung cancer (NSCLC) both in vitro and in vivo.( 31 ) Cisplatin‐mediated up‐regulation of PPAR‐γ expression induced a synergistic effect between TGZ and cisplatin in NSCLC both in vitro and in vivo.( 17 ) The combination of RGZ pretreatment with cisplatin showed significant antineoplastic activity in breast cancer in vivo.( 32 ) A novel high‐affinity PPAR‐γ agonist (RS5444) itself and in combination with paclitaxel inhibited human anaplastic thyroid carcinoma tumor growth via p21waf1/cip1. RS5444 did not induce apoptosis but, combined with paclitaxel, doubled the apoptotic index compared to that of only paclitaxel.( 33 ) Our results also suggest that there is a strong possibility of combining TGZ with another anticancer agent as a successful treatment for refractory tumors.
In conclusion, a PPAR‐γ agonist, TGZ, inhibited EHMES‐10 cell growth via induction of cell cycle arrest resulting in the overexpression of p21waf1/cip1 and apoptosis. Troglitazone (TGZ), alone or in combination with cisplatin, may provide therapeutic benefit to patients with MPM.
Abbreviations
- MPM
malignant pleural mesothelioma
- SCID
severe combined immunodeficient
Acknowledgments
The authors thank Dr S. Yano (Kanazawa University, Ishikawa, Japan) and Dr K. Kameda (Ehime University, Ehime, Japan) for technical assistance with this study.
References
- 1. Pistolesi M, Rusthoven J. Malignant pleural mesothelioma: update, current management, and newer therapeutic strategies. Chest 2004; 126: 1318–29. [DOI] [PubMed] [Google Scholar]
- 2. Tsao AS, Wistuba I, Roth JA, Kindler HL. Malignant pleural mesothelioma. J Clin Oncol 2009; 27: 2081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berghmans T, Paesmans M, Lalami Y et al. Activity of chemotherapy and immunotherapy on malignant mesothelioma: a systematic review of the literature with meta‐analysis. Lung Cancer 2002; 38: 111–21. [DOI] [PubMed] [Google Scholar]
- 4. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990; 347: 645–50. [DOI] [PubMed] [Google Scholar]
- 5. Lehmann JM, Moore LB, Smith‐Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome roliferator‐activated receptor gamma (PPAR gamma). J Biol Chem 1995; 270: 12953–6. [DOI] [PubMed] [Google Scholar]
- 6. Koeffler HP. Peroxisome proliferator‐activated receptor gamma and cancers. Clin Cancer Res 2003; 9: 1–9. [PubMed] [Google Scholar]
- 7. Tontonoz P, Singer S, Forman BM et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator‐activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci USA 1997; 94: 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sarraf P, Mueller E, Jones D et al. Differentiation and reversal of malignant changes in colon cancer through PPAR gamma. Nat Med 1998; 4: 1046–52. [DOI] [PubMed] [Google Scholar]
- 9. Mueller E, Sarraf P, Tontonoz P et al. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell 1998; 1: 465–70. [DOI] [PubMed] [Google Scholar]
- 10. Elstner E, Müller C, Koshizuka K et al. Ligands for peroxisome proliferator‐activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci USA 1998; 95: 8806–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu J, Qiao L, Zimmermann L et al. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology 2006; 43: 134–43. [DOI] [PubMed] [Google Scholar]
- 12. Chaffer CL, Thomas DM, Thompson EW, Williams ED. PPARgamma‐independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer 2006; doi 10.1186/1471-2407-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shiau CW, Yang CC, Kulp SK et al. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl‐xL/Bcl‐2 functions independently of PPARgamma. Cancer Res 2005; 65: 1561–9. [DOI] [PubMed] [Google Scholar]
- 14. Yokoyama A, Kohno N, Fujino S et al. Origin of heterogeneity of interleukin‐6 (IL‐6) levels in malignant pleural effusions. Oncol Rep 1994; 1: 507–11. [DOI] [PubMed] [Google Scholar]
- 15. Nakataki E, Yano S, Matsumori Y et al. Novel orthotopic implantation model of human malignant pleural mesothelioma (EHMES‐10 cells) highly expressing vascular endothelial growth factor and its receptor. Cancer Sci 2006; 97: 183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 17. Reddy RC, Srirangam A, Reddy K et al. Chemotherapeutic drugs induce PPAR‐gamma expression and show sequence‐specific synergy with PPAR‐gamma ligands in inhibition of non‐small cell lung cancer. Neoplasia 2008; 10: 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watkins PB, Whitcomb RW. Hepatic dysfunction associated with troglitazone. N Engl J Med 1998; 338: 916–7. [DOI] [PubMed] [Google Scholar]
- 19. Gale EA. Lessons from the glitazones: a story of drug development. Lancet 2001; 357: 1870–5. [DOI] [PubMed] [Google Scholar]
- 20. Graham DJ, Green L, Senior JR, Nourjah P. Troglitazone‐induced liver failure: a case study. Am J Med 2003; 114: 299–306. [DOI] [PubMed] [Google Scholar]
- 21. Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator‐activated receptor gamma agonists. Lancet Oncol 2004; 5: 419–29. [DOI] [PubMed] [Google Scholar]
- 22. Han S, Sidell N, Fisher PB, Roman J. Up‐regulation of p21 gene expression by peroxisome proliferator‐activated receptor gamma in human lung carcinoma cells. Clin Cancer Res 2004; 10: 1911–9. [DOI] [PubMed] [Google Scholar]
- 23. Koga H, Harada M, Ohtsubo M et al. Troglitazone induces p27Kip1‐associated cell‐cycle arrest through down‐regulating Skp2 in human hepatoma cells. Hepatology 2003; 37: 1086–96. [DOI] [PubMed] [Google Scholar]
- 24. Li Q, Yano S, Ogino H et al. The therapeutic efficacy of anti‐vascular endothelial growth factor antibody, bevacizumab, and pemetrexed against orthotopically implanted human pleural mesothelioma cells in severe combined immunodeficient mice. Clin Cancer Res 2007; 13: 5918–25. [DOI] [PubMed] [Google Scholar]
- 25. Sorenson CM, Eastman A. Mechanism of cis‐diamminedichloroplatinum(II)‐induced cytotoxicity: role of G2 arrest and DNA double‐strand breaks. Cancer Res 1988; 48: 6703–7. [PubMed] [Google Scholar]
- 26. Sorenson CM, Barry MA, Eastman A. Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J Natl Cancer Inst 1990; 82: 749–55. [DOI] [PubMed] [Google Scholar]
- 27. Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin‐induced cell death always produced by apoptosis? Mol Pharmacol 2001; 59: 657–63. [DOI] [PubMed] [Google Scholar]
- 28. DiPaola RS. To arrest or not to G(2)‐M Cell‐cycle arrest : commentary re: A. K. Tyagi et al., Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin‐induced growth inhibition, G(2)‐M arrest, and apoptosis. Clin Cancer Res 2002; 8: 3311–4. [PubMed] [Google Scholar]
- 29. Kulke MH, Demetri GD, Sharpless NE et al. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy‐resistant metastatic colorectal cancer. Cancer J 2002; 8: 395–9. [DOI] [PubMed] [Google Scholar]
- 30. Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator‐activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat 2003; 79: 391–7. [DOI] [PubMed] [Google Scholar]
- 31. Girnun GD, Naseri E, Vafai SB et al. Synergy between PPARgamma ligands and platinum‐based drugs in cancer. Cancer Cell 2007; 11: 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tikoo K, Kumar P, Gupta J. Rosiglitazone synergizes anticancer activity of cisplatin and reduces its nephrotoxicity in 7, 12‐dimethyl benz{a}anthracene (DMBA) induced breast cancer rats. BMC Cancer 2009; 9: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Copland JA, Marlow LA, Kurakata S et al. Novel high‐affinity PPARgamma agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene 2006; 25: 2304–17. [DOI] [PubMed] [Google Scholar]