

Abstract
Tumor hypoxia is an obstacle to radiotherapy. Radiosensitivity under hypoxic conditions is determined by molecular oxygen levels, as well as by various biological cellular responses. The insulin‐like growth factor (IGF) signaling pathway is a widely recognized survival signal that confers radioresistance. However, under hypoxic conditions the role of IGF signaling in radiosensitivity is still poorly understood. Here, we demonstrate that IGF‐II stimulation decreases clonogenic survival under hypoxic conditions in the pancreatic cancer cell lines AsPC‐1 and Panc‐1, and in the human breast cancer cell line MCF‐7. IGF treatment under hypoxic conditions suppressed increased radiation sensitivity in these cell lines by pharmacologically inhibiting the phosphoinositide 3‐kinase–mammalian target of rapamycin pathway, a major IGF signal‐transduction pathway. Meanwhile, IGF‐II induced the endoplasmic reticulum stress response under hypoxia, including increased protein levels of CHOP and ATF4, mRNA levels of CHOP, GADD34, and BiP, as well as splicing levels of XBP‐1. The response was suppressed by inhibiting phosphoinositide 3‐kinase and mammalian target of rapamycin activity. Overexpression of CHOP in AsPC‐1 cells increased radiation sensitivity by IGF‐II simulation under hypoxic conditions, whereas suppression of CHOP expression levels with small hairpin RNA or a dominant negative form of a proline‐rich extensin‐like receptor protein kinase in hypoxia decreased IGF‐induced radiosensitivity. IGF‐induced endoplasmic reticulum stress contributed to radiosensitization independent of cell cycle status. Taken together, IGF stimulation increased radiosensitivity through the endoplasmic reticulum stress response under hypoxic conditions. (Cancer Sci 2008; 99: 2395–2401)
Tumor hypoxia is an obstacle to radiotherapy, because radiosensitivity is reduced when the oxygen partial pressure is low (known as the oxygen‐enhancement effect).( 1 ) Therefore, numerous studies have been done to increase the radiosensitivity of hypoxic cells in solid tumors, although a widely accepted treatment has not yet emerged.( 2 ) Radioresistance under hypoxic conditions is attributed to the absence of molecular oxygen, as DNA damage produced by radiation‐induced free radicals can be permanent under the presence of molecular oxygen. Meanwhile, cancer cells are able to survive in severely deteriorated microenvironments, such as hypoxia, by means of various biological adaptation responses.( 3 ) Therefore, absence of molecular oxygen cannot be the sole contributor to radioresistance under hypoxic conditions; the biological responses of cancer cells are also likely to be involved.
Two ligands belonging to the insulin‐like growth factor (IGF) family, IGF‐I and IGF‐II, interact with the receptor IGF‐IR, a transmembrane tyrosine kinase.( 4 ) The role of IGF‐IR in malignant transformation is well documented.( 4 , 5 ) IGF‐IR signaling can induce many effects, including mitogenesis, transformation, and cell survival. Radioresistance in cancer cells is reportedly enhanced by IGF signaling in normal fibroblasts( 6 ) and in cancer cell lines.( 7 , 8 , 9 ) However, most of these experiments were conducted in normoxic conditions. Thus, the role of IGF signaling in the radiosensitivity of cancer cells under hypoxic conditions is still poorly understood.
Downstream molecules in the IGF pathway, such as Akt, phosphoinositide 3‐kinase (PI3K), mammalian target of rapamycin (mTOR), and mitogen‐activated protein kinase (MAPK), have been reported to play an important role in cellular radioresistance.( 10 , 11 ) mTOR is a serine–threonine protein kinase that phosphorylates a series of substrates involved in protein translation, including the eukaryotic initiation factor 4E‐binding protein‐1 and ribosomal p70 S6 kinase.( 12 ) Myriad growth factors act as upstream activators of mTOR, including IGF. Sustained mTOR activation is important for tumor growth.( 12 ) Given that hypoxia is a common characteristic of solid tumors, it is paradoxical that mTOR signaling is downregulated in hypoxia.( 13 )
We previously reported that aberrant activation of mTOR, a downstream IGF signal‐transduction molecule, accelerated cell death in Lewis lung carcinoma cells under hypoxic conditions( 14 ) and also that activation of IGF signaling induces apoptotic cell death in the human pancreatic cancer cell line AsPC‐1 under severe hypoxia, by enhancing the endoplasmic reticulum (ER) stress response.( 15 ) In a later report, drastic cell death in the AsPC‐1 cell line was induced by IGF‐I treatment under anoxic conditions, but not under hypoxic conditions (1% oxygen), in spite of the substantial induction of ER stress.( 15 ) Therefore, we speculated that additional cellular damage‐inducing stress, such as irradiation under hypoxic conditions, might have synergistic effects when combined with IGF treatment.
In the present study, we examined the role of IGF‐II and downstream PI3K–mTOR signaling in radiation sensitivity under hypoxic conditions. We demonstrate that stimulation of IGF and the consequent activation of the PI3K–mTOR pathway under hypoxic conditions induces radiosensitization through enhancement of the ER stress response.
Materials and Methods
Cells and cell culture. The pancreatic cancer cell lines AsPC‐1 and Panc‐1, and the breast cancer cell line MCF‐7, were obtained from the American Type Culture Collection (Rockville, MD, USA). AsPC‐1 and MCF‐7 cells were cultured in RPMI‐1640 with 10% serum. Panc‐1 cells were cultured in Dulbecco's modified Eagle's medium with 10% serum. Cells were cultured in a humidified atmosphere of 5% CO2/95% air at 37°C; these conditions were termed ‘normoxia’. Hypoxic conditions were achieved using a Multigas Incubator (Astec, Fukuoka, Japan). Cells were incubated with 1% O2 and 5% CO2 at 37°C in the humidified chamber, and these conditions were termed ‘hypoxia’.
Reagents. Recombinant IGF‐II was purchased from R&D Systems (Minneapolis, MN, USA). LY294002 and rapamycin were from Wako Pure Chemical Industries (Osaka, Japan). Tunicamycin was from Sigma (St Louis, MO, USA).
Colony‐forming assay. Radiosensitivity was determined by colony‐forming assay, as described elsewhere, with some modifications.( 1 ) Briefly, cells were seeded at a density of 2 × 105 cells (AsPC‐1), 7 × 105 cells (MCF‐7), or 4 × 105 cells (Panc‐1) per 25‐cm2 tissue culture flask with double‐sealed cap (Iwaki, Chiba, Japan) in each medium with 10% fetal bovine serum, and cultured in normoxia. After 2 days incubation for AsPC‐1 cells, and 18 h incubation for MCF‐7 and Panc‐1 cells, the medium was changed to serum‐free medium, with or without IGF‐II 100 ng/mL, and they were transferred immediately to the hypoxic chamber. After 17 h incubation in hypoxia, flasks were sealed under a hypoxic workbench (INVIVO2 400, Ruskinn Technology, Bridgend, UK) and cells were γ‐irradiated with the indicated doses using a MBR‐1505R irradiator (Hitachi, Tokyo, Japan) at a dose rate of 0.3–0.4 Gy/min. After 2 h incubation in hypoxia, the cells were detached from the plates and replated at clonal densities. For AsPC‐1 cells, 2 × 105 feeder cells were cocultured in 6‐cm culture dishes. The feeder cells were prepared from the AsPC‐1 cells, seeded at 1 × 106 cells/6‐cm dish, cultured in normoxic conditions for 2 days, and γ‐irradiated at 35 Gy. Colony formation was scored after 2 weeks in normoxic culture. The surviving fraction was calculated using the following formula:
| surviving fraction = (number of colonies counted/number of cells seeded) × 1/PE, |
where PE is defined as the plating efficiency of non‐irradiated control cells, given by the following formula:
| PE = number of colonies counted/number of cells seeded. |
Immunoblot analysis. Western blotting analysis was carried out as described previously, with some modifications.( 15 ) Cells were prepared as in the colony‐forming assay. The cells were harvested 24 h after being transferred to a hypoxic chamber with or without stimulation of IGF. Primary antibodies against GADD153/CHOP and CREB‐2/ATF4, and horseradish peroxidase‐conjugated secondary antirabbit and antimouse IgG antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Plasmids and transfection. The coding region of CHOP was amplified by polymerase chain reaction (PCR) using cDNA from tunicamycin‐treated AsPC‐1 cells as a template, sequenced entirely, and subcloned into the retroviral vector pMXspuro to generate pMXs/CHOP. The coding region of a proline‐rich extensin‐like receptor protein kinase (PERK) lacking its C‐terminal region (i.e. nucleotides 1–1746 of PERK) was amplified by PCR using cDNA from AsPC‐1 cells as a template and subcloned into pGEMTe plasmid (Promega Japan, Tokyo, Japan), and the entire sequence was confirmed. After adding a Flag‐tag sequence to the C‐terminus, the Flag‐tagged fragment of PERK was subcloned into pMXspuro to generate pMXs/dnPERK. Gene silencing of CHOP was carried out using the pSuperRetro plasmid purchased from Oligoengine (Seattle, WA, USA).( 15 ) Retroviral transfection was carried out as described previously.( 15 )
Reverse transcription–polymerase chain reaction. Semiquantitative reverse transcription‐PCR for CHOP, XBP‐1, GADD34, BiP, and β‐actin were carried out as described previously.( 15 )
Cell cycle analysis. AsPC‐1 cells were pulsed with 10 µmol/L BrdU for 1 h. The cells were then harvested, washed with phosphate‐buffered saline (PBS), and resuspended in ice‐cold 70% ethanol in PBS. Cells were then treated with 2 mol/L HCl, neutralized with boric acid, and labeled with α‐BrdU antibody (BD). After washing, cells were treated with 5 µg/mL propidium iodide (Invitrogen Japan, Tokyo, Japan) and 10 µg/mL RNase (Promega), and analyzed by flow cytometry (FACS‐Calibur) using CELLQuest software (BD Biosciences, Franklin Lakes, NJ, US).
Statistical analysis. Statistical analysis was carried out with GraphPad Prism 4 (GraphPad Software, San Diego, CA, USA). The statistical significance of the results was tested with the unpaired t‐test. A value of P < 0.05 was considered to be statistically significant.
Results
Radiosensitivity was increased by insulin‐like growth factor‐II stimulation under hypoxic conditions in various cancer cell lines. To examine the role of IGF on radiation sensitivity under hypoxic conditions, a clonogenic assay was carried out with AsPC‐1 cells cultured in hypoxic as well as normoxic conditions, with or without IGF‐II treatment (Fig. 1a). Consistent with reports in other cell types, AsPC‐1 cells were more resistant to radiation under hypoxic conditions than under normoxic conditions. When the cells were treated with IGF‐II, radiosensitivity increased under hypoxic conditions, but not under normoxic conditions. The increase in radiosensitivity under hypoxic conditions with IGF‐II treatment was dose‐dependent (Fig. 1b). The cell viability at the time of irradiation and the plating efficiency made no difference (Fig. 1c), indicating that IGF‐II treatment alone under hypoxic conditions was not cytotoxic, in contrast to anoxic conditions under which the IGF‐II‐treated cells underwent significant cell death, as reported previously.( 15 ) Involvement of apoptosis in IGF‐induced radiosensitivity under hypoxic conditions was unlikely, as neither caspase‐3 activation nor poly (ADP‐ribose) polymerase (PARP) cleavage was observed. This result was in contrast to the observation that IGF‐treated cells in anoxic conditions underwent massive apoptosis (Fig. 1d). To generalize these findings, another pancreatic cancer cell line, Panc‐1, and the human breast cancer cell line MCF‐7 were subjected to similar experiments. These cells also demonstrated increased radiosensitivity under hypoxic conditions when treated with IGF‐II (Fig. 2a,b).
Figure 1.
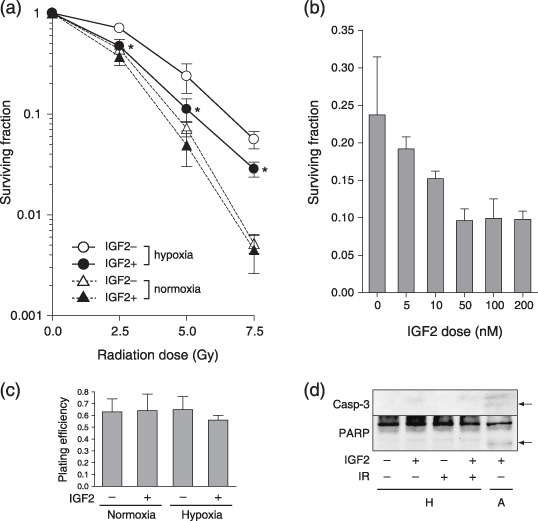
Radiosensitivity of a pancreatic cancer cell line is increased by insulin‐like growth factor (IGF)‐II stimulation under hypoxic conditions. (a) The surviving fraction of AsPC‐1 cells, irradiated with the indicated doses. In normoxic conditions (dotted lines), the cells were treated with (closed triangle) or without (open triangle) IGF‐II at 100 ng/mL. Under hypoxic conditions (solid lines), the cells were treated with (closed circle) or without (open circle) IGF‐II. IGF2 indicates IGF‐II. Experiments were repeated three times; data shown are the mean ± SD. *P < 0.05, versus control (non‐treated) under hypoxic conditions. (b) The surviving fraction of AsPC‐1 cells, irradiated with 5 Gy and treated with indicated doses of IGF‐II. (c) Plating efficiency of AsPC‐1 cells in the indicated conditions. IGF2, 100 ng/mL IGF‐II. (d) Western blot of caspase‐3 and poly (ADP‐ribose) polymerase. Samples were prepared 2 h after irradiation, with the exception of IGF‐II‐treated cells in anoxic conditions. Arrows indicate the cleaved active form of each molecule. A, anoxia 48 h; H, hypoxia; IGF2, 100 ng/mL IGF‐II; IR, 5 Gy irradiation.
Figure 2.
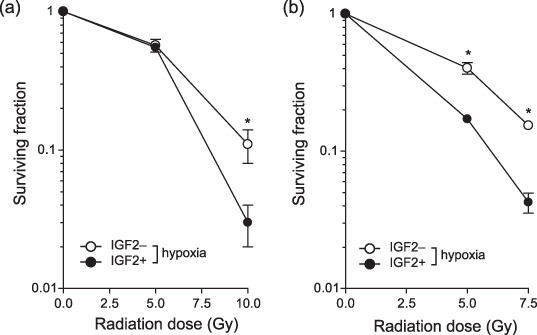
Radiosensitization by insulin‐like growth factor (IGF)‐II under hypoxia in various cancer cell lines. The surviving fraction of (a) Panc‐1 and (b) MCF‐7 cells under hypoxic conditions, treated with (closed circle) or without (open circle) IGF‐II at 100 ng/mL. Experiments were repeated three times, and the mean ± SD are shown. *P < 0.05, versus control (non‐treated).
Insulin‐like growth factor increased radiosensitivity under hypoxic conditions though the phosphoinositide 3‐kinase–mammalian target of rapamycin pathway. Next we investigated the contribution of the PI3K–mTOR pathway, a central IGF signal‐transduction pathway, to IGF‐induced radiosensitization under hypoxic conditions. The increased radiation sensitivity that was induced by IGF treatment under hypoxic conditions was suppressed in both AsPC‐1 and MCF‐7 cell lines by treatment with the PI3K inhibitor LY294002 or the mTOR inhibitor rapamycin (Fig. 3a,c). Radiosensitivity in Panc‐1 cells was also decreased by mTOR inhibition, whereas PI3K inhibition increased radiosensitivity (Fig. 3b). Together, IGF treatment under hypoxic conditions increased radiosensitivity via the mTOR pathway in general, but the contribution of PI3K might be cell‐type specific.
Figure 3.
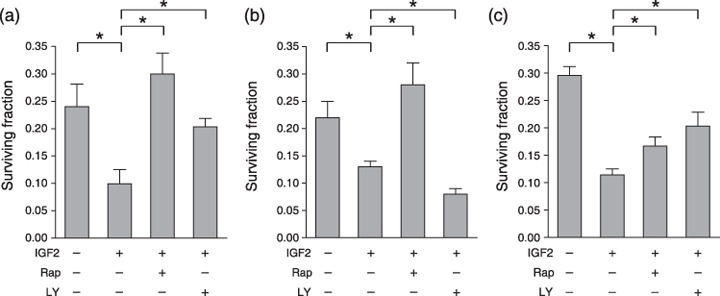
Insulin‐like growth factor (IGF) increases radiosensitivity under hypoxic conditions though the phosphoinositide 3‐kinase–mammalian target of rapamycin pathway. The surviving fraction of (a) AsPC‐1, (b) Panc‐1, and (c) MCF‐7 cells in the conditions shown above each panel. IGF2, IGF‐II, 100 ng/mL; LY, LY294002, 10 µmol/L; Rap, rapamycin, 100 nmol/L. Experiments were repeated three times, and the mean ± SD are shown. *P < 0.05.
Insulin‐like growth factor generated endoplasmic reticulum stress in AsPC‐1 cells under hypoxic conditions via the phosphoinositide 3‐kinase–mammalian target of rapamycin pathway. We next assessed the levels of ER stress induced by IGF‐II under hypoxic conditions. Consistent with a previous report regarding IGF‐I,( 15 ) IGF‐II treatment under hypoxic conditions remarkably increased the protein levels of CHOP and ATF4 (Fig. 4a), and the mRNA levels of CHOP, GADD34, and BiP (Fig. 4b). Splicing of XBP‐1 was also increased (Fig. 4b). When PI3K or mTOR activity was inhibited by LY294002 or rapamycin, respectively, the IGF‐induced unfolded protein response (UPR) was suppressed (Fig. 4a,b). Thus, activation of the PI3K and mTOR pathways under hypoxic conditions induced radiosensitivity in parallel with ER stress.
Figure 4.
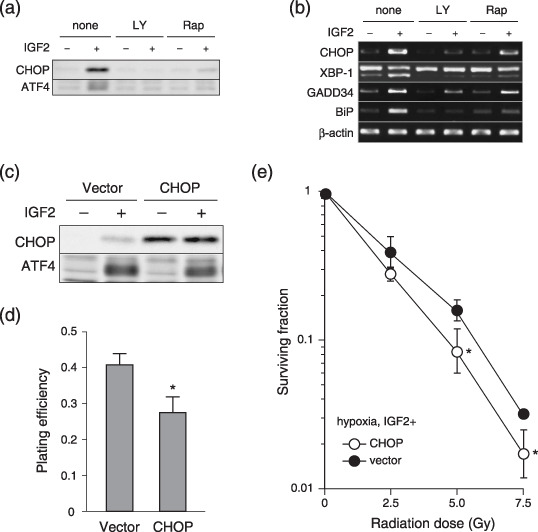
Insulin‐like growth factor (IGF)‐induced endoplasmic reticulum stress is involved in radiosensitivity under hypoxic conditions. (a) Western blotting and (b) Reverse transcription–polymerase chain reaction of AsPC‐1 cells for the indicated gene products. The conditions are shown above each panel. (c) Western blotting of AsPC‐1 cells, transfected with control vector (Vector) or CHOP expression vector (CHOP). (d) Plating efficiency of the cells in (c,e). Surviving fraction of the cells under hypoxic conditions, transfected with control vector (closed circle) or with CHOP expression vector (open circle). The cells were treated with IGF‐II at 100 ng/mL. Experiments were repeated three times, and the mean ± SD are shown. *P < 0.05, versus control. LY, LY294002; Rap, rapamycin.
Insulin‐like growth factor‐induced endoplasmic reticulum stress was involved in radiosensitivity under hypoxic conditions. We examined whether IGF‐induced ER stress in hypoxia directly influenced radiosensitivity. First, CHOP, a downstream molecule of UPR, was overexpressed in AsPC‐1 cells. Forced expression of CHOP was confirmed by western blotting (Fig. 4c). The plating efficiency was decreased in CHOP‐overexpressing cells (Fig. 4d), probably because of the ability of CHOP to induce apoptosis.( 16 ) The dose–response curve of radiation corrected by PE revealed that CHOP overexpression significantly increased radiation sensitivity (Fig. 4e).
To further investigate the direct role of ER stress in IGF‐induced radiosensitivity, we perturbed the UPR in AsPC‐1 cells by introducing CHOP small hairpin RNA (shRNA/CHOP), or the dominant negative form of PERK (dnPERK). The levels of CHOP protein in shRNA/CHOP‐transfected and dnPERK‐transfected cells decreased remarkably in comparison with CHOP protein levels in the control vector‐transfected cells, even after treatment with IGF‐II under hypoxic conditions (Fig. 5a,c). The radiosensitivity of IGF‐treated shRNA/CHOP‐transfected and dnPERK‐transfected cells under hypoxic conditions was also decreased in comparison with the control vector‐transfected cells (Fig. 5b,d). The expression levels of dnPERK were confirmed by Flag‐tag detection. Taken together, IGF‐induced radiosensitivity under hypoxic conditions was decreased by UPR suppression, indicating that IGF‐induced ER stress is involved directly in radiosensitization.
Figure 5.
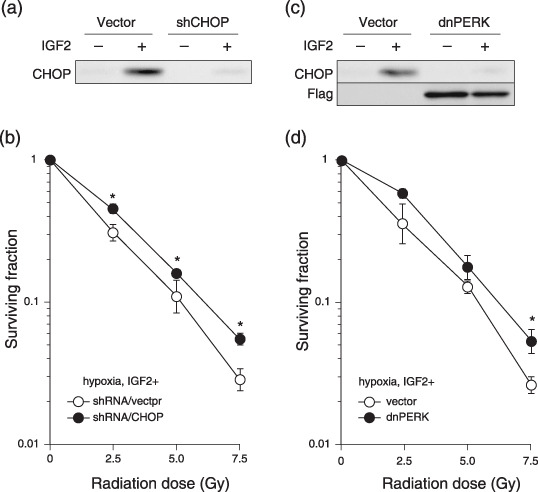
Suppression of the endoplasmic reticulum stress response in hypoxia decreases insulin‐like growth factor (IGF)‐induced radiosensitivity. (a,c) Western blotting of AsPC‐1 cells, transfected with control vector (Vector), short hairpin RNA/CHOP (shCHOP), or the dominant negative form of PERK (dnPERK). Immunoblotting was carried out with (a,c) anti‐CHOP antibody or (c) anti‐Flag antibody. (b,d) Surviving fraction of the cells under hypoxic conditions, transfected with control vector (open circle), (b) shCHOP (closed circle), or (d) dnPERK. The cells were treated with IGF‐II at 100 ng/mL. Experiments were repeated three times, and the mean ± SD are shown. *P < 0.05, versus control.
Radiosensitization under hypoxic conditions by insulin‐like growth factor‐induced endoplasmic reticulum stress occurred independently of cell cycle status. IGF are known to stimulate cell proliferation( 4 ) and radiosensitivity is known to increase with increasing rates of cell division.( 17 ) Therefore, we examined the cell cycle status of AsPC‐1 cells under hypoxic conditions with or without IGF treatment. Stimulation with IGF‐II resulted in an increased population in S phase and a decreased population in G1–G0 phase (Fig. 6a,b). Therefore, IGF‐induced cell cycle activation could also be a mechanism of radiosensitization. Nonetheless, the cells with suppressed CHOP expression or with impaired PERK activity demonstrated similar cell cycle profiles to that of control vector‐transfected cells when treated with IGF‐II. Therefore, IGF‐induced ER stress is an independent parameter from cell cycle status.
Figure 6.
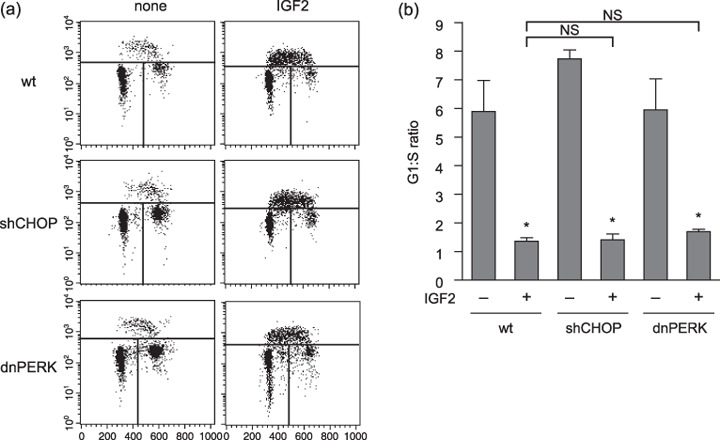
Insulin‐like growth factor (IGF)‐induced endoplasmic reticulum stress and cell cycle status are independent factors in radiosensitizaion under hypoxia. (a) Flow cytometric analysis of the cells under the indicated conditions. The parameters were propidium iodide, PI (x‐axis) and BrdU incorporation (y‐axis). Cells were divided into three regions: upper region (S phase), left‐lower region (G1–G0), and right‐lower region (G2–M). (b) The G1–G0 : S ratio was calculated from the results in (a) by dividing the percentage of the cells in G1–G0 phase by that in S phase. The experiments were repeated three times, the mean ± SD are shown. *P < 0.05, versus each non‐treated control. dnPERK, dominant negative form of PERK; NS, not significant versus wild‐type (wt) cells; shCHOP, short hairpin RNA/CHOP.
Discussion
We have demonstrated that activation of IGF‐IR signaling enhances radiosensitivity under hypoxic conditions, through increasing ER stress. The IGF‐IR pathway is a widely recognized survival signal, and confers radioresistance by activating downstream survival pathways.( 18 ) It should be noted that most of the studies were carried out in normoxic conditions. Kaneko et al. recently reported that decreased radiosensitivity in HeLa cells overexpressing IGF‐IR in normoxic conditions in vitro were conversely increased in vivo. ( 19 ) They attributed the discrepancy to the reduction of hypoxic area in tumors, caused by increased angiogenesis resulting from vascular endothelial growth factor production driven by IGF‐IR signaling. Our results suggest another possibility: that IGF‐IR signaling increases radiosensitivity in cancer cells in the hypoxic area of tumors in vivo. In terms of the intracellular signaling, the experiments using inhibitors of mTOR or PI3K (Fig. 3) suggest that activation of mTOR is the common entry of IGF‐induced ER stress. It should be noted that inhibition of PI3K increased IGF‐induced radiosensitivity exceptionally in Panc‐1 cells. As PI3K phosphorylates a substantial number of molecules other than mTOR, the contribution of PI3K to IGF‐induced radiosensitivity would be cell‐type specific.
Recently, hypoxia has been shown to generate ER stress( 20 ) and we have clarified that ER stress was involved in IGF‐induced radiosensitivity in hypoxia by demonstrating that the attenuation of PERK or CHOP results in radiation resistance. Although the role of ER stress in radiation sensitivity has been conceptually proposed,( 21 ) it has not been well proven until now. Indeed, other ER stress inducers, such as tunicamycin and thapsigargin, decrease the radiation sensitivity of AsPC‐1 cells under hypoxia (H. Endo and M. Inoue, 2007, unpublished observation). The decrease in radiosensitivity caused by these ER stress inducers might be explained by drastic cell cycle arrest after treatment with both reagents, as these reagents did not induce apparent cell death in AsPC‐1 cells under hypoxia. Thus, the IGF‐induced radiosensitivity under hypoxic conditions is not likely due to ER stress alone.
The pathways downstream of PERK and CHOP that led to increased radiosensitivity are unclear. ER stress response is initially cell protective, and is crucial for cell survival under stress conditions. However, apoptosis is induced when cells are challenged by overwhelming ER stress.( 22 ) We previously reported that IGF induces robust apoptosis in AsPC‐1 cells through ER stress in anoxia.( 15 ) Moreover, CHOP is characterized as an apoptosis inducer.( 16 ) Therefore, it is reasonable to speculate that apoptosis is involved in the radiosensitivity induced by IGF treatment under hypoxic conditions. We did not observe any evidence of apoptosis after IGF treatment or additional irradiation under hypoxic conditions (Fig. 1c). Therefore, apoptosis is not likely to be a major cause of IGF‐induced radiation sensitivity in hypoxia.
Autophagy is characterized by an accumulation of cytoplasmic double‐membrane autophagic vacuoles called autophagosomes that degrade and turn over intracellular organelles.( 23 ) When cells encounter environmental stresses such as nutrient starvation and pathogen infection, autophagy takes place, resulting in either adaptation and survival or death. Autophagic cell death, also called type 2 cell death, differs from apoptosis by the presence of autophagosomes, autolysosomes, and an intact nucleus in the cell. It has recently been reported that ER stress induces autophagy( 24 , 25 , 26 ) and autophagy is involved in radiation sensitivity.( 27 , 28 , 29 ) Therefore, autophagy may be involved in the radiosensitivity induced by IGF treatment under hypoxic conditions. Indeed, LC3 protein was clearly upregulated when the cells were treated with IGF‐II under hypoxic conditions, and that upregulation is blocked by inhibition of PI3K as well as mTOR (H. Endo and M. Inoue, 2007, unpublished observation), in contrast to reports that autophagy is initiated by mTOR inhibition.( 30 ) Further studies are required to elucidate the mechanism of autophagy, as well as the contribution of autophagy to IGF‐induced radiosensitivity under hypoxic conditions.
Our results suggest that forced activation of IGF signals under hypoxic conditions can be a means to enhance radiosensitivity. From a therapeutic standpoint, however, the stimulation of IGF‐IR is no simple matter, as the IGF‐IR signal may support the survival and proliferation of cancer cells in well‐oxygenated tumor regions. Hypoxia‐targeting strategies must be used to activate IGF‐IR signals solely in hypoxic regions.( 31 ) Activating downstream cell death signals, such as enhanced ER stress, rather than IGF‐IR itself, may be an alternative way to enhance radiosensitivity under hypoxic conditions.
Acknowledgments
M.I. and H.E. received grant support from a Grant‐in‐Aid for Scientific Research from the Japanese Society for the Promotion of Science.
References
- 1. Hall EJ, Giaccia AJ. Radiobiology for the Radiologist, 6th edn. Philadelphia: Lippincott, Williams & Wilkins, 2006. [Google Scholar]
- 2. Kaanders JH, Bussink J, Van Der Kogel AJ. Clinical studies of hypoxia modification in radiotherapy. Seminars Radiation Oncol 2004; 14: 233–40. [DOI] [PubMed] [Google Scholar]
- 3. Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Seminars Radiation Oncol 2004; 14: 198–206. [DOI] [PubMed] [Google Scholar]
- 4. LeRoith D, Roberts CT Jr. The insulin‐like growth factor system and cancer. Cancer Lett 2003; 195: 127–37. [DOI] [PubMed] [Google Scholar]
- 5. Baserga R, Peruzzi F, Reiss K. The IGF‐1 receptor in cancer biology. Int J Cancer 2003; 107: 873–7. [DOI] [PubMed] [Google Scholar]
- 6. Yu D, Watanabe H, Shibuya H, Miura M. Redundancy of radioresistant signaling pathways originating from insulin‐like growth factor I receptor. J Biol Chem 2003; 278: 6702–9. [DOI] [PubMed] [Google Scholar]
- 7. Macaulay VM, Salisbury AJ, Bohula EA, Playford MP, Smorodinsky NI, Shiloh Y. Downregulation of the type 1 insulin‐like growth factor receptor in mouse melanoma cells is associated with enhanced radiosensitivity and impaired activation of Atm kinase. Oncogene 2001; 20: 4029–40. [DOI] [PubMed] [Google Scholar]
- 8. Cosaceanu D, Budiu RA, Carapancea M, Castro J, Lewensohn R, Dricu A. Ionizing radiation activates IGF‐1R triggering a cytoprotective signaling by interfering with Ku‐DNA binding and by modulating Ku86 expression via a p38 kinase‐dependent mechanism. Oncogene 2007; 26: 2423–34. [DOI] [PubMed] [Google Scholar]
- 9. Allen GW, Saba C, Armstrong EA et al . Insulin‐like growth factor‐I receptor signaling blockade combined with radiation. Cancer Res 2007; 67: 1155–62. [DOI] [PubMed] [Google Scholar]
- 10. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene 2003; 22: 5885–96. [DOI] [PubMed] [Google Scholar]
- 11. Kim IA, Bae SS, Fernandes A et al . Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res 2005; 65: 7902–10. [DOI] [PubMed] [Google Scholar]
- 12. Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med 2005; 11: 353–61. [DOI] [PubMed] [Google Scholar]
- 13. Arsham AM, Howell JJ, Simon MC. A novel hypoxia‐inducible factor‐independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem 2003; 278: 29 655–60. [DOI] [PubMed] [Google Scholar]
- 14. Hamanaka Y, Mukai M, Shimamura M et al . Suppression of PI3K/mTOR pathway rescues LLC cells from cell death induced by hypoxia. Biochem Biophys Res Commun 2005; 330: 318–26. [DOI] [PubMed] [Google Scholar]
- 15. Endo H, Murata K, Mukai M, Ishikawa O, Inoue M. Activation of insulin‐like growth factor signaling induces apoptotic cell death under prolonged hypoxia by enhancing endoplasmic reticulum stress response. Cancer Res 2007; 67: 8095–103. [DOI] [PubMed] [Google Scholar]
- 16. Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S. Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett 1996; 395: 143–7. [DOI] [PubMed] [Google Scholar]
- 17. Halperin EC, Perez CA, Brady LW. Principles and Practice of Radiation Oncology, 5th edn. Philadelphia: Lippincott, Williams & Wilkins, 2008. [Google Scholar]
- 18. Chinnaiyan P, Allen GW, Harari PM. Radiation and new molecular agents, part II: targeting HDAC, HSP90, IGF‐1R, PI3K, and Ras. Seminars Radiation Oncol 2006; 16: 59–64. [DOI] [PubMed] [Google Scholar]
- 19. Kaneko H, Yu D, Miura M. Overexpression of IGF‐I receptor in HeLa cells enhances in vivo radioresponse. Biochem Biophys Res Commun 2007; 363: 937–41. [DOI] [PubMed] [Google Scholar]
- 20. Koumenis C, Wouters BG. ‘Translating’ tumor hypoxia. unfolded protein response (UPR)‐dependent and UPR‐independent pathways. Mol Cancer Res 2006; 4: 423–36. [DOI] [PubMed] [Google Scholar]
- 21. Moretti L, Cha YI, Niermann KJ, Lu B. Switch between apoptosis and autophagy: radiation‐induced endoplasmic reticulum stress? Cell Cycle 2007; 6: 793–8. [DOI] [PubMed] [Google Scholar]
- 22. Ferri KF, Kroemer G. Organelle‐specific initiation of cell death pathways. Nat Cell Biol 2001; 3: E255–63. [DOI] [PubMed] [Google Scholar]
- 23. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5: 726–34. [DOI] [PubMed] [Google Scholar]
- 24. Kouroku Y, Fujita E, Tanida I et al . ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine‐induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14: 230–9. [DOI] [PubMed] [Google Scholar]
- 25. Ogata M, Hino SI, Saito A et al . Autophagy is activated for cell survival after ER stress. Mol Cell Biol 2006; 26: 9220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yorimitsu T, Nair U, Yang Z, Klionsky DJ. ER stress triggers autophagy. J Biol Chem 2006; 281: 30299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paglin S, Hollister T, Delohery T et al . A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 2001; 61: 439–44. [PubMed] [Google Scholar]
- 28. Paglin S, Lee NY, Nakar C et al . Rapamycin‐sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF‐7 cells. Cancer Res 2005; 65: 11 061–70. [DOI] [PubMed] [Google Scholar]
- 29. Daido S, Yamamoto A, Fujiwara K, Sawaya R, Kondo S, Kondo Y. Inhibition of the DNA‐dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res 2005; 65: 4368–75. [DOI] [PubMed] [Google Scholar]
- 30. Ravikumar B, Vacher C, Berger Z et al . Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585–95. [DOI] [PubMed] [Google Scholar]
- 31. Kizaka‐Kondoh S, Inoue M, Harada H, Hiraoka M. Tumor hypoxia: a target for selective cancer therapy. Cancer Sci 2003; 94: 1021–8. [DOI] [PMC free article] [PubMed] [Google Scholar]