

Abstract
Pancreatic ductal adenocarcinoma (PDA) is one of the most debilitating malignancies in humans. A thorough understanding of the cytogenesis of this disease will aid in establishing successful treatments. We have developed an animal model which uses adult HrasG12V and KrasG12V transgenic rats in which oncogene expression is regulated by the Cre/loxP system and neoplastic lesions are induced by injection of adenovirus‐expressing Cre recombinase. When adenovirus with Cre recombinase under the control of the CMV enhancer/chicken β‐actin (CAG) promoter (Ad‐CAG‐Cre) is injected into the pancreatic duct of these animals, pancreatic neoplasias develop. Pathologically, the origin of these lesions is duct, intercalated duct, and centroacinar cells, but not acinar cells. The present study was undertaken to test the effect of acinar cell‐specific oncogenic ras expression. Adult transgenic rats were injected with adenovirus with Cre recombinase under the control of the acinar cell‐specific promoters amylase (Ad‐Amy‐Cre) and elastase‐1 (Ad‐Ela‐Cre) or under the control of the non‐specific CAG promoter. Injection of either Ad‐Amy‐Cre or Ad‐Ela‐Cre into the pancreatic ducts of transgenic animals in which oncogenic Kras is tagged with hemagglutinin (HA), HA‐KrasG12V rats resulted in expression of oncogenic ras in acinar cells but not in duct, intercalated duct, or centroacinar cells. Notably, injected animals did not develop any observable proliferative or neoplastic lesions. In marked contrast, injection of Ad‐CAG‐Cre resulted in pancreatic cancer development within 4 weeks. These results indicate that adult acinar cells are refractory to Ras oncogene activation and do not develop neoplasia in this model. (Cancer Sci2009)
Pancreatic ductal adenocarcinoma (PDA) is a highly lethal disease, which is usually diagnosed in an advanced state. Most patients die within 1 year of diagnosis, ( 1 ) and the 5‐year survival rate is <5%.( 2 ) Understanding of the cytogenesis of PDA offers new directions for targeted therapeutic approaches to combat this disease.
Previously, we reported on an animal model in which pancreatic neoplasia was induced in adult HrasG12V transgenic rats by injection of adenovirus with Cre recombinase under the control of the CMV enhancer/chicken β‐actin (CAG) promoter into the pancreatic duct.( 3 ) In these animals, it was shown that duct, intercalated duct, centroacinar, and acinar cells were all infected with the adenovirus, but induced pre‐neoplastic and neoplastic lesions were shown to express only duct cell‐specific characteristics and not acinar cell‐specific characteristics. Moreover, proliferative lesions were not observed in acinar cells. Therefore, we hypothesized that PDA does not develop from adult pancreatic acinar cells in this model.
The present study was undertaken to directly test the capability of mature acinar cells to develop into a neoplastic lesion. Transgenic rats with an Hras or hemagglutinin (HA)‐tagged Kras oncogene were injected with Cre recombinase expressing adenoviruses in which Cre expression was under the control of promoters specifically active in acinar cells. Mature acinar cells in injected rats did express active Ras proteins, but did not develop any proliferative or neoplastic lesions.
Materials and Methods
Generation of transgenic rats. For the generation of transgenic rats conditionally expressing human KrasG12V, we first made a cDNA fragment encoding the human Kras4BG12V with a 3× HA tag sequence at its 5′ end (HA‐KrasG12V). The HA‐KrasG12V cDNA was subcloned into the SacI/KpnI site of pCALNL5 (DNA Bank, RIKEN Bio Resource Center, Ibaraki, Japan)( 4 , 5 ) to produce pCALNLHAKras. pCALNLHAKras was digested with SalI/HindIII. The purified cassette (Fig. 1A) was injected into the pronuclei of Sprague–Dawley rats (CLEA Japan, Tokyo, Japan). Techniques used for the generation of transgenic rats were the same as those reported previously.( 3 , 6 ) A total of 265 injected eggs were transplanted into pseudo‐pregnant Sprague–Dawley rats. Of 37 potential transgenic rats screened, four male and one female rat were shown by PCR to carry the transgene. Transgenic founder rats were mated with Sprague–Dawley rats, and offspring were screened for the presence of the transgene by PCR analysis of genomic DNA isolated from tail biopsies at the age of 3 weeks. The following primers were used: 5′‐TCTGGATCAAATCCGAACGC‐3′, 5′‐TGACCTGCTGTGTCGAGAAT‐3′. Two founder rats carrying a CALNLHAKrasG12V transgene transmittable to descendent generations (Kras301and Kras327) and two founder rats (Kras409 and Kras417) carrying a non‐tagged KrasG12V transgene were established using the same cassette (data not shown). In this study, we used Kras301 and Kras327. Hras250 rats conditionally expressing human HrasG12V were generated as previously described. ( 3 ) They were maintained in plastic cages in an air‐conditioned room with a 12‐h light/12‐h dark cycle. All experiments were conducted according to the Guidelines for Animal Experiments of the Nagoya City University Graduate School of Medical Sciences.
Figure 1.
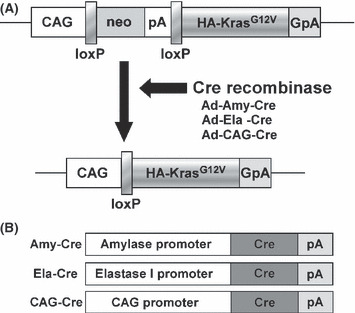
Conditional expression of KrasG12V transgene. (A) The CALNLHAKrasG12V transgene is comprised of a hybrid CMV enhancer/chicken β‐actin (CAG) promoter, a cassette for the neomycin resistance gene flanked by loxP sites, and a sequence containing a human KrasG12V with a hemagglutinin (HA)‐tag. Infection with the Cre recombinase‐expressing adenovirus results in Cre‐mediated recombination of the transgene and removal of the neo‐coding region and its associated mRNA polyadenylation signal, generating a functional HA‐KrasG12V gene expression unit. GpA, rabbit‐ β‐globin poly(A) site; pA, SV40 early poly(A) site. (B) Cre recombinase with nuclear localization signal expressing adenovirus in which Cre expression is under the control of three different promoters: the amylase promoter and the elastase‐1 promoter which are active in acinar cells, and the CAG promoter which is a nonspecific promoter.
Preparation of adenovirus vectors. Adenoviruses in which either the mouse amylase‐2 or the rat elastase‐1 promoter drove the expression of Cre recombinase (Ad‐Amy‐Cre or Ad‐Ela‐Cre) (Fig. 1B) were prepared as described previously.( 7 ) Recombinant adenovirus vectors carrying the Cre gene (Ad‐CAG‐Cre) (Fig. 1B) and empty adenovirus vector were prepared as described previously.( 3 ) Recombinant adenovirus vectors were amplified in HEK‐293 cells and then purified using Vivapure Adenopack (Vivascience, Hannover, Germany). The titer of the adenovirus was determined by using the Rapid titer kit (Clontech, Mountain View, CA, USA). The virus stock was concentrated to 1.0 × 1010 pfu/mL.
Induction of active Ras in the pancreas. Adenovirus vectors were injected into the pancreatic ducts of 12‐week‐old adult male rats through the common duct as previously reported. To induce active Ras specifically in acinar cells, adenoviruses (6 × 108 pfu/rat) in which the expression of Cre recombinase was under the control of acinar cell specific promoters, either the amylase‐2 (Ad‐Amy‐Cre) or elastase‐1 (Ad‐Ela‐Cre) promoter, were used. To induce active Ras non‐specifically, adenoviruses (6 × 108 pfu/rat) in which the expression of Cre recombinase was under the control of the non‐specific CAG promoter were used.
Western blotting. Western blot analysis and detection of activated Ras protein was performed using a Ras Activation Assay kit (Upstate, Lake Placid, NY, USA) as described previously. ( 3 , 8 ) Concentrations of the proteins were determined by Bio‐Rad Protein assay. Proteins were separated by SDS‐PAGE. After transfer to a polyviniliden defluoride membrane, the membrane was blocked with 5% nonfat milk and then incubated for 1 h at room temperature with primary antibodies. The following antibodies were used: anti‐Ras, clone Ras10 (Upstate) diluted 1:4000; HA‐probe (Y‐11; Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:1,000; and monoclonal anti‐β‐actin (A5441; Sigma, St Louis, MO) diluted 1:10 000. The primary antibodies were detected using HRP‐conjugated secondary antibodies (Southern Biotechnology Associates, Birmingham, AL, USA) and ECL plus (GE Heallthcare UK, Buckinghamshire, UK).
Immunostaining. Tissues were fixed in 10% formalin or 4% paraformaldehyde fixative and embedded in paraffin. For Ki67, proliferating cell nuclear antigen (PCNA), and HA‐tag staining, sections were boiled for 10 min in a 10‐mM citrate buffer (pH 6.0) and then allowed to cool in PBS for 30 min before incubation with antibodies. For anti‐α‐amylase staining, section slides were incubated for 10 min in a 0.1% trypsin solution at 37°C and then washed in PBS for 5 min before incubation with antibodies.
Before staining, each section was blocked with 10% goat serum (Nichirei Bio Science, Tokyo, Japan) for 5 min at room temperature. The slides were incubated overnight at 4°C with primary antibodies against Ki67antigen (NCL‐Ki67‐p; Novocastra Laboratories, Newcastle, UK), diluted 1:3000; PCNA (clone PC10; DakoCytomation, Glostrup, Denmark), diluted 1:50; HA‐Tag (6E2; Cell Signaling, Danvers, MA, USA), diluted 1:100; or anti‐α‐amylase (A8273; Sigma, St Louis, MO, USA), diluted 1:200. Slides were incubated with secondary antibodies conjugated with Alexa Fluor488, 546, and 647 (Invitrogen, Carlsbad, CA, USA), and images were obtained with a FLUOVIEW FV300 confocal microscope (Olympus, Tokyo, Japan) or a BZ‐9000 fluorescence microscope (Keyence, Osaka, Japan).
Results
Targeted activation of HA‐KrasG12V transgenes in mature acinar cells. Injection of transgenic rats with Cre recombinase expressing adenovirus resulted in excision of the stuffer DNA between the CAG promoter and the transgene and consequent expression of the transgene in infected cells (Fig. 1A). Kras301/ 327 rats were injected with Ad‐Amy‐Cre or Ad‐Ela‐Cre. Expression of HA‐KrasG12V was observed only in amylase‐positive acinar cells and not in duct, centroacinar, intercalated duct, or islet cells (Table 2) (Fig. 2A; data for Ad‐Ela‐Cre is identical to that of Ad‐Amy‐Cre). Some acinar cells with nuclei with an “owl‐eye” or “ground glass” appearance, which are generally used for identification of virus‐infected cells,( 9 ) in rats treated with Ad‐Amy‐Cre or Ela‐Cre were also positive for both amylase and HA (Fig. 2A‐d,e). All acinar cells positive for HA were entirely negative for Ki67 (Fig. 2A).
Table 2.
Target cell and tumor type in HrasG12Vand hemagglutinin (HA)‐KrasG12Vtransgenic rats
| Virus vector | Target cells | Tumor yield | |||
|---|---|---|---|---|---|
| Acinar cells | Centroacinar cells | Duct cells | Acinar cells | Duct cells | |
| Amy‐Cre | + | − | − | − | − |
| Ela‐Cre | + | − | − | − | − |
| CAG‐Cre( 3 ) | + | + | + | − | + |
Figure 2.
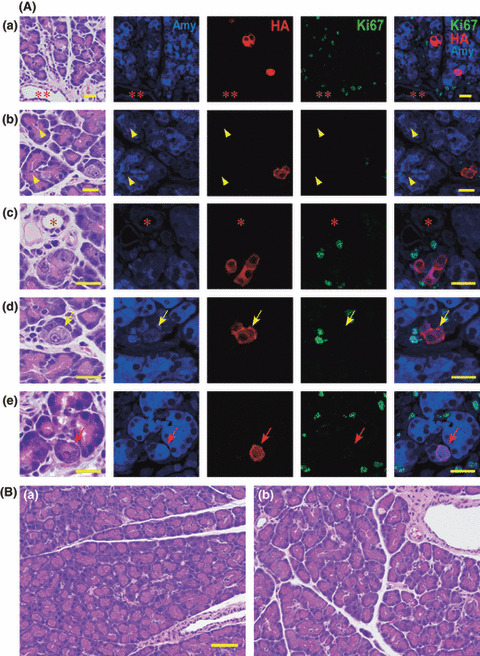
Acinar cell‐specific expression of hemagglutinin (HA)‐KrasG12V. (A) Localization of amylase (blue) protein, HA‐KrasG12V (red) and Ki67 (green) at 2 days after injection of virus with Ad‐Amy‐Cre (a, b, c, d) and Ela‐Cre (e). All the HA‐KrasG12V positive cells (red) were acinar cells; expression was not observed in duct cells (**), centroacinar cells (yellow arrowhead), or small duct cells (*). Most virally infected acinar cells positive for HA‐KrasG12V were indistinguishable from non‐infected acinar cells by hematoxylin–eosin staining. Some infected acinar cells have nuclei with a so‐called “owl‐eye” (yellow arrows) or “ground glass” (red arrows) appearance. Ki67 (green) is not present in the nuclei of the cells expressing HA‐KrasG12V (red). Bar, 20 μm. (B) None of the Ad‐Amy‐Cre (a) or the Ad‐Ela‐Cre (b) groups displayed any pancreatic lesions, even after 8 weeks. Bar, 50 μm.
Lack of PDA development by targeted activation of RasG12V in mature acinar cells. None of the Kras301/327 rats injected with Ad‐Amy‐Cre or Ela‐Cre developed pancreatic lesions (Ad‐Amy‐Cre, 0 out of 5; Ad‐Ela‐Cre, 0 out of 7) after 8 weeks (Fig. 2B, Table 1). Similarly, none of the Hras250 rats injected with Ad‐Amy‐Cre or Ad‐Ela‐Cre (6 × 108 pfu/rat) developed pancreatic lesions (Ad‐Amy‐Cre, 0 out of 7; Ad‐Ela‐Cre, 0 out of 8) after 8 weeks (Table 1). In addition, Kras301/327 rats injected with higher titers of Ad‐Amy‐Cre (6 × 109 pfu/rat) did not develop pancreatic lesions (data not shown). Finally, tumor induction was not observed in injected Kras301/327 rats even after 6 months (data not shown).
Table 1.
Pancreas tumor induction by activation of HrasG12Vor hemagglutinin (HA)‐KrasG12Voncogene after Cre‐adenovirus injection
| Oncogene | Virus vector | Number of rats with tumor (%) |
|---|---|---|
| HrasG12V | Amylase‐Cre | 0/7 (0) |
| Elastase‐Cre | 0/8 (0) | |
| CAG‐Cre | 30/35 (87.5) | |
| HA‐KrasG12V | Amylase‐Cre | 0/5 (0) |
| Elastase‐Cre | 0/7 (0) | |
| CAG‐Cre | 22/22 (100) |
Neoplasia development by activation of RasG12V transgenes in ductular cells. Both Kras301/327 and Hras250 rats injected with Ad‐CAG‐Cre (6 × 108 pfu/rat) developed pancreatic neoplasias: 22 of 22 Kras301/327 rats and 30 of 35 and Hras250 rats after 2 to 4 weeks (Table 1), as observed in our previous report.( 3 ) Pancreatic neoplasias were also observed in Kras301/327 rats injected with lower titers of Ad‐CAG‐Cre (6 × 107 pfu/rat) (data not shown). Activation of the transgene in the pancreatic ductal lesions of Kras301/327 rats was shown by Western blotting using anti‐HA antibody (Fig. 3). The expression of HA‐KrasG12V was detected in pancreatic intraepithelial neoplasia (PanIN) and neoplastic lesions, but not in normal‐looking pancreatic duct cells or stromal cells (Fig. 4A). Ki67 or PCNA and HA were positive in PanIN lesions (Fig. 4B) and in many neoplastic cells (Fig. 4C).
Figure 3.
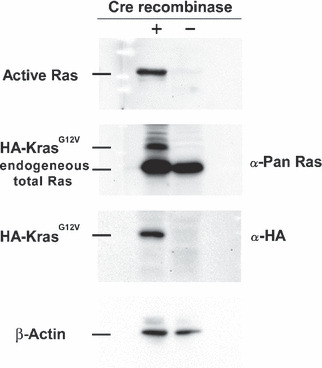
Transgene activation in Kras301 and 327 by Western blotting. A high level of active Ras and hemagglutinin (HA)‐KrasG12V were detected in the pancreas of the Ad‐CAG‐Cre‐treated rats. The amount of active Ras was analyzed by RBD (Ras‐binding domain of Raf‐1) pull‐down assay followed by Western blotting with anti‐pan Ras antibody. HA‐KrasG12V and endogeneous total Ras was detected using anti‐Pan Ras antibody. HA‐KrasG12V was detected using anti‐HA antibody. β‐Actin was used as a loading control.
Figure 4.
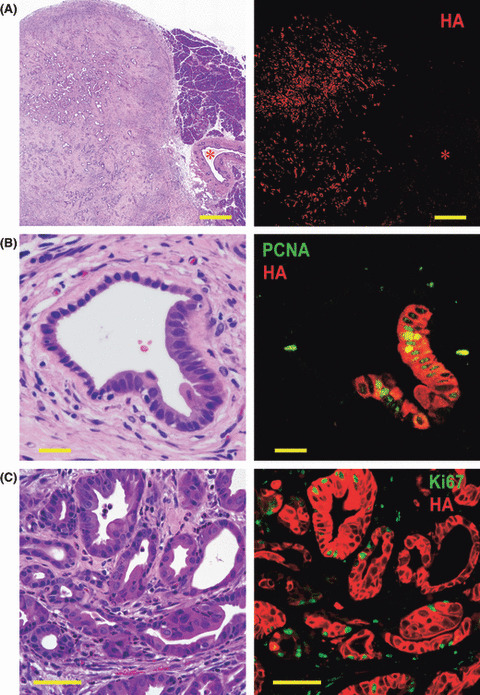
Pancreatic ductal adenocarcinoma (PDA) induced by injection of Ad‐CAG‐Cre in Kras301/327 rats. (A) The expression of hemagglutinin (HA)‐KrasG12V (red) was seen only in PDA lesions (on the left of photo), and not in stromal cells, acinar cells (on the right of photo), or normal pancreatic duct cells (*). Bar, 500 μm. (B) A pancreatic intraepithelial neoplasia (PanIN) lesion was surrounded by fibrous tissue with some infiltration of inflammatory cells. Expression of proliferating cell nuclear antigen (PCNA) (green) and HA protein (red) in a PanIN lesion in rats of the CAG‐Cre group. PCNA is preferentially expressed in PanIN cells. Bar, 20 μm. (C) Expression of Ki67 (green) and HA protein (red) in PDA cells. Many PDA cells (red) are simultaneously positive for Ki67. Bar, 50 μm.
Discussion
The morphological and molecular signatures associated with human pancreas tumors suggests that duct epithelium is responsible for the development of PDA, but it remains unclear whether other pancreatic cells might also contribute to the cytogenesis of these lesions. In our previous study using the Hras250 rat, 4 weeks after injection of adenovirus with Cre recombinase under the control of the constitutive CAG promoter, proliferative lesions in the duct epithelium, intercalated ducts, and centroacinar cells were widespread, but we could not detect any proliferative lesions in acinar cells; moreover, subsequent neoplastic lesions expressed only duct cell‐specific characteristics and not acinar cell‐specific ones.( 3 ) We have obtained essentially identical results with Kras transgenic rats as we did with Hras250 rats (data not shown). These results suggest that PDAs may arise from centroacinar cells, intercalated duct, or pancreatic duct epithelium, but not from acinar cells.
The current study was undertaken to clarify whether mature acinar cells in adult rats could be induced to develop to PDA by targeted activation of oncogenic ras. Activation of oncogenic ras in acinar cells did not lead to the development of any observable pancreatic lesions, while nonspecific activation of oncogenic ras in the pancreas resulted in rapid development of pancreatic neoplasias. Our results clearly show that conditional expression of oncogenic ras in acinar cells in fully developed pancreas tissue does not result in induction of neoplasia in this model (Table 1).
Previous reports in which Kras was activated in immature acinar cells during embryonic development( 10 , 11 ) suggested that acinar‐ductal metaplasia played a role in the development of PDA. In these models, premalignant acinar‐ductal metaplasia and acinar tumor mixed with duct‐like lesions developed in transgenic mice. This acinar‐ductal metaplasia, however, may have occurred before the pancreas fully developed. Our model, on the other hand, targets mature acinar cells which express digestive enzymes, amylase and/or elastase, and these cells do not undergo acinar‐ductal metaplasia in response to ras activation.
Our results are in agreement with a recent study in which the distribution of K‐RAS2 gene mutations was extensively examined in surgically resected pancreata from human patients and which concluded that ductal neoplasms of the human pancreas did not appear to arise from acinar cells.( 12 )
Kras mutations were not observed in pancreatic acinar cell carcinoma (ACC) induced in mature rats by administration of azaserine.( 13 ) Furthermore, alterations in the APC/β‐catenin pathway were detected in 23.5% of human ACC,( 14 ) but mutation of Kras was not observed.( 15 , 16 ) Thus, it is possible that APC/β‐catenin or another pathway, but not necessarily Kras activation, is involved in ACC development.
While pancreas cancer in the hamster model is also believed to arise from ductal epithelial hyperplasia,( 17 , 18 ) several studies using transgenic mice( 11 , 19 , 20 , 21 , 22 , 23 , 24 ) suggest that PDA may develop from acinar cells. In most of these studies, however, oncogenic stimuli are present during embryonic development, prior to the development of a mature pancreas. Therefore, the acinar cells which were activated and developed into neoplasias in these models could very well be at a different developmental stage to the acinar cells which are present in a mature pancreas. This is important because the majority of PDA patients are 60 years of age or older. It is highly unlikely that an oncogenic insult occurring in the uterus is the root cause of most of these PDAs. Moreover, epidemiological studies indicate the incidence of PDA is closely related to lifestyle.( 25 ) Therefore, PDA most likely develops from cells in the mature pancreas. Consequently, pancreas tumor models in which the oncogenic insult occurs during embryonic development are unlikely to be appropriate for determining the cytogenesis of human PDA.
Two models( 22 , 23 , 24 ) use conditional activation of Cre recombinase to active oncogenic ras in adult animals: one model( 22 ) uses the tet‐off system to control expression of Cre recombinase and the other model( 23 , 24 ) uses the tamoxifen‐estrogen receptor system to control nuclear localization of Cre recombinase. These studies had slightly conflicting results. In one study, expression of oncogenic ras in adult acinar cells did not by itself induce pancreatic lesions; additional treatment causing chronic pancreatitis was also needed.( 22 ) In the other study, expression of oncogenic ras was sufficient to induce PanIN‐like lesions.( 23 , 24 ) In our model, we clearly showed that while expression of oncogenic ras is sufficient to induce duct, intercalated duct, and/or centroacinar cells to develop into pancreatic cancers, it is not sufficient to induce acinar cells to develop into pancreatic cancers. Whether these discrepancies are due to experimental procedures, the nature of the Cre recombinase constructs used, or differences between mice and rats remains to be resolved. There are however, a few readily apparent differences. In our rat system, there is no expression of Cre recombinase in the animal until injection of adenovirus‐expressing Cre recombinase, and the expression of Cre recombinase is transient. In the model which uses tamoxifen, on the other hand, Cre recombinase is expressed during embryonic development, but nuclear localization is regulated by tamoxifen.( 23 , 24 ) In this model, however, there was a low level of tamoxifen‐independent recombination events resulting in expression of oncogenic ras in embryonic acinar cells.( 24 ) It is possible that embryonic acinar cells expressing oncogenic ras did not fully differentiate in the adult pancreas; for example, in the mouse colon expression of KrasG12V inhibits differentiation.( 26 ) Therefore, it is possible that in the tamoxifen‐estrogen regulated model,( 23 , 24 ) the acinar cells which were activated to undergo metaplasia to duct‐like cells in the adult were not actually mature acinar cells. The other obvious difference is that in the model regulated by the tet‐off system, two events were required to induce pancreas cancer: activation of oncogenic Kras and chronic pancreatitis.( 22 ) Chronic pancreatitis would very likely result in the death of mature acinar cells and their replacement from a proliferative compartment. It is possible that these replacement cells are not fully mature acinar cells, again suggesting the possibility that the acinar cells which underwent metaplasia to duct‐like cells were not actually mature acinar cells.
The primary aim of this study was to determine whether activation of oncogenic Kras in mature, digestive enzyme‐secreting acinar cells would lead to pancreatic lesions. Our findings support our earlier hypothesis that PDA does not develop from Kras activation in mature acinar cells. It is possible, however, that PDA could develop from Kras activation in immature acinar cells, and in this regard we would like to emphasize the results of Guerra et al. ( 22 ) in which activation of Kras in the mature pancreas accompanied by chronic pancreatitis resulted in induction of PDA in transgenic mice. Importantly, chronic pancreatitis has been shown to be one of the main risk factors for PDA development in humans.( 27 , 28 )
Other factors which may influence PDA development in our model are inflammation and fibrosis. Shortly after infection of pancreatic tissue with Cre recombinase carrying adenovirus to activate the Kras transgene, infiltration of macrophages and lymphocytes could be observed. This infiltration is presumably in response to viral infection. A moderate degree of inflammation, however, was still observed in the stromal tissue surrounding the tumors when PDA developed. These finding suggest that inflammation may play a role in PDA development in this model. However, interaction between the immune system and tumors is complex and whether inflammation actually promotes PDA development in this model remains to be examined.
A current study has demonstrated that the fibrous element accompanying inflammation can also play an important role in cancer development.( 29 ) This aspect of PDA development in our model also remains to be examined.
In summary, while there are discrepancies between different animal models of pancreatic cancer, our results indicate that expression of oncogenic ras in fully mature acinar cells does not induce cell proliferation or result in development of any pancreatic lesions. Thus, we conclude that mature acinar cells are not the origin of PanIN or pancreatic neoplasia in this model.
Acknowledgments
We thank Dr T. Shirai (Nagoya City University) for his kind advice for histological examination and assistance in histological specimen preparation. This work was supported in part by a Grant‐in‐Aid for Scientific Research (C) from Japan Society for the Promotion of Science; a Grant‐in‐Aid for Cancer Research (15‐2, 16‐13, 17S‐6, 20S‐8) from the Ministry of Health, Labour and Welfare, Japan; a Grant‐in‐Aid for Research on Nanotechnical Medical (H19‐Nano‐Ippan‐014) from the Ministry of Health, Labour, and Welfare of Japan; a Grant‐in‐Aid for Research on Risk of Chemical Substances (H19‐Kagaku‐Ippan‐006) from the Ministry of Health, Labour, and Welfare of Japan; and a Grant‐in‐Aid for Nagoya Ohjinkai Young Investigators Medical Research Award from Nagoya City University, Japan.
References
- 1. Beger HG, Rau B, Gansauge F, Poch B, Link KH. Treatment of pancreatic cancer: challenge of the facts. World J Surg 2003; 27: 1075–84. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin 2007; 57: 43–66. [DOI] [PubMed] [Google Scholar]
- 3. Ueda S, Fukamachi K, Matsuoka Y et al. Ductal origin of pancreatic adenocarcinomas induced by conditional activation of a human Ha‐ras oncogene in rat pancreas. Carcinogenesis 2006; 27: 2497–510. [DOI] [PubMed] [Google Scholar]
- 4. Kanegae Y, Lee G, Sato Y et al. Efficient gene activation in mammalian cells by using recombinant adenovirus expressing site‐specific Cre recombinase. Nucleic Acids Res 1995; 23: 3816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Niwa H, Yamamura K, Miyazaki J. Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene 1991; 108: 193–9. [DOI] [PubMed] [Google Scholar]
- 6. Asamoto M, Ochiya T, Toriyama‐Baba H et al. Transgenic rats carrying human c‐Ha‐ras proto‐oncogenes are highly susceptible to N‐methyl‐N‐nitrosourea mammary carcinogenesis. Carcinogenesis 2000; 21: 243–9. [DOI] [PubMed] [Google Scholar]
- 7. Minami K, Okuno M, Miyawaki K et al. Lineage tracing and characterization of insulin‐secreting cells generated from adult pancreatic acinar cells. Proc Natl Acad Sci U S A 2005; 102: 15116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukamachi K, Imada T, Ohshima Y, Xu J, Tsuda H. Purple corn color suppresses Ras protein level and inhibits 7,12‐dimethylbenz[a]anthracene‐induced mammary carcinogenesis in the rat. Cancer Sci 2008; 99: 1841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi TK, Sato S, Tsubota K, Takamura E. Cytological evaluation of adenoviral follicular conjunctivitis by cytobrush. Ophthalmologica 1991; 202: 156–60. [DOI] [PubMed] [Google Scholar]
- 10. Schmid RM. Acinar‐to‐ductal metaplasia in pancreatic cancer development. J Clin Invest 2002; 109: 1403–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res 2003; 63: 2016–9. [PubMed] [Google Scholar]
- 12. Shi C, Hong SM, Lim P et al. KRAS2 mutations in human pancreatic acinar‐ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin. Mol Cancer Res 2009; 7: 230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Longnecker DS, Curphey TJ. Adenocarcinoma of the pancreas in azaserine‐treated rats. Cancer Res 1975; 35: 2249–58. [PubMed] [Google Scholar]
- 14. Abraham SC, Wu TT, Hruban RH et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta‐catenin pathway. Am J Pathol 2002; 160: 953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Terhune PG, Memoli VA, Longnecker DS. Evaluation of p53 mutation in pancreatic acinar cell carcinomas of humans and transgenic mice. Pancreas 1998; 16: 6–12. [DOI] [PubMed] [Google Scholar]
- 16. Van Kranen HJ, Vermeulen E, Schoren L et al. Activation of c‐K‐ras is frequent in pancreatic carcinomas of Syrian hamsters, but is absent in pancreatic tumors of rats. Carcinogenesis 1991; 12: 1477–82. [DOI] [PubMed] [Google Scholar]
- 17. Tsutsumi M, Kondoh S, Noguchi O et al. K‐ras gene mutation in early ductal lesions induced in a rapid production model for pancreatic carcinomas in Syrian hamsters. Jpn J Cancer Res 1993; 84: 1101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsutsumi M, Konishi Y. Precancerous conditions for pancreatic cancer. J Hepatobiliary Pancreat Surg 2000; 7: 575–9. [DOI] [PubMed] [Google Scholar]
- 19. Tuveson DA, Zhu L, Gopinathan A et al. Mist1‐KrasG12D knock‐in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res 2006; 66: 242–7. [DOI] [PubMed] [Google Scholar]
- 20. Hingorani SR, Petricoin EF, Maitra A et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003; 4: 437–50. [DOI] [PubMed] [Google Scholar]
- 21. Wagner M, Luhrs H, Kloppel G, Adler G, Schmid RM. Malignant transformation of duct‐like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology 1998; 115: 1254–62. [DOI] [PubMed] [Google Scholar]
- 22. Guerra C, Schuhmacher AJ, Canamero M et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K‐Ras oncogenes in adult mice. Cancer Cell 2007; 11: 291–302. [DOI] [PubMed] [Google Scholar]
- 23. Habbe N, Shi G, Meguid RA et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 2008; 105: 18913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De La OJ, Emerson LL, Goodman JL et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A 2008; 105: 18907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiao L, Mitrou PN, Reedy J et al. A combined healthy lifestyle score and risk of pancreatic cancer in a large cohort study. Arch Intern Med 2009; 169: 764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haigis KM, Kendall KR, Wang Y et al. Differential effects of oncogenic K‐Ras and N‐Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet 2008; 40: 600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lowenfels AB, Maisonneuve P, Cavallini G et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med 1993; 328: 1433–7. [DOI] [PubMed] [Google Scholar]
- 28. Malka D, Hammel P, Maire F et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 2002; 51: 849–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sangai T, Ishii G, Kodama K et al. Effect of differences in cancer cells and tumor growth sites on recruiting bone marrow‐derived endothelial cells and myofibroblasts in cancer‐induced stroma. Int J Cancer 2005; 115: 885–92. [DOI] [PubMed] [Google Scholar]