

Abstract
Leukocyte trafficking, which is critically regulated by chemokines and their receptors, shares many of the characteristics of tumor cell infiltration and metastasis. Expression of CC chemokine receptor 4 (CCR4) by tumor cells is associated with skin involvement, but CCR4 also has an important role in normal and tumor immunity. In a subset of patients with CCR4+ T‐cell leukemia/lymphoma, the tumor cells themselves function as regulatory T (Treg) cells, contributing to tumor survival in the face of host antitumor immune responses. In other types of cancers, the chemokines TARC/CCL17 and MDC/CCL22, specific ligands for CCR4 that are produced by tumor cells and the tumor microenvironment, attract CCR4+ Treg cells to the tumor, where they create a favorable environment for tumor escape from host immune responses. A novel humanized anti‐CCR4 monoclonal antibody (mAb) has been developed, the Fc region of which is defucosylated to enhance antibody‐dependent cellular cytotoxicity by increasing its binding affinity to Fc receptor on effector cells. We are now conducting a phase I clinical trial of this anti‐CCR4 mAb in patients with CCR4+ T‐cell leukemia/lymphoma in Japan (clinical trials gov. identifier: NCT00355472). Anti‐CCR4 mAb could be an ideal treatment modality for many different cancers, not only to directly kill the CCR4+ tumor cells, but also to overcome the suppressive effect of CCR4+ Treg cells on the host immune response to tumor cells. (Cancer Sci 2006; 97: 1139–1146)
Chemokines are a group of structurally related small cytokines (8–14 kDa) that commonly induce directed migration of different leukocyte populations. Based on the arrangement of the N‐terminal conserved cysteine residues, chemokines are classified into four subfamilies: CC, CXC, C and CX3C. In contrast to other cytokines, chemokines interact with and signal via a group of seven transmembrane G protein‐coupled receptors. Chemokines play essential roles in migration and homing of lymphocyte subpopulations, which express specific sets of chemokine receptors in accordance with their lineage and functional maturation. Thus, individual T‐cell subsets express a specific set of chemokine receptors imparting unique migration and tissue homing properties. It is now recognized that chemokine receptors can serve as useful markers for different T‐cell subsets.( 1 ) Manipulating a specific chemokine network leads to altered behavior of the corresponding functional T‐cell subset.
Role of CC chemokine receptor 4 in human immunity
The human immune system is regulated by balanced cytokine production controlled by two distinct T helper (Th) cell subsets, Th1 and Th2 cells.( 2 ) Th1 cells produce interferon (IFN)‐γ and interleukin (IL)‐2, whereas Th2 cells produce IL‐4 and IL‐5; Th1 cells play a critical role in cellular immunity, whereas Th2 cells are involved in humoral immunity. In addition to Th1 and Th2 cells, regulatory T (Treg) cells also play an important role in maintaining immune balance. Treg populations described to date include the FOXP3+ naturally occurring CD4+CD25+ Treg subset (CD4+CD25+ Treg), IL‐10‐producing Treg (IL‐10‐Treg), and transforming growth factor‐β (TGF‐β)‐producing Treg (TGF‐β‐Treg).( 3 , 4 ) Disruption of the balance between these immune cells leads to various diseases, including cancer. Because most cancer patients do not develop a satisfactory antitumor immune response, effective activation of such a response is a critical factor in cancer immunotherapy in order to overcome strong immunosuppression in the cancer‐bearing host.( 5 , 6 ) CC chemokine receptor 4 (CCR4) is important for regulating immune balance and is known to be expressed selectively on Th2 cells and Treg cells.( 7 , 8 , 9 ) With special regard to the relationship between CD4+CD25+ Treg and CCR4, Yagi et al. reported that human FOXP3‐transduced CD4+CD25− naive T cells upregulate the expression of CCR4 and acquire the functions of CD4+CD25+ Treg cells.( 10 ) This observation indicates a significant relationship between FOXP3 and CCR4. Furthermore, we have additionally demonstrated two important characteristics of the relationship between CCR4 and Th2 and Treg cells in human immunity. First, FOXP3+ naturally occurring CD4+CD25+ Treg cells are mainly present in the CD4+CD25highCCR4+ population, but not in the CD4+CD25highCCR4− population. Second, human peripheral CCR4+ T cells can be divided into two separate functional subpopulations (Th2 and Treg cells) according to their level of expression of CD25; that is, the CCR4+CD25−&low population is greatly enriched in Th2 cells, whereas the CCR4+CD25high population is greatly enriched in Treg cells (data not shown). The chemokines thymus‐ and activation‐regulated chemokine/CC chemokine ligand 17 (TRC/CCL17) and macrophage‐derived chemokine (MDC)/CCL22 exert their effects via CCR4.( 7 )
Role of CCR4 in cancer and viral infection
Adult T‐cell leukemia/lymphoma
Most adult T‐cell leukemia/lymphoma cells are positive for CCR4. Adult T‐cell leukemia/lymphoma (ATLL), a peripheral T‐cell neoplasm most often composed of highly pleomorphic lymphoid cells, is caused by the human retrovirus human T‐cell lymphotropic virus type‐1 (HTLV‐1).( 11 ) Yoshie et al. first reported frequent expression of CCR4 in ATLL cells in 2002. Using reverse transcription–polymerase chain reaction (RT‐PCR), they analyzed peripheral blood mononuclear cell (PBMC) samples from healthy donors (n = 13) and ATLL patients (n = 24) and found a highly elevated expression of CCR4 in 22 of 24 ATLL cases.( 12 ) Subsequently, we reported an extensive immunohistochemical study using tissue biopsy samples from 103 ATLL patients. We have demonstrated that tumor cells from a large majority of patients with ATLL express CCR4 (91/103, 88.3%).( 13 )
CCR4 expression in ATLL is significantly associated with skin involvement. ATLL cells frequently infiltrate into a variety of organs such as the lymph nodes, spleen, liver and skin.( 11 ) Leukocyte trafficking, which is critically regulated by chemokines and their receptors, shares many characteristics with tumor cell infiltration and metastasis. For example, CXCR4, a chemokine receptor, is expressed more abundantly in breast cancer tissues than in normal breast tissues; its ligand, CXCL12, is present in organs such as the lymph nodes, bone marrow and lung, where breast cancer cells tend to metastasize.( 14 ) It is thus conceivable that chemokines and their receptors play important roles in tissue infiltration of ATLL cells, analogous to the assumed mechanism in metastatic breast cancer. We have shown that the extent of CCR4 expression is significantly associated with skin involvement. Importantly, of the organs infiltrated by ATLL cells, only the skin was TARC/CCL17‐positive or MDC/CCL22‐positive by RT‐PCR. In addition, cutaneous lymphocyte antigen (CLA)‐positive ATLL showed significantly more skin involvement than CLA‐negative cases. Thus, the representative ATLL cells infiltrating into skin expressed both CCR4 and CLA.( 13 ) The molecular interactions that facilitate ATLL migration to and residence in the skin are fundamental to the normal physiology of immunosurveillance. ATLL cells also home to the skin by virtue of interactions with dermal capillary endothelial cells; CCR4 and CLA on the ATLL cells recognize TARC/CCL17 and E‐selectin on endothelial cells in the skin, respectively, and ATLL cells subsequently extravasate into the dermis. ATLL cells often display an affinity for epidermal cells and cluster around Langerhans’ cells, forming Pautrier's microabscesses, which can be observed by histological examination (Fig. 1). This process is principally guided by the interactions of CCR4, αEβ7 and the T‐cell receptor (TCR) expressed on ATLL cells with MDC/CCL22, E‐cadherin and major histocompatibility complex (MHC) class II molecules expressed on Langerhans’ cells in the epidermis, respectively. This skin‐homing capacity of ATLL cells is almost identical to that seen in cases of mycosis fungoides.( 15 )
Figure 1.
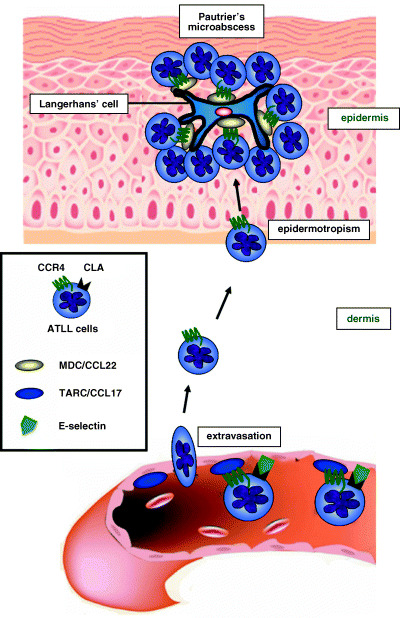
Skin homing capacity of adult T‐cell leukemia/lymphoma (ATLL) cells. CC chemokine receptor 4 (CCR4) and cutaneous lymphocyte antigen (CLA) on ATLL cells recognize TARC/CCL17 and E‐selectin on endothelial cells in the skin, respectively, and ATLL cells subsequently extravasate into the dermis. ATLL cells often display an affinity for epidermal cells and cluster around Langerhans’ cells, forming Pautrier's microabscesses, principally caused by the interaction of CCR4 on ATLL cells with MDC/CCL22 expressed on Langerhans’ cells in the epidermis.
CCR4‐expressing ATLL cells may function as CD4+CD25+ Treg cells. ATLL has a very poor prognosis, with the median survival time of patients with the acute or lymphoma subtype of ATLL being less than 1 year. ATLL patients are in a severely immunocompromised state, leading to frequent and severe infectious complications and to an unfavorable outcome.( 11 ) Although the frequency of opportunistic infections is much higher in patients with ATLL than other hematological malignancies, the underlying mechanisms responsible for this remain obscure. Recently, several investigators including ourselves have reported that FOXP3, which is a master control gene of naturally occurring CD4+CD25+ Treg cells, is expressed in the tumor cells from a subset of patients with ATLL.( 16 , 17 , 18 , 19 , 20 ) Because ATLL cells from a subset of patients have a CD4+CD25+CCR4+FOXP3+ phenotype, these tumors might originate from CD4+CD25+CCR4+FOXP3+ Treg cells, based on their phenotypic characteristics. However, whether ATLL cells actually function as Treg cells has not been clearly demonstrated. We have recently demonstrated that CCR4‐positive ATLL cells from a subset of patients do indeed function as CD4+CD25+ Treg cells in an autologous setting (data not shown). This finding provides significant insights into understanding the immunopathogenesis of ATLL, that is, how HTLV‐1‐infected cells can survive in the face of host immune responses (Fig. 2). It also adds to our understanding of the cause of ATLL patients’ severely immunocompromised state.
Figure 2.
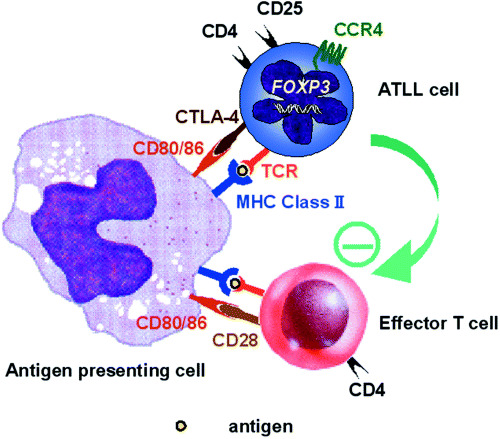
CD4+CD25+ regulatory T cell (Treg) capacity of adult T‐cell leukemia/lymphoma (ATLL) cells. CD4+CD25+CCR4+ ATLL cells from a subset of patients suppress the proliferation of CD4+ autologous non‐ATLL cells in the presence of autologous antigen‐presenting cell in response to T‐cell receptor (TCR) stimulation.
T‐cell and NK‐cell neoplasms other than ATLL
Differential expression patterns of chemokine receptors in different T‐cell lymphomas were reported for the first time in 2000 by Jones et al. and this may explain, in part, the distinctive patterns of spread associated with each tumor subtype.( 21 ) Subsequently, we carried out immunohistochemical analysis of CCR4 expression, evaluating 169 cases of T‐cell and natural killer (NK)‐cell lymphomas other than ATLL. CCR4 was rarely expressed in the three well‐defined subtypes, namely, precursor T‐lymphoblastic lymphoma, ALK‐positive anaplastic large cell lymphoma (ALCL) and extranodal NK/T‐cell lymphoma. CCR4 was expressed in tumor cells from a subset of patients with angioimmunoblastic T‐cell lymphoma (34.8%), mycosis fungoides in transformation (41.2%), ALK‐negative ALCL (66.7%) and peripheral T‐cell lymphoma (PTCL) unspecified (38.0%). We next focused on PTCL unspecified and analyzed the clinical significance of CCR4 expression and its association with FOXP3. Multivariate analysis showed that CCR4 expression was an independent and significant unfavorable prognostic factor in patients with PTCL unspecified.( 22 ) Ohshima et al. also reported that CCR4 expression was an unfavorable prognostic factor for this disease.( 23 ) A significant correlation was found between the presence of CCR4 and FOXP3 mRNA in affected lymph node cells obtained from patients with PTCL unspecified,( 22 ) suggesting a possible association of CCR4‐positive tumors with Treg cells, and thereby with an immunocompromised state. The reason why CCR4 expression is an unfavorable prognostic factor not only for ATLL( 13 ) but also for PTCL unspecified( 22 , 23 ) remains unclear, so further investigations are needed.
Diffuse large B‐cell lymphoma
CC chemokine receptor 4 is known to be expressed in a subset of CD4‐positive T‐cells( 7 ) and in certain subtypes of T‐cell lymphomas.( 13 , 15 , 21 , 22 ) We have reported one apparently unique case of a CCR4‐positive B‐cell lymphoma. This patient with diffuse large B‐cell lymphoma (DLBCL) presented with a giant skin ulcer of the right axilla. Immunohistochemical and quantitative RT‐PCR analysis showed that the tumor B‐cells were positive for CCR4, and that TARC/CCL17 was expressed at extremely high levels in the lymphoma‐affected skin.( 24 ) These observations suggest that the interaction between CCR4 and TARC/CCL17 played a significant role in the involvement of this DLBCL in the skin, similar to what has been observed in certain subsets of T‐cell malignancies.( 13 , 15 )
Hodgkin's lymphoma
Hodgkin's lymphoma (HL) is characterized by the presence of a small number of tumor cells in a rich background of T and B cells, macrophages and other inflammatory cells.( 25 ) The contribution of these abundant non‐tumor cells to the pathogenesis of HL had been poorly understood for a long time. Because a small number of HL tumor cells can survive the host anticancer immune response, it seems likely that HL cells may have developed mechanisms that inhibit effective immune responses by the host against the tumor cells. We have demonstrated that HL tumor cells produce TARC/CCL17 and MDC/CCL22, and migratory CD4+CCR4+ cells induced by HL cells are hyporesponsive to TCR stimulation and suppress the activation and proliferation of effector CD4+ T‐cells in an autologous setting. Using double immunostaining, we further demonstrated that HL cells in the affected lymph nodes are surrounded by a large number of lymphocytes expressing both CCR4 and FOXP3. These findings indicate that the migratory cells induced by HL cells function as Treg cells to create a favorable environment for the tumor cells to escape from host immunity (Fig. 3).( 26 ) These observations are consistent with reports that HL tumor cells express high levels of TARC/CCL17 and MDC/CCL22,( 25 , 27 , 28 , 29 ) and that an elevated serum level of TARC/CCL17 is an unfavorable prognostic factor in patients with HL.( 28 )
Figure 3.
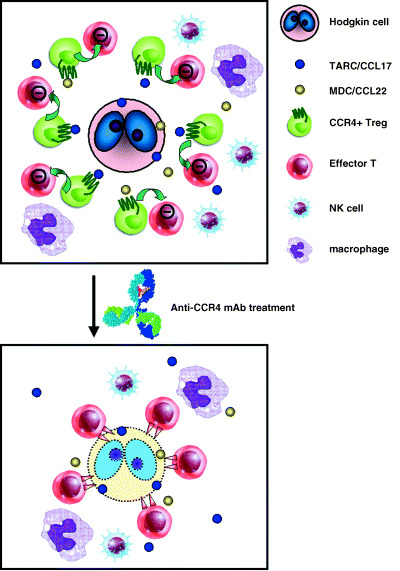
Specific recruitment of CC chemokine receptor 4 (CCR4)‐positive regulatory T (Treg) cells in Hodgkin's lymphoma (HL) fosters immune privilege, making CCR4 on Treg cells a novel target for cancer immunotherapy. HL tumor cells produce TARC/CCL17 andr MDC/CCL22, and migratory CD4+CCR4+ cells induced by HL cells function as Treg cells, so that these cells create a favorable environment for tumor immune escape (upper panel). The recognition of the importance of Treg cells not only in HL but also in other cancers will allow the rational design of more effective treatments. The depletion of Treg cells using an anti‐CCR4 monoclonal antibody in patients with cancers including HL could become a promising strategy for overcoming the suppressive effect of CCR4+ Treg cells on the host's immune response to tumor cells, thereby boosting antitumor immunity (lower panel).
B‐cell chronic lymphocytic leukemia
B‐cell chronic lymphocytic leukemia (B‐CLL) is characterized by the accumulation of monoclonal lymphocytes that appear to originate from mature, antigen‐experienced B‐cells. The clinical course of CLL is usually indolent, and most patients with CLL are asymptomatic. Ghia et al. reported that B‐CLL cells produce MDC/CCL22 and attract CCR4+ T‐cells.( 30 ) Furthermore, Beyer et al. reported that patients with B‐CLL show significantly increased frequencies of CD4+CD25high Treg cells in their peripheral blood compared to healthy individuals.( 31 ) These findings indicate that B‐CLL cells also create a favorable environment for the tumor cells to escape from host immunity by recruiting CCR4+ Treg cells, as observed in HL.
Solid tumors
A higher frequency of Treg cells in peripheral blood was reported in patients with various solid tumors including breast cancer, colorectal cancer, esophageal cancer, gastric cancer, hepatocellular carcinoma, lung cancer, melanoma, ovarian cancer and pancreatic cancer.( 5 ) In an elegant study, Curiel et al. showed how Treg cells migrate to tumors − under the influence of the interaction between CCR4 and MDC/CCL22 – and provided evidence that Treg cells create a favorable environment for tumors and tumor growth in humans.( 9 ) Their first important finding stemmed from experiments with tumor ascites from patients with tumors positive for HER2. Treg cells isolated from this tumor ascites inhibited the proliferation of autologous T effector cells stimulated in vitro with myeloid dendritic cells pulsed with HER2 peptides. Moreover, the Treg cells reversed the inhibition of tumor growth induced by adoptive transfer of tumor‐specific effector T‐cells. Their second important finding was that MDC/CCL22 produced in the tumor microenvironment mediates Treg trafficking through CCR4. They showed that malignant ascites induced CD4+CD25+ T cells to migrate in vitro, and that this migration was specifically blocked by antibody to MDC/CCL22. High levels of MDC/CCL22 were detected in tumor ascites, and MDC/CCL22 mRNA was expressed in tumor tissues and macrophages in the ascites. Moreover, Treg cells expressed high levels of CCR4. The role of MDC/CCL22 in tumor trafficking was validated in vivo in immunodeficient mice transplanted with primary human ovarian tumors. Treg cells transplanted into these mice migrated into the tumor, but treatment of the mice with antibody to MDC/CCL22 decreased this migration. Their final important finding was a strong inverse correlation between Treg content and survival in patients with ovarian carcinoma.
Epstein–Barr virus (EBV) infection
Nakayama et al. have recently reported that EBV‐infected B cells acquire the ability to produce TARC/CCL17 and MDC/CCL22, and suggested that the production of these factors, which attract CCR4+ Treg cells, may help EBV‐infected B cells evade immune surveillance by the host immune system.( 32 ) This finding indicates that specific recruitment of CCR4+ Treg cells plays an important role not only for cancers as described in the present manuscript, but also for viruses to survive in the face of immune responses.
CCR4 as a novel molecular target for antibody‐based therapy
Overview of antibody‐based therapy The use of monoclonal antibody (mAb) for the treatment of cancer has become a promising approach over the last few years, as exemplified by the great success of mAb against HER2, trastuzumab, used for the treatment of metastatic breast cancer overexpressing HER2;( 33 ) mAb against CD20, rituximab, used for DLBCL;( 34 ) mAb against vascular endothelial growth factor (VEGF), bevacizumab, used for metastatic colorectal cancer;( 35 ) and mAb against epidermal growth factor receptor (EGFR), cetuximab, used for locoregionally advanced squamous‐cell carcinoma of the head and neck.( 36 ) Recombinant mAb of the human IgG1 isotype, including mouse/human chimeric, humanized, and human IgG1, are commonly used therapeutically. Through their human constant region (Fc), therapeutic antibodies can mediate antibody‐dependent cellular cytotoxicity (ADCC), or complement‐dependent cytotoxicity.( 37 ) Genetic analysis of Fc receptor (FcR) polymorphisms has clearly shown that ADCC is one of the critical effector functions responsible for the clinical efficacy of therapeutic antibodies. A superior clinical response of patients carrying the FcRIIIa allotype with higher affinity to anti‐CD20 IgG1 rituximab was observed in contrast to patients with the low‐affinity allotype.( 38 ) Breast cancer patients that responded to anti‐HER2 IgG1 trastuzumab with complete or partial remission were found to mediate in vitro ADCC with trastuzumab better than non‐responders.( 39 )
Absence of fucose from human IgG1 complex‐type oligosaccharides is critical for enhancing ADCC
One IgG molecule contains two asparagine N‐linked oligosaccharide sites in its Fc region.( 40 ) The general structure of IgG N‐linked oligosaccharides is complex‐type, characterized by a mannosyl‐chitobiose core with or without bisecting N‐acetylglucosamine (GlcNAc)/L‐fucose and other chain variants including the presence or absence of galactose and sialic acid. Several groups have reported that ADCC enhancement can be achieved by manipulating human IgG1 subclass antibody oligosaccharides. ADCC requires the presence of oligosaccharides in the Fc region and is sensitive to change in the oligosaccharide structure.( 41 ) Of all the sugar components in the oligosaccharide, galactose( 42 ), bisecting‐GlcNAc( 43 ), and fucose( 44 , 45 ) have been reported to affect ADCC (Fig. 4a). Shinkawa et al. reported that fucose was the most important of these sugar components; defucosylation of humanized anti‐IL‐5 receptor antibody or chimeric anti‐CD20 antibody enhanced their ADCC >50‐fold. Compared with fucose, the involvement of bisecting‐GlcNAc in ADCC was minimal, and galactose did not contribute to ADCC.( 45 ) The influence of non‐fucosylated oligosaccharide on ADCC has also been reported by Shields et al. using humanized anti‐HER2 IgG1 and humanized anti‐IgE IgG1.( 44 ) They showed that the improved ADCC of defucosylated IgG1 resulted from its improved binding to FcRIIIa.
Figure 4.
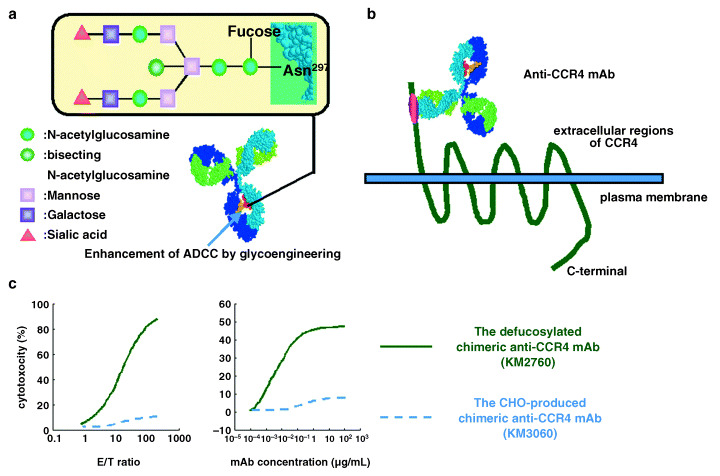
The defucosylated anti‐CC chemokine receptor 4 (CCR4) monoclonal antibody (mAb). (a) Structure of oligosaccharide in the Fc region of human IgG1 is shown. Antibody‐dependent cellular cytotoxicity (ADCC) requires the presence of oligosaccharides in the Fc region and is sensitive to change in the oligosaccharide structure. Of all the sugar components in the oligosaccharide, fucose has the most important influence on ADCC. Defucosylation of human IgG1‐type antibody is one of the most powerful ways to improve antibody effector function. (b) Anti‐CCR4 mAb, whose Fc region is defucosylated to enhance ADCC activity by increasing its binding affinity to Fc receptor on effector cells, was developed. The defucosylated anti‐CCR4 mAb recognizes the N‐terminal portion of the CCR4 molecule. (c) The defucosylated chimeric anti‐CCR4 IgG1 mAb KM2760 mediates much higher ADCC against CCR4/EL4 cells in the presence of human peripheral blood mononuctear cells as effector cells compared with the highly fucosylated, but otherwise identical, Chinese hamster ovary (CHO)‐produced anti‐CCR4 IgG1 KM3060 in standard 4‐h 51Cr release assays. KM2760 needed far fewer effector cells to achieve the same level of ADCC as KM3060 (left panel). KM2760 needed much lower mAb concentrations to achieve the same ADCC as KM3060 (right panel).
We further investigated the relationship between enhanced ADCC and antigen density on target cells using IgG1 antibodies with reduced fucose. Using mouse T‐cell leukemia/lymphoma cell line EL4‐derived transfectants with different levels of expression of exogenous human CD20 as target cells, ADCC of fucose variants of chimeric IgG1 antibodies specific for these antigens was measured. We also investigated IgG1 binding to NK cells and NK cell activation during ADCC induction to elucidate the mechanism by which low‐fucose IgG1 induces ADCC against target cells with low antigen expression. Low‐fucose IgG1 showed potent ADCC at low antigen densities at which their corresponding high‐fucose counterparts could not induce measurable ADCC. Quantitative analysis revealed that fucose depletion reduced by 3‐fold the amount of antigen required on target cells for the same level of ADCC induction. IgG1 binding to NK cells was increased by ligating IgG1 with clustered antigen, especially for low‐fucose IgG1. Upregulation of an activation marker, CD69, on NK cells, particularly the CD56dim subset, in the presence of the antibody and target cells was much greater for the low‐fucose antibody. In conclusion, fucose removal from IgG1 could reduce the amount of antigen required for ADCC induction via efficient recruitment and activation of NK cells.( 46 )
CCR4 is a suitable target for antibody‐based therapy in patients with CCR4‐positive neoplasms
We have developed the defucosylated chimeric anti‐CCR4 IgG1 mAb KM2760 (Fig. 4b).( 47 ) KM2760 shares the same variable region with mouse mAb KM2160, with which we have carried out extensive immunohistochemical studies on a variety of leukemias/lymphomas.( 13 , 22 , 24 ) Because CCR4 is usually a selective marker of T‐cell leukemia/lymphoma as described in the present manuscript, the effectiveness of KM2760 for T‐cell malignancy was evaluated. Mouse EL4 cells were transduced with human CCR4 cDNA and the highest CCR4‐expressing clone was selected and designated CCR4/EL4. KM2760 mediated much higher ADCC against CCR4/EL4 cells in the presence of human PBMC as effector cells compared with the highly fucosylated, but otherwise identical, Chinese hamster ovary (CHO)‐produced anti‐CCR4 IgG1 KM3060 in standard 4‐h 51Cr release assays. KM2760 needed far fewer effector cells to achieve the same ADCC level as KM3060 (Fig. 4c). This feature should be therapeutically beneficial because the number of effector cells capable of penetrating into large tumor masses could be limiting, as effectors would be outnumbered by tumor cells in clinical settings. In addition, KM2760 needed much lower mAb concentrations to achieve the same ADCC as KM3060 (Fig. 4c).( 47 ) This feature also should be clinically beneficial because defucosylated mAb could help to overcome the problem of cost, which is currently one of the major disadvantages of antibody drugs.
Next, in order to compare the antitumor activity of KM2760 and KM3060 in vivo, we constructed a human PBMC‐engrafted T acute lymphoblastic leukemia (T‐ALL) mouse model using CCR4/EL4 to determine ADCC efficacy with human effector cells. In this model, KM2760 showed significantly higher antitumor efficacy than KM3060, indicating that it retains its high potency in vivo.( 47 ) KM2760 also induced a robust ADCC activity against CCR4‐positive human ATLL and T‐cell leukemia/lymphoma lines in the presence of PBMC from healthy individuals in a dose‐dependent manner in vitro.( 17 , 47 )
Unlike established cell lines, tumor cells present in patients with ATLL or PTCL may be heterogeneous and may behave differently in KM2760‐induced ADCC. Furthermore, the ultimate goal of immunotherapy is to obtain sufficient tumoricidal activity by simply administering mAb in vivo. However, the therapeutic effect may be hampered by the immunocompromised state common in patients with ATLL or PTCL. Therefore, we next examined KM2760‐induced ADCC in several patients with ATLL or PTCL in an autologous setting. ADCC mediated by autologous effector cells was generally lower than that mediated by allogeneic effector cells from healthy individuals, although in some cases the former was comparable to the latter. This suggests that the ATLL or PTCL patients’ PBMC retain substantial KM2760‐induced ADCC‐effector potential.( 17 ) KM2760 also demonstrated promising antitumor activity in the CCR4‐positive ATLL‐ or PTCL‐bearing mouse xenograft models (data not shown).
We also note the characteristics of CCR4 as a target antigen for antibody‐based therapy. Soluble forms of CCR4 have been detected in the culture supernatants of a variety of CCR4‐positive T‐cell lines (data not shown). This characteristic should be therapeutically beneficial because anti‐CCR4 mAb can directly bind to CCR4 expressed on tumor cells without competition from soluble forms of CCR4. In addition, we evaluated CCR4 expression in a variety of human tissues by both quantitative RT‐PCR and immunohistochemistry and confirmed its selective expression in CD4+ lymphocytes (data not shown). Because CCR4 protein seems not to be expressed in normal human tissue parenchymal cells, direct organ injury caused by administration of anti‐CCR4 mAb in humans would not be expected.
In conclusion, the CCR4 molecule could be a suitable target for novel antibody‐based therapy for patients with CCR4‐positive T‐cell neoplasms.
CCR4 on Treg cells is a novel target for immunotherapy in patients with different cancers
Awareness of the importance of Treg cells in different cancers will allow the rational design of more effective treatments; increased frequencies of Treg cells are now accepted as an important mechanism for tumor escape from host immuniy in several different types of cancer, as described in the present manuscript. The depletion of Treg cells in patients with tumors could become a promising strategy for boosting tumor‐associated antigen‐specific immunity (Fig. 3).( 6 ) For instance, experimental depletion of Treg cells in mice with tumors improved immune‐mediated tumor clearance,( 48 ) and enhanced the response to immune‐based therapy.( 49 ) Also, a clinical trial of a fully human mAb that blocks CTLA‐4 has already established the possibility of effecting substantial tumor destruction, although at the expense of some autoimmunity.( 50 ) We have previously reported that in vitro KM2760 treatment significantly reduced the proportion of CD4+CCR4+ T‐cells in PBMC obtained from healthy individuals.( 26 ) We have also reported that in vitro KM2760 treatment reduced the level of FOXP3 mRNA expression in parallel with the CCR4 mRNA level in PBMC.( 17 ) Collectively, these findings suggest that anti‐CCR4 mAb could function as a novel inducer of effective anticancer immunity by depleting the CCR4+ Treg cells, and therefore could be used as a novel strategy for treatment of many different cancers. However, it should be noted that anti‐CCR4 mAb might result in induction of autoimmunity, as reported in the anti‐CTLA‐4 mAb study.( 50 ) With this in mind, before clinical use, anti‐CCR4 mAb underwent extensive evaluation in cynomolgus monkeys. Because the binding affinity of this mAb to monkey and human CCR4 was almost identical, we believe that the former are suitable for evaluating safety in humans. The anti‐CCR4 mAb was found not to cause any notable acute or chronic clinical toxicity in cynomolgus monkeys (unpublished data from Kyowa Hakko Kogyo, Tokyo, Japan).
Conclusions and future directions
Based on the promising results of our previous work regarding CCR4,( 13 , 17 , 22 , 24 , 26 , 46 , 47 ) we are currently conducting a phase I clinical trial of defucosylated humanized anti‐CCR4 mAb (KW‐0761) in patients with CCR4‐positive T‐cell leukemia/ lymphoma in Japan (clinical trials gov. identifier: NCT00355472). We believe that anti‐CCR4 mAb could be an ideal treatment modality for patients with CCR4‐positive neoplasms, and could also be used as a novel strategy for treatment of many other diseases, such as HL, B‐CLL, ovarian cancer and EBV‐associated disease, to overcome the suppressive effect of CCR4+ Treg cells on the host's immune response to tumor or virus‐infected cells. Furthermore, combination treatment with this mAb and conventional chemotherapy seems to be very promising, in analogy to what has been observed for trastuzumab,( 33 ) rituximab,( 34 ) bevacizumab( 35 ) and cetuximab.( 36 )
However, we must be especially alert to the dangers of manipulating the human immune system, as observed in clinical trials of TGN1412,( 51 ) a humanized mAb designed as an agonist of the CD28 receptor on T lymphocytes that stimulates the production and activation of these cells. It was hoped that this product would benefit patients with B‐CLL or autoimmune diseases such as multiple sclerosis or rheumatoid arthritis. At this juncture, in light of the events in the TGN1412 trial, we should carefully reconsider how preclinical studies for evaluating the safety of newly developed drugs in humans are to be carried out. Academia, the pharmaceutical and biotechnology industries, and regulators must work together to prevent such clinical‐research nightmares from happening in the future, because they (of course including ourselves) have a responsibility to the public to develop novel medical treatments for patients suffering from incurable diseases.
Acknowledgments
We are grateful to all the members of the CCR4 research group, Drs Toshihiko Ishii, Atsushi Inagaki, Hiroki Yano, Masaki Ri, Shigeru Kusumoto, Mikinori Miyazaki, Hirokazu Komatsu and Shinsuke Iida at the Department of Internal Medicine and Molecular Science, Nagoya City University Graduate School of Medical Sciences, Dr Hiroshi Inagaki at the Department of Clinical Pathology, Nagoya City University Graduate School of Medical Sciences, Drs Atae Utsunomiya, Yoshifusa Takatsuka and Shogo Takeuchi at the Department of Hematology, Imamura Bun‐in Hospital, Drs Toshitada Takahashi and Yoshiki Akatsuka at the Division of Immunology, Aichi Cancer Center Research Institute, Dr Shigeo Nakamura at the Department of Pathology and Clinical Laboratories, Nagoya University Hospital, Drs Takashi Uchiyama and Kazunori Imada at the Department of Haematology and Oncology, Graduate School of Medicine, Kyoto University, and Drs Kenya Shitara, Shiro Akinaga, Rinpei Niwa and Toshio Ota at Tokyo Research Laboratories, Kyowa Hakko Kogyo, for continuing fruitful collaborations for many years. We wish to thank Ms Chiori Fukuyama, Mr Seizo Nagaya and Ms Satomi Ozeki for their skillful technical assistance. This work was supported by a Grant‐in‐Aid for General Scientific Research (RU), a Grant‐in‐Aid for Scientific Research on Priority Areas (RU) from the Ministry of Education, Culture, Science, Sports, and Technology, and a Grant‐in‐Aid for Cancer Research (RU) from the Ministry of Health, Labor, and Welfare, Japan.
The authors have declared that no conflict of interest exists.
References
- 1. Yoshie O, Imai T, Nomiyama H. Chemokines in immunity. Adv Immunol 2001; 78: 57–110. [DOI] [PubMed] [Google Scholar]
- 2. Mosmann TR, Sad S. The expanding universe of T‐cell subsets: Th1, Th2 and more. Immunol Today 1996; 17: 138–46. [DOI] [PubMed] [Google Scholar]
- 3. O’Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med 2004; 10: 801–5. [DOI] [PubMed] [Google Scholar]
- 4. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299: 1057–61. [DOI] [PubMed] [Google Scholar]
- 5. Zou W. Regulatory T cells, tumor immunity and immunotherapy. Nat Rev Immunol 2006; 6: 295–307. [DOI] [PubMed] [Google Scholar]
- 6. Zou W. Immunosuppressive networks in the tumor environment and their therapeutic relevance. Nat Rev Cancer 2005; 5: 263–74. [DOI] [PubMed] [Google Scholar]
- 7. Imai T, Nagira M, Takagi S et al. Selective recruitment of CCR4‐bearing Th2 cells toward antigen‐presenting cells by the CC chemokines thymus and activation‐regulated chemokine and macrophage‐derived chemokine. Int Immunol 1999; 11: 81–8. [DOI] [PubMed] [Google Scholar]
- 8. Iellem A, Mariani M, Lang R et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+CD25+ regulatory T cells. J Exp Med 2001; 194: 847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Curiel TJ, Coukos G, Zou L et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10: 942–9. [DOI] [PubMed] [Google Scholar]
- 10. Yagi H, Nomura T, Nakamura K et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol 2005; 16: 1643–56. [DOI] [PubMed] [Google Scholar]
- 11. Kikuchi M, Jaffe ES, Ralfkiaer E. Adult T‐cell leukaemia/lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. Pathology and Genetics Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001, 200–3. [Google Scholar]
- 12. Yoshie O, Fujisawa R, Nakayama T et al. Frequent expression of CCR4 in adult T‐cell leukemia and human T‐cell leukemia virus type 1‐transformed T cells. Blood 2002; 99: 1505–11. [DOI] [PubMed] [Google Scholar]
- 13. Ishida T, Utsunomiya A, Iida S et al. Clinical significance of CCR4 expression in adult T‐cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res 2003; 9: 3625–34. [PubMed] [Google Scholar]
- 14. Müller A, Homey B, Soto H et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001; 410: 50–6. [DOI] [PubMed] [Google Scholar]
- 15. Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides. N Engl J Med 2004; 350: 1978–88. [DOI] [PubMed] [Google Scholar]
- 16. Karube K, Ohshima K, Tsuchiya T et al. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T‐cell leukaemia/lymphoma cells. Br J Hematol 2004; 126: 81–4. [DOI] [PubMed] [Google Scholar]
- 17. Ishida T, Iida S, Akatsuka Y et al. The CC chemokine receptor 4 as a novel specific molecular target for immunotherapy in adult T‐cell leukemia/lymphoma. Clin Cancer Res 2004; 10: 7529–39. [DOI] [PubMed] [Google Scholar]
- 18. Kohno T, Yamada Y, Akamatsu N et al. Possible origin of adult T‐cell leukemia/lymphoma cells from human T lymphotropic virus type‐1‐infected regulatory T cells. Cancer Sci 2005; 96: 527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roncador G, Garcia JF, Garcia JF et al. FOXP3, a selective marker for a subset of adult T‐cell leukaemia/lymphoma. Leukemia 2005; 19: 2247–53. [DOI] [PubMed] [Google Scholar]
- 20. Chen S, Ishii N, Ine S et al. Regulatory T cell‐like activity of Foxp3+ adult T cell leukemia cells. Int Immunol 2006; 18: 269–77. [DOI] [PubMed] [Google Scholar]
- 21. Jones D, O’Hara C, Kraus MD et al. Expression pattern of T‐cell‐associated chemokine receptors and their chemokines correlates with specific subtypes of T‐cell non‐Hodgkin lymphoma. Blood 2000; 96: 685–90. [PubMed] [Google Scholar]
- 22. Ishida T, Inagaki H, Utsunomiya A et al. CXCR3 and CCR4 expression in T‐cell and NK‐cell lymphomas with special reference to clinicopathological significance for peripheral T‐cell lymphoma, unspecified. Clin Cancer Res 2004; 10: 5494–500. [DOI] [PubMed] [Google Scholar]
- 23. Ohshima K, Karube K, Kawano R et al. Classification of distinct subtypes of peripheral T‐cell lymphoma unspecified, identified by chemokine and chemokine receptor expression: analysis of prognosis. Int J Oncol 2004; 25: 605–13. [PubMed] [Google Scholar]
- 24. Ishida T, Inagaki H, Kusumoto S et al. CC chemokine receptor 4‐positive diffuse large B‐cell lymphoma involving the skin: a case report. Int J Hematol 2005; 82: 148–51. [DOI] [PubMed] [Google Scholar]
- 25. Stein H, Mann R, Delsol G et al. Classical Hodgkin lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, eds. Pathology and Genetics Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001, 244–53. [Google Scholar]
- 26. Ishida T, Ishii T, Inagaki A et al. Specific recruitment of CCR4‐positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res 2006; 66: 5716–22. [DOI] [PubMed] [Google Scholar]
- 27. Van Den Berg A, Visser L, Poppema S. High expression of the CC chemokine in Reed–Sternberg cells. A possible explanation for the characteristic T‐cell infiltrate in Hodgkin's lymphoma. Am J Pathol 1999; 154: 1685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weihrauch MR, Manzke O, Beyer M et al. Elevated serum levels of CC thymus and activation‐related chemokine (TARC) in primary Hodgkin's disease: potential for a prognostic factor. Cancer Res 2005; 65: 5516–19. [DOI] [PubMed] [Google Scholar]
- 29. Maggio EM, Van Den Berg A, Visser L et al. Common and differential chemokine expression patterns in rs cells of NLP, EBV positive and negative classical Hodgkin lymphomas. Int J Cancer 2002; 99: 665–72. [DOI] [PubMed] [Google Scholar]
- 30. Ghia P, Strola G, Granziero L et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol 2002; 32: 1403–13. [DOI] [PubMed] [Google Scholar]
- 31. Beyer M, Kochanek M, Darabi K et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood 2005; 106: 2018–24. [DOI] [PubMed] [Google Scholar]
- 32. Nakayama T, Hieshima K, Nagakubo D et al. Selective Induction of Th2‐attracting chemokines CCL17 and CCL22 in human B cells by latent membrane protein 1 of Epstein–Barr virus. J Virol 2004; 78: 1665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slamon DJ, Leyland‐Jones B, Shak S et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–92. [DOI] [PubMed] [Google Scholar]
- 34. Coiffier B, Lepage E, Briere J et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large‐B‐cell lymphoma. N Engl J Med 2002; 346: 235–42. [DOI] [PubMed] [Google Scholar]
- 35. Hurwitz H, Fehrenbacher L, Novotny W et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 36. Bonner JA, Harari PM, Giralt J et al. Radiotherapy plus cetuximab for squamous‐cell carcinoma of the head and neck. N Engl J Med 2006; 354: 567–78. [DOI] [PubMed] [Google Scholar]
- 37. Carter P. Improving the efficacy of antibody‐based cancer therapies. Nat Rev Cancer 2001; 1: 118–29. [DOI] [PubMed] [Google Scholar]
- 38. Cartron G, Dacheux L, Salles G et al. Therapeutic activity of humanized anti‐CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcRIIIa gene. Blood 2002; 99: 754–8. [DOI] [PubMed] [Google Scholar]
- 39. Gennari R, Menard S, Fagnoni F et al. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin Cancer Res 2004; 10: 5650–5. [DOI] [PubMed] [Google Scholar]
- 40. Rademacher TW, Homans SW, Parekh RB, Dwek RA. Immunoglobulin G as a glycoprotein. Biochem Soc Symp 1986; 51: 131–48. [PubMed] [Google Scholar]
- 41. Jefferis R, Lund J, Pound JD. IgG‐Fc‐mediated effector functions: molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol Rev 1998; 163: 59–76. [DOI] [PubMed] [Google Scholar]
- 42. Kumpel BM, Wang Y, Griffiths HL, Hadley AG, Rook GA. The biological activity of human monoclonal IgG anti‐D is reduced by β‐galactosidase treatment. Hum Antibodies Hybridomas 1995; 6: 82–8. [PubMed] [Google Scholar]
- 43. Umana P, Jean‐Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody‐dependent cellular cytotoxic activity. Nat Biotechnol 1999; 17: 176–80. [DOI] [PubMed] [Google Scholar]
- 44. Shields RL, Lai J, Keck R et al. Lack of fucose on human IgG1 N‐linked oligosaccharide improves binding to human FcRIII and antibody‐dependent cellular toxicity. J Biol Chem 2002; 277: 26 733–40. [DOI] [PubMed] [Google Scholar]
- 45. Shinkawa T, Nakamura K, Yamane N et al. The absence of fucose but not the presence of galactose or bisecting N‐acetylglucosamine of human IgG1 complex‐type oligosaccharides shows the critical role of enhancing antibody‐dependent cellular cytotoxicity. J Biol Chem 2003; 278: 3466–73. [DOI] [PubMed] [Google Scholar]
- 46. Niwa R, Sakurada M, Kobayashi Y et al. Enhanced natural killer cell binding and activation by low‐fucose IgG1 antibody results in potent antibody‐dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res 2005; 11: 2327–36. [DOI] [PubMed] [Google Scholar]
- 47. Niwa R, Shoji‐Hosaka E, Sakurada M et al. Defucosylated chimeric anti‐CCR4 IgG1 with enhanced antibody‐dependent cellular cytotoxicity shows potent therapeutic activity to T cell leukemia and lymphoma. Cancer Res 2004; 64: 2127–33. [DOI] [PubMed] [Google Scholar]
- 48. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD4+CD25+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol 1999; 163: 5211–18. [PubMed] [Google Scholar]
- 49. Steitz J, Bruck J, Lenz J, Knop J, Tuting T. Depletion of CD25+CD4+ T cells and treatment with tyrosinase‐related protein 2‐transduced dendritic cells enhance the interferon α‐induced, CD8+ T‐cell‐dependent immune defense of B16 melanoma. Cancer Res 2001; 61: 8643–6. [PubMed] [Google Scholar]
- 50. Sanderson K, Scotland R, Lee P et al. Autoimmunity in a phase I trial of a fully human anti‐cytotoxic T‐lymphocyte antigen‐4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol 2005; 23: 741–50. [DOI] [PubMed] [Google Scholar]
- 51. Wood AJ, Darbyshire J. Injury to research volunteers − The clinical‐research nightmare. N Engl J Med 2006; 354: 1869–71. [DOI] [PubMed] [Google Scholar]