

Abstract
The DNA repair system surveys the genome, which is always suffering from exposure to both exogenous as well as endogenous mutagens, to maintain the genetic information. The fact that the basis of this DNA repair system is highly conserved, from prokaryote to mammalian cells, suggests the importance of precise genome maintenance mechanisms for organisms. In the past 15 years, considerable progress has been made in understanding how repair processes interact and how disruptions of these mechanisms lead to the accumulation of mutations and carcinogenesis. In 1993, two groups reported that DNA mismatch repair could be associated with hereditary non‐polyposis colorectal cancer, indicating a connection between faulty DNA repair function and cancer. More recently, an inherited disorder of DNA glycosylase, which removes mutagenic oxidized base from DNA, has been reported in individuals with a predisposition to multiple colorectal adenomas and carcinomas. This is the first report that directly indicates the role of the repair of oxidative DNA in human inherited cancer. Studies from gene knockout mice have elucidated the principal role of these repair systems in the process of carcinogenesis. Moreover, clinical samples derived from cancer patients have shown the direct involvement. This review focuses on the function of DNA mismatch repair and oxidative DNA/nucleotide repair among various DNA repair systems in cells, both of which are essentially involved in the carcinogenesis of gastrointestinal tract cancer. (Cancer Sci 2008; 99: 451–458)
Abbreviations:
- 8‐oxoG
8‐oxo‐7,8‐dihydroguanine
- APC
adenomatous polyposis coli
- CRC
colorectal cancer
- FAP
familial adenomatous polyposis
- HNPCC
hereditary non‐polyposis colorectal cancer
- LOH
loss of heterozygosity
- MAP
MUTYH‐associated polyposis
- MMR
mismatch repair
- MSI
microsatellite instability
- OR
odds ratio
- PCNA
proliferating cell nuclear antigen
- RPA
replication protein A
- SCC
squamous cell carcinoma
Considering the huge spectrum of damage that the genome can suffer from either spontaneously or from exposure to genotoxic environmental agents, it is quite reasonable that cells possess a multitude of mechanisms to confront these events. For example, mutagenic nucleotide substrate and endogenous DNA lesions generated during normal cellular metabolism as well as errors made during replication of undamaged template DNA could be sources for spontaneous mutation. Initially, the molecular mechanisms of spontaneous mutagenesis were elucidated by genetic analyses of Escherichia coli mutator strains, and further research has also demonstrated that the basic mechanisms are well conserved evolutionally among various organisms. However, the most definitive difference between a prokaryote and a mammalian is the response to lethal DNA damage. The prokaryote can tolerate lethal damage by adaptation due to the SOS response; in contrast, the mammal might remove highly damaged cells using a specific self‐avoiding system called apoptosis.
Recently, considerable progress has been made in understanding how the repair process interacts and how disruptions of these mechanisms lead to the accumulation of mutations and carcinogenesis. In particular, important processes have been elucidated from syndromes that predispose some patients to develop cancer and from inductive methods using gene‐targeting approaches, which thus make it possible to investigate those functions in vivo in mice. hMSH2 is the human homolog of bacterial MutS, which is responsible for the correction of base/base mismatch as well as insertion/deletion misalignment with other proteins. In 1993, Fishel et al. reported that mutations of hMSH2 could be identified in the familial lines of patients with HNPCC.( 1 ) At the same time, Leach et al. cloned the MutS homolog using linkage analysis and detected germline mutation of this gene in HNPCC kindreds.( 2 ) These are reports that proved the connection of the disorder of DNA repair function and cancer, particularly in colorectal cancer. Although, oxidative DNA damage has been implicated in cancer etiology, there were no reported human inherited disorders attributed to the repair of oxidative DNA damage by 2002. Al‐Tassan et al. reported that bi‐allelic germline mutations in MUTYH, which removes mutagenic oxidized bases from DNA, are present in individuals with a predisposition to multiple colorectal adenomas and carcinomas.( 3 ) This is the first observation to directly elucidate the role of the repair of oxidative DNA in human inherited cancer.
To date, numerous reports from investigations of the role of DNA repair in counteracting the carcinogenesis process have been published. This review article focuses on the MMR and repair against oxidative DNA/nucleotide damage to clarify their correlations to human gastrointestinal cancers and elucidate their roles in avoiding mutation accumulation and carcinogenesis using gene knockout mice in vivo.
DNA mismatch repair
In mammalian cells, the MMR system is involved in the correction of errors that arise during DNA replication, DNA damage surveillance, and the prevention of recombination between non‐identical sequences.( 4 ) MMR was initially identified for correcting errors made during replication of undamaged template DNA; however, it has been demonstrated that it is involved in the repair or removal of oxidative DNA damage( 5 ) as well as DNA modified by chemicals such as alkylating agent, cisplatin, and 5‐fluorouracil.( 6 ) MLH1 and MSH2 are the MMR proteins that are most frequently implicated in an MMR deficiency.( 7 ) MMR genes (MSH2, MSH3, MSH6, MLH1, MLH3, PMS1, PMS2) are mutated or inactivated by hypermethylation of the promoter, as a somatic, epigenetic phenomenon.( 8 )
Cells with a defective MMR system generate mutations at a rate up to 100‐fold higher than the rate observed in normal cells, both in mammalian cells and tissues of mice in vivo. ( 9 , 10 , 11 ) Loeb proposed that this increased mutation frequency (i.e. mutator phenotype) may facilitate the occurrence of mutations in other genes that govern genetic stability, and regulate the cell cycle and apoptosis, therefore causing carcinogenesis.( 12 ) Changes in the length of nucleotide repeats at microsatellite loci, that is, MSI, were observed in cancer cells, and regarded as an important phenotype that reflects the mutator phenotype of cancer cells with MMR deficiencies.( 13 )
The MMR repair gene knockout mouse was first reported in 1995, 2 years after the MSH2 gene was cloned. Msh2 −/– mice were viable but susceptible to malignancies, particularly lymphomas, at an early age.( 14 , 15 ) Reitmair et al. reported that intestinal carcinomas occurred at an older age while the majority of mice developed lymphoma at an early age.( 16 ) Thereafter, other genes involved in MMR were disrupted by a gene‐targeting technique and phenotypes were reported.( 17 , 18 , 19 , 20 , 21 , 22 ) As summarized in Table 1, among seven genes involved in mismatch recognition in mammalian cells, all but the Pms1 gene knockout mice showed the predisposition of cancer development at a more rapid or slower duration from birth.( 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 ) In general, Msh2 −/–, Mlh1 −/– and Pms2 −/– mice developed lymphoma at an early stage and Msh2 −/–, Mlh1 −/– mice also tended to generate intestinal carcinomas at a relatively later stage. In contrast, Msh3 −/–, Msh6 −/– and Mlh3 −/– mice developed lymphoma and gastrointestinal cancer later than Msh2 −/– and Mlh1 −/– developed lymphomas.
Table 1.
Phenotype of mismatch repair‐deficient mice
| Targeted gene | Increase in mutation frequency † | Cancer susceptibility | Sites of cancer | Reference no. |
|---|---|---|---|---|
| Msh2 | 7–160 | Cancer prone | lymphoma | ( 14, 15, 16 ) |
| GI tract | ||||
| Skin | ||||
| Msh3 | 2 | Low ‡ | GI tract | ( 21 ) |
| Msh6 | 2–13 | Cancer prone | Lymphoma | ( 19 ) |
| GI tract | ||||
| Mlh1 | 7–100 | Cancer prone | GI tract | ( 20, 22 ) |
| Lymphoma | ||||
| Mlh3 | – | Cancer prone | GI tract | ( 17 ) |
| Lymphoma | ||||
| Pms1 | – | No | ( 20 ) | |
| Pms2 | 5–100 | Cancer prone | Lymphoma | ( 20 ) |
| Sarcoma |
Tissue specimens derived from MMR showed increased mutation frequency. The effect of this elevation differs from the target sequence of monitor genes, for example, in the presence or absence of mononucleotide repeat. In general, Msh2 −/–, Mlh1 −/– and Pms2 −/– mice show a high frequency of mutation, whereas Msh3 −/– and Msh6 −/– mice showed relatively low levels of increased mutation frequencies.( 9 , 10 , 11 ) Considering the mismatch processing, the loss of MSH2 or MLH1 function results in a complete depletion of the MutSα/MutLα (and β) and MutSβ/MutLα (and β) hetero‐complex, therefore inactivating the repair of both deletion/insertion misalignment and base/base mismatch. Cells mutated in either one of these two genes therefore show strong mutator phenotypes. In contrast, the mutation of MSH3 or MSH6 results in the depletion of MutSβ/MutLα (and β) or MutSα/MutLα (and β) hetero‐complex, respectively. MutSβ/MutLα (and β) recognizes not only deletion/insertion misalignment but also partially recognizes base/base mismatch, and similarly MutSα/MutLα (and β) recognizes not only base/base mismatch but partially recognizes deletion/insertion misalignment. These overlapping recognition activities suppress the mutator effects. Interestingly, mice that showed a high frequency of spontaneous mutation in their tissue developed tumors in an early stage; in contrast, mice with a mild mutator phenotype developed tumors at a late stage. These observations strongly support the idea of the ‘mutator phenotype in cancer’ proposed by Loeb.( 12 ) However, it is important to note that these observations apply in the case of ‘care taker’ genes. There was a report that the ‘gate keeper’ gene, such as p53‐deficient mice, didn't show elevated mutation frequency.( 23 )
An MSI analysis is indeed an efficient approach for detecting defective MMR, because MMR include proteins from several gene products and MMR genes have no marked hot spots for mutation in their long coding region. However, the reported frequency for MSI‐positive tumors in each malignancy differs widely in the literature.( 24 ) As these discrepancies may relate to problems in the methods used,( 13 ) the authors have used fluorescence‐labeled primers and an auto‐sequencer for laser scanning to detect precisely the alteration of microsatellite loci.( 13 ) As summarized in Table 2a, the frequencies of MSI in esophageal, stomach, and colorectal cancer analyzed using fluorescent primers and an auto‐sequencer were 8%, 24%, and 34.7%, respectively.( 24 , 25 , 26 ) Using this precise fluorescence system, Oda et al. pointed out the qualitative difference in the form of alterations at microsatellite loci.( 27 ) In brief, one alteration was drastic and was frequently observed in more than two analyzed loci, whereas the other was subtle and often only observed in one locus. Therefore, the former and latter alterations might be closely connected to MSI‐H and MSI‐L, respectively. The relationship between MSI and defective MMR may be more complicated than has been suspected, which is fully described in the review article.( 24 ) Interestingly, an extremely high frequency of MSI (all alterations were subtle and might be categorized as MSH‐L) was observed in tumors from MSH2‐deficient mice (A. Egashira, T. Tsuzuki, Y. Maehara, unpublished data, 2007), whereas the reported frequency of MSI in tumors from MMR deficient animals is not always high.( 28 )
Table 2.
Mismatch repair dysfunction in sporadic gastrointestinal tract cancer
|
(a) Frequencies of MSI observed in gastric, stomach, and colorectal cancer | |||
|---|---|---|---|
| MSI † | Reference no. | Frequencies cited in the literature ‡ | |
| (i) Esophagus | 8% | ( 26 ) | 5–25% |
| (MSI‐L 8%) | |||
| (MSI‐H 0%) | |||
| (ii) Stomach | 24% | ( 24 ) | 13–44% |
| (MSI‐L 13%) | |||
| (MSI‐H 11%) | |||
| (iii) Colon | 34.7% | ( 25 ) | 15–50% |
| (MSI‐L 23.1%) | |||
| (MSI‐H 11.6%) | |||
|
(b) Involvement of MMR genes in esophageal, gastric, and colorectal cancer | |||
|---|---|---|---|
| MMR gene | Proportion | Method used for analysis | Reference no. of alteration |
| (i) Esophagus | |||
| MSH2, MLH1, MSH6 | 14% | In vitro MMR assay′ | ( 31 ) |
| MSH3, PMS2 | |||
| MLH1 | 33% | MSP | ( 32 ) |
| MSH2, MLH1 | 28.7% | IHC | ( 33 ) |
| (ii) Stomach | |||
| MLH1 | 31% | MSP, IHC, RT‐PCR | ( 36 ) |
| MLH1 | 32% | MSP, IHC, WB | ( 37 ) |
| MSH2, MLH1 | 0% | IHC | ( 40 ) |
| MSH2, MLH1 | 32–36% | IHC | ( 39 ) |
| (iii) Colon | |||
| MSH2, MLH1 | 7% | Mutation or LOH | ( 43 ) |
| (among MSI) | |||
| MLH1 | 25–64% | IHC (64%), MSP (42%) | ( 46 ) |
| (among MSI‐H,MSS) | Mutation (26%) | ||
| MSH2 (among MSI) | <1% | Mutation | ( 44 ) |
| MSH2, MLH1 | 12% | IHC | ( 45 ) |
| MSH6 (among MSI‐L) | 7–17% | IHC (17%) | ( 47 ) |
| Mutation (7%) | |||
| MSH2, MLH1, MSH6 | 0–12% | IHC | ( 48 ) |
| PMS2 | |||
MSI is analyzed using fluorescent primers and an auto sequencer.( 13 )
Frequencies of MSI are reviewed in the article.( 24 ) MSI, microsatellite instability.
IHC, immunohistochemistry; LOH, loss of heterozygosity; MMR, mismatch repair; MSI, microsatellite instability; MSP, methylation‐specific PCR; RT‐PCR, reverse transcription‐polymerase chain reaction; WB, western blotting.
Because mutations of the MMR genes were identified in the families of patients with HNPCC,( 1 , 2 , 29 ) which are among the most common hereditary human cancers, many studies have reported both the hereditary and sporadic cancers. Germline mutations in one of four major HNPCC‐associated MMR genes, MSH2, MLH1, MSH6 and PMS2, are detected in up to 70–80% of such families. Among all mutations reported as predisposing MMR gene mutations, mutations of MSH2 or MLH1 account for more than 80%, whereas MSH6 and PMS2 mutations account for less than 15% and MLH3 mutations account for only a small percentage.( 8 ) The mutation of the PMS1 gene is questionable, because a re‐examination of the originally reported HNPCC‐like family with a PMS1 mutation showed this family to have an additional MSH2 mutation that co‐segregated with colon cancer in the family.( 30 ) In addition to the relatively lower frequencies of MSH6, PMS2 and MLH1 mutations, mutations in these genes often occurred in the families with atypical HNPCC who showed late onset disease. These characteristics are in line with the phenotype observed in analogous gene‐disrupted mice. Pms1‐knockout mice do not show cancer susceptibility; in contrast to Msh6‐, Pms2‐ and Mlh3‐knockout mice, which show a mild elevation of the mutation frequency and later develop tumors in older age.( 17 , 19 , 20 )
DNA mismatch repair dysfunction in gastrointestinal tract cancers
Table 2b shows the results from several studies analyzing the rate of impaired MMR genes and MSI in sporadic esophageal, gastric and colorectal cancers. Esophageal cancer is the sixth most common cause of cancer death in the world. There are two major histological types of esophageal cancer: SCC and adenocarcinoma. Although the latter type has been increasing, particularly in the USA and Europe, the former histological type is predominant worldwide. Although esophageal cancer is not a tumor associated with the HNPCC spectrum, there are several reports analyzing MSI and/or MMR function. Araki et al. reported that the frequency of MSI was 8% and 4%, respectively, in patients with esophageal SCC in Japan and China.( 26 ) One represented a relatively high frequency of MSI in esophageal carcinoma.( 24 ) Those authors excluded the suspicious MSI/LOH, which could not be distinguished from LOH by the method used. The suspicious MSI/LOH should not be included when considering the high frequencies of LOH observed in esophageal cancer. Uchida et al. reported that no mutation or aberrant expression was found in the MMR genes in esophageal SCC cell lines, but the MMR activity was somehow reduced in 3 of 22 cell lines analyzed.( 31 ) Recently, Guo et al. reported MLH1 hypermethylation in esophageal SCC with MSI.( 32 ) However, neither the frequency of MSI, particularly MSI‐H, nor the deficient expression of MLH1 protein is necessarily high.( 33 ) Furthermore, the role of MMR in carcinogenesis of the esophagus might be low, and these studies are supported by few reports in which MMR‐deficient mice develop esophageal cancer.
Gastric cancer is the second most common extra‐colonic malignancy in HNPCC.( 34 ) A large cohort study in Germany showed that either a MSH2 or MLH1 germline mutation carrier would develop gastric cancer at the rate of 5.2% and 4.3%, respectively.( 35 ) These reports clearly show a connection between germline mutation of the MMR gene and gastric cancer. However, the role of MMR in avoiding sporadic gastric carcinoma is different from that of hereditary disease. The frequencies of MSI in sporadic gastric cancer varies in the literature, from approximately 13–44%.( 24 ) Using precise fluorescent analysis, the frequencies for MSI‐H and MSI‐L were determined to be 11% and 13%, respectively.( 24 ) Sporadic mutations of MMR genes have not been comprehensively explored in gastric cancers, whereas inactivation of MLH1 due to hypermethylation of a promoter lesion are frequently observed in the gastric cancers with MSH‐H.( 36 , 37 ) These alterations of MLH1 are also detected in the adenomas of the stomach.( 38 ) The expression of MMR protein was investigated in several reports, but the frequency also differs in the literature.( 39 , 40 )
HNPCC family members have a lifetime risk of 60–80% for developing CRC,( 35 ) whereas HNPCC might account for 2–5% of all colorectal cancers.( 41 , 42 ) Among all mutations reported in HNPCC, mutations of MSH2 or MLH1 account for more than 80%.( 8 ) Most tumors observed in HNPCC families showed MSI‐H.( 7 ) Therefore, CRC from HNPCC families appears to be characterized by a high frequency of MSI‐H and MMR gene mutation, particularly the MSH2 and MLH1 genes. However, the situation is somewhat different with sporadic CRC. Although the percentages of MSI range from 15 to 50% in the literature,( 24 ) Ikeda et al. reported the frequency of MSI‐L and MSI‐H observed in CRC as 23% and 12%, respectively, using the precise fluorescent system.( 25 ) Table 2b shows that the overall percentage of MMR gene alteration is not high.( 43 , 44 , 45 , 46 , 47 , 48 ) In contrast to the HNPCC families in which tumors with germline mutation of MSH2 or MLH1 are closely connected with MSI‐H, those who develop sporadic CRC with MSI‐H harbor a few somatic MSH2 or MLH1 mutations. Sporadic CRC with MSI‐H frequently shows inactivation of MLH1 due to hypermethylation of a promoter region, which is also observed in the gastric cancer with MSH‐H. Jass et al. emphasized that there might be differences in the setting of tumors in HNPCC families and sporadic CRC with MSI‐H.( 49 ) In addition, the relationship between MSI, in particular MSI‐H, and defective MMR may be highly complicated.( 24 )
Repair against oxidative DNA/nucleotide damage and the pathogenesis of its deficiency in mouse models
Cells are continuously attacked by reactive species. Oxygen radicals produced endogenously in the course of normal cellular metabolism, as well as in response to various environmental mutagens and ionizing radiation, can damage DNA and its precursors. Among a large variety of DNA lesions caused by oxygen radicals, an oxidized form of guanine, 8‐oxoG, is thought to be a key lesion due to its abundance and its possible role in carcinogenesis and aging.( 50 ) 8‐OxoG nucleotides formed in DNA as well as 8‐oxodGTP formed in the nucleotide pool induce mutations. 8‐OxoG has a propensity to form base pairs with adenine as well as cytosine, therefore 8‐OxoG in DNA causes a G:C to T:A transversion, if not repaired, while 8‐oxo‐dGTP incorporated into DNA during DNA replication causes both G:C to T:A and A:T to C:G transversions.( 51 ) From E. coli to higher eukaryotes, organisms are equipped with elaborate mechanisms to counteract the mutagenesis caused by the oxidized nucleotides in both DNA and nucleotide pools. Three enzymes, MTH1, OGG1, and MUTYH, have been shown to play important roles in counteracting the accumulation of 8‐oxoG in cellular genomes of human and rodent cells (Fig. 1).( 52 ) MTH1 hydrolyzes 8‐oxo‐dGTP and 2‐OH‐dATP to their monophosphate forms and pyrophosphates, thereby preventing the incorporation of 8‐oxo‐dGTP and 2‐OH‐dATP into DNA during replication.( 53 ) OGG1, an 8‐oxoG DNA glycosylase, excises 8‐oxoG opposite cytosine in DNA, which minimizes the formation of a pre‐mutagenic base pair, A‐8‐oxoG. MUTYH, a mammalian homolog of E. coli MutY, is a DNA glycosylase that has been shown to excise 2‐hydroxyadenine incorporated opposite guanine and adenine incorporated opposite 8‐oxoG.( 54 ) As a result, MUTYH is considered to play a crucial role in preventing G:C to T:A transversion in mammals.( 52 )
Figure 1.
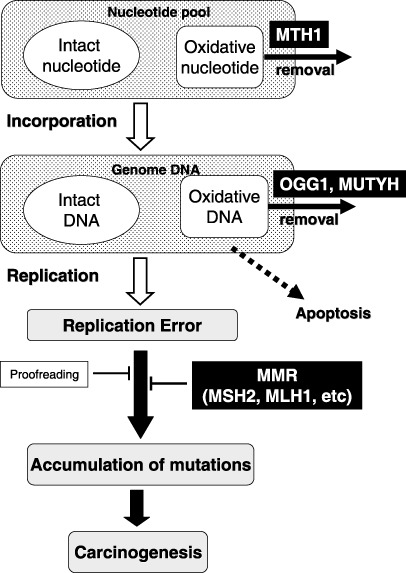
Proposed process of carcinogenesis due to the accumulation of mutations. There is a considerable amount of oxidative DNA/nucleotides in the genome DNA or nucleotide pool. MTH1 catalyzes 8‐oxodGTP to 8‐oxodGMP, thus avoiding the incorporation of this mutagenic nucleotide into the genome. Oxidative DNA can be generated in the genome DNA or oxidative nucleotides can be incorporated into genome DNA from the nucleotide pool. OGG1 eliminate 8‐oxo‐7,8‐dihydroguanine (8‐oxoG) paired with cytosine, whereas MUTYH efficiently removes adenine paired with 8‐oxoG in the genome DNA. In contrast, replicative DNA polymerase causes the mis‐incorporation of nucleotides at a low frequency during replication using non‐damaged nucleotides. Mismatch repair (MMR) is responsible for correcting these replication errors, which the proofreading activity of the DNA polymerase itself is unable to correct. If the capacity of these repair systems is impaired, then mutations may thus accumulate in an accelerated manner, thereby facilitating the process of carcinogenesis.
To analyze the function of these proteins in vivo, mutant mice were generated with each of these genes by targeted disruption (Table 1). Klungland et al. reported that Ogg1‐deficient mice accumulated 8‐oxoG in DNA. (55) Minowa et al. reported that Ogg1‐deficient mice showed increased mutation frequency.( 56 ) However, neither group observed the development of malignancies in these mice. However, Sakumi et al. reported that spontaneous lung adenoma/carcinomas developed in Ogg1‐deficient mice 1.5 years after birth.( 57 ) Tsuzuki et al. reported that Mth1‐deficient mice developed malignancies in some organs, proving the direct connection between oxidative DNA damage and carcinogenesis in a mouse model.( 58 ) Interestingly, the overall mutation frequency showed no apparent increase (less than twofold) in Mth1−/– mice, while MutT‐deficient E. coli shows a 1000‐fold increased mutation frequency.( 10 ) Nevertheless, the frequency of 1‐base pairs frameshift mutations at the mononucleotide runs in the reporter gene was 5.7‐fold higher in the spleens of Mth1‐null mice than in those of wild‐type mice. Because the elevated incidence of single‐base frameshifts at mononucleotide runs is a hallmark of a defect in the Msh2‐dependent MMR system, this weak site‐specific mutator effect of Mth1‐deficiency could be attributed to the involvement of MMR function that may act to correct mispairs with the oxidized nucleotides. Consistent with this hypothesis, a significant increase in the frequency of G:C to T:A transversions was observed in Mth1/Msh2 double mutants compared with either mutant alone, It is interesting to note that mismatch repair may participate in the avoidance of 8‐oxoG‐related mutagenesis in mammalian cells, as a deficiency of MMR in Msh2‐null mouse embryonic cells leads to an accumulation of 8‐oxoG in the genome.( 59 ) Xie et al. reported that no significant differences in tumor incidence occur between Mutyh‐null and wild‐type mice within a 12‐month period.( 60 ) However, our examination of tumor development using a large cohort of wild‐type and Mutyh‐null mice at the age of approximately 18 months revealed an increased occurrence of tumors in various internal organs of Mutyh‐null mice compared with wild‐type mice.( 61 ) The elevation of mutation frequencies observed in these mice was lower than those observed in E. coli, which demonstrate a 10–1000‐fold higher frequency of mutation in comparison to wild type.( 51 ) This difference might be due to the involvement of multiple anti‐mutagenic pathways including other repair systems, such as a mismatch recognition mechanism, for the mutagenesis caused by oxidative DNA and nucleotide damage.( 10 ) Xie et al. reported that Ogg1 −/–/Mutyh −/– mice are predisposed to lung adenoma/carcinoma, lymphoma and gastrointestinal adenoma/carcinoma, whereas the single mutant did not show predisposition for tumors.( 60 ) Furthermore, it is of interest that the MMR system seems to be involved in the avoidance of mutagenesis caused by oxidative DNA damage.( 5 , 10 ) Ogg1‐, Mth1‐ or Mutyh‐deficient mice develop tumors more frequently than wild‐type mice at a later stage.( 57 , 58 , 61 ) These observations might be correlated with the findings that the elevated mutation frequencies observed in these mice were not extremely high, in contrast to the Msh2‐deficient mice that showed a strongly elevated mutation frequency and highly developed tumors at an earlier stage.( 9 , 14 , 15 ) It should be mentioned that in Saccharomyces cerevisiae in which the orthologue of MutY does not exist, MMR and error‐free trans‐lesion DNA synthesis could prevent the mutagenic effect of 8‐oxoG in cooperation with Ogg1.( 62 , 63 ) It is also of great interest that DNA polymerase η seems to efficiently promote the error‐free incorporation of cytosine opposite 8‐oxoG, whereas high‐fidelity replicative DNA polymerase δ frequently mis‐incorporate adenine opposite 8‐oxoG, even in mammalian cells.( 63 ) Furthermore, the fact that the accuracy of polymerase η was enhanced in the addition of PCNA and/or RPA, which is part of the replication machinery, thus suggesting how complicated it is to maintain the fidelity during replication containing 8‐oxoG residues.( 64 )
Dysfunction of oxidative DNA/nucleotide damage repair in gastrointestinal tract cancer
Al‐Tassan et al. reported a unique somatic mutation pattern of the APC gene and MUTYH hypomorphic mutation.( 3 ) They analyzed a family that is affected with multiple colorectal adenomas and carcinomas but lacks the germline APC gene that is associated with FAP. They showed a high incidence of somatic G:C to T:A transversion mutations in the APC gene that is uncommon in tumors from classic FAP individuals and also demonstrated siblings with tumors that were compound heterozygous for MUTYH gene mutations (Tyr165Cys and Gly382Asp). Thereafter, the attenuated phenotype of inherited polyposis is proposed as MAP, which is an autosomal recessive disease with germline mutations in the MUTYH gene.( 65 , 66 ) Carriers with a bi‐allelic germline mutation of the MUTYH gene had an increased risk of colorectal cancer. Furthermore, carriers with a mono‐allelic mutation also seem to have an increased risk of colorectal cancer;( 67 , 68 ) however, further study with a large cohort is necessary to verify these observations. There are few reports that investigate the role of somatic MUTYH mutation in carcinogenesis (Table 4). Meanwhile, Halford et al. found no somatic mutations of MUTYH in any of 75 unselected CRC and CRC cell lines.( 69 ) However, Kim et al. demonstrated that 2 of 95 sporadic gastric cancers had bi‐allelic disruption of the MUTYH gene with somatic mutation of one allele and LOH of the remaining allele.( 70 )
Table 4.
Association between oxidative DNA/nucleotide repair dysfunction and sporadic gastrointestinal tract cancer
| Gene | Mutation/polymorphism | Reference no. |
|---|---|---|
| (a) Esophagus | ||
| MTH1 | – | |
| OGG1 | Ser326Cys | ( 75 ) |
| MUTYH | – | |
| (b) Stomach | ||
| MTH1 | Val83Met | ( 78 ) |
| OGG1 | Arg154His | ( 71 ) |
| Ser326Cys | ( 76 ) | |
| MUTYH | Pro391Ser, Gln400Arg | ( 70 ) |
| (c) Colon | ||
| MTH1 | no correlation | ( 79 ) |
| OGG1 | Arg154His | ( 73 ) |
| Ser326Cys | ( 77 ) | |
| MUTYH | Tyr165Cys, Gly382Asp | ( 67, 68 ) |
Other reports have demonstrated a correlation between OGG1 gene polymorphism and cancer susceptibility (Table 4). The Arg154His mutation was initially observed in a gastric cancer cell line( 71 ) and this mutation was shown to alter the activity of this enzyme.( 72 ) Kim et al. analyzed 625 CRC (including 29 FAP, 19 HNPCC, and 86 suspected HNPCC) and 527 normal control cases for OGG1 Arg154His. They showed that Arg154His was a rare polymorphism associated with sporadic CRC (P = 0.053).( 73 ) The OGG1 protein encoded by the Ser326 allele exhibited substantially higher activity than the Cys326 variants in an in vitro E. coli complementation activity assay.( 74 ) Xing et al. investigated the association between Ser326Cys polymorphism and esophageal SCC among 201 normal controls and 196 patients with esophageal cancer in China. They found that individuals homozygous for the Cys/Cys genotype had a significantly increased risk of developing esophageal SCC, with an OR adjusted for age, sex, and smoking of 1.9.( 75 ) Takezaki et al. investigated the association of the OGG1 gene mutation with stomach cancer risk using 101 stomach cancer patients and 198 controls. In that investigation, the OGG1 Ser326Cys polymorphism did not alter the overall ORs for stomach cancers; however, subgroup analyses revealed increased ORs with a frequent drinking habit in Cys/Cys carriers.( 76 ) Kim et al. investigated 125 colon cancer patients and 247 controls. There was no significant difference in Ser326Cys genotype distribution between the patients and controls. Subgroup analysis revealed increased ORs with smoking or with frequent consumption of meat in Cys/Cys carriers, although the statistical significance of the former factor is marginal.( 77 )
For MTH1, a polymorphism of Val83Met has been found and studies have reported that Met83‐MTH1 expressed in E. coli is more thermolabile than Val83‐MTH1, with both its secondary structure and 8‐oxodGTPase activity.( 52 ) Kimura et al. demonstrated that this polymorphic variation of MTH1 in gastric cancer patients occurs significantly more frequently than in healthy individuals. Furthermore, the p53 mutation correlated with the variant form of MTH1. The frequency of the p53 mutation was significantly higher in tumors harboring at least one Met83 allele than in those without the Met83 allele (P = 0.034).( 78 )
It is critical to carefully interpret the results of an association between polymorphism and cancer susceptibility. Recently, Schafmayer et al. investigated the variation of several repair genes in sporadic colorectal cancer and reported no association with cancer susceptibility, even in cases with the previously indicated risk variant.( 79 ) This result could be affected by race or cohort scale, or even by the statistical methods used. Because the etiology of esophageal cancer is deeply connected with exposure to both tobacco and alcohol consumption, environmental factors as well as genetic factors are thus considered to play an important role in the process of carcinogenesis.( 80 )
Mutator phenotype in DNA repair‐deficient cells and mice
An elevation of the mutation frequency could play a role in carcinogenesis in MMR‐deficient cells and it might occur when there is a defect of some repair function against oxidative DNA/nucleotide damage. It is important to determine whether the proto‐oncogene or the tumor suppressor gene could be mutated in DNA repair‐deficient cells. As Al‐Tassan et al. reported, germline MUTYH mutations seem to be correlated with APC tumor suppressor gene mutations in colonic adenoma and adenocarcinoma.( 3 ) Because Mutyh‐deficient mice were susceptible to intestinal tumor development, as observed in MAP patients, current experiments of analyzing mutations in the tumor‐associated genes, such as the Apc gene, by amplifying genomic DNA derived from the intestinal tumors, found in Mutyh‐deficient mice, would provide significant insights into the involvement of Mutyh function in oxidation‐induced carcinogenesis. In addition, Xie et al. reported the tumors in Ogg1 −/–/Mutyh −/– mice to have mutations in the K‐ras oncogene.( 60 ) Kimura et al. showed a correlation between MTH1 hypomorphic polymorphism and p53 mutation in gastric cancer.( 78 )
Insertion/deletion mutations are observed mostly in the mononucleotide run within the cording region of several genes, such as TGFβRII, BAX, MSH3, etc. There might be base substitutions in the proto‐oncogene or tumor suppressor gene. Oda et al. demonstrated that a p53 gene mutation is strongly associated with a certain subtype of MSI,( 27 ) in accordance with several reports that noted the connection between MSI‐L and mutation of the proto‐oncogene or tumor suppressor gene.( 43 ) The authors observed several germline base substitutions within the cording region of proto‐oncogenes and tumor suppressor genes in MSH2‐deficient mouse tumors (A. Egashira, T. Tsuzuki, Y. Maehara, unpublished data, 2007). Although the possibility that the mutations observed merely resulted from phenotypical advantage could not be ruled out, these observations might support the idea of the mutator phenotype, which has been proposed by Loeb.
Perspectives
The detection of defective DNA repair in tumor tissue property may provide important information that can be used to guide the clinical management of patients. For instance, MMR‐deficient cells are tolerant to DNA methylating agents, anti‐metabolites, and intra‐strand cross‐linking agents,( 4 ) which are commonly used for cancer treatment. There have been several reports that have demonstrated a correlation between MSI status and the effect of adjuvant chemotherapy. Furthermore, oxidative DNA/nucleotide damages could be generated by some therapeutic agents. DNA repair properties not only play an important role in avoiding carcinogenesis in gastrointestinal cancers, but may also control the effects of some drugs even if the tumor was completely removed macroscopically.( 81 ) Understanding the DNA repair capacity of an individual may enable the personalized choice of effective therapeutic agents, most of which function by producing specific types of DNA damage in cancer cells. For this purpose, it is efficient to use animal models that can mimic the phenotype of the cancer by administering some drugs that specially eliminate or reduce specific repair functions. To elucidate the mechanism of DNA repair in carcinogenesis, the results from mice could be applied to humans, and these approaches enable us to achieve a comprehensive understanding of these complicated mechanisms discussed above.
References
- 1. Fishel R, Lescoe MK, Rao MR et al . The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. (Published erratum appears in Cell 1994; 77: 167.) Cell 1993; 75: 1027–38. [DOI] [PubMed] [Google Scholar]
- 2. Leach FS, Nicolaides NC, Papadopoulos N et al . Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993; 75: 1215–25. [DOI] [PubMed] [Google Scholar]
- 3. Al‐Tassan N, Chmiel NH, Maynard J et al . Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet 2002; 30: 227–32. [DOI] [PubMed] [Google Scholar]
- 4. Jiricny J. The multifaceted mismatch‐repair system. Nat Rev Mol Cell Biol 2006; 7: 335–46. [DOI] [PubMed] [Google Scholar]
- 5. Russo MT, Blasi MF, Chiera F et al . The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair‐deficient cells. Mol Cell Biol 2004; 24: 465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buermeyer AB, Deschenes SM, Baker SM, Liskay RM. Mammalian DNA mismatch repair. Annu Rev Genet 1999; 33: 533–64. [DOI] [PubMed] [Google Scholar]
- 7. Liu B, Parsons R, Papadopoulos N et al . Analysis of mismatch repair genes in hereditary non‐polyposis colorectal cancer patients. Nat Med 1996; 2: 169–74. [DOI] [PubMed] [Google Scholar]
- 8. Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 2003; 21: 1174–9. [DOI] [PubMed] [Google Scholar]
- 9. Andrew SE, Xu XS, Baross‐Francis A et al . Mutagenesis in PMS2‐ and MSH2‐deficient mice indicates differential protection from transversions and frameshifts. Carcinogenesis 2000; 21: 1291–5. [PubMed] [Google Scholar]
- 10. Egashira A, Yamauchi K, Yoshiyama K et al . Mutational specificity of mice defective in the MTH1 and/or the MSH2 genes. DNA Repair (Amst) 2002; 1: 881–93. [DOI] [PubMed] [Google Scholar]
- 11. Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM. Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6. Carcinogenesis 2006; 27: 2402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Loeb LA. A mutator phenotype in cancer. Cancer Res 2001; 61: 3230–9. [PubMed] [Google Scholar]
- 13. Maehara Y, Oda S, Sugimachi K. The instability within: problems in current analyses of microsatellite instability. Mutat Res 2001; 461: 249–63. [DOI] [PubMed] [Google Scholar]
- 14. De Wind N, Dekker M, Berns A, Radman M, Te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995; 82: 321–30. [DOI] [PubMed] [Google Scholar]
- 15. Reitmair AH, Schmits R, Ewel A et al . MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet 1995; 11: 64–70. [DOI] [PubMed] [Google Scholar]
- 16. Reitmair AH, Redston M, Cai JC et al . Spontaneous intestinal carcinomas and skin neoplasms in Msh2‐deficient mice. Cancer Res 1996; 56: 3842–9. [PubMed] [Google Scholar]
- 17. Chen PC, Dudley S, Hagen W et al . Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res 2005; 65: 8662–70. [DOI] [PubMed] [Google Scholar]
- 18. Edelmann W, Cohen PE, Kane M et al . Meiotic pachytene arrest in MLH1‐deficient mice. Cell 1996; 85: 1125–34. [DOI] [PubMed] [Google Scholar]
- 19. Edelmann W, Yang K, Umar A et al . Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell 1997; 91: 467–77. [DOI] [PubMed] [Google Scholar]
- 20. Prolla TA, Baker SM, Harris AC et al . Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet 1998; 18: 276–9. [DOI] [PubMed] [Google Scholar]
- 21. Edelmann W, Umar A, Yang K et al . The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res 2000; 60: 803–7. [PubMed] [Google Scholar]
- 22. Edelmann W, Yang K, Kuraguchi M et al . Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res 1999; 59: 1301–7. [PubMed] [Google Scholar]
- 23. Sands AT, Suraokar MB, Sanchez A, Marth JE, Donehower LA, Bradley A. p53 deficiency does not affect the accumulation of point mutations in a transgene target. Proc Natl Acad Sci USA 1995; 92: 8517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oda S, Zhao Y, Maehara Y. Microsatellite instability in gastrointestinal tract cancers: a brief update. Surg Today 2005; 35: 1005–15. [DOI] [PubMed] [Google Scholar]
- 25. Ikeda Y, Oda S, Abe T, Ohno S, Maehara Y, Sugimachi K. Features of microsatellite instability in colorectal cancer: comparison between colon and rectum. Oncology 2001; 61: 168–74. [DOI] [PubMed] [Google Scholar]
- 26. Araki K, Wang B, Miyashita K et al . Frequent loss of heterozygosity but rare microsatellite instability in oesophageal cancer in Japanese and Chinese patients. Oncology 2004; 67: 151–8. [DOI] [PubMed] [Google Scholar]
- 27. Oda S, Maehara Y, Ikeda Y et al . Two modes of microsatellite instability in human cancer: differential connection of defective DNA mismatch repair to dinucleotide repeat instability. Nucleic Acids Res 2005; 33: 1628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lowsky R, Magliocco A, Ichinohasama R et al . MSH2‐deficient murine lymphomas harbor insertion/deletion mutations in the transforming growth factor beta receptor type 2 gene and display low not high frequency microsatellite instability. Blood 2000; 95: 1767–72. [PubMed] [Google Scholar]
- 29. Papadopoulos N, Nicolaides NC, Wei YF et al . Mutation of a mutL homolog in hereditary colon cancer. Science 1994; 263: 1625–9. [DOI] [PubMed] [Google Scholar]
- 30. Liu T, Yan H, Kuismanen S et al . The role of hPMS1 and hPMS2 in predisposing to colorectal cancer. Cancer Res 2001; 61: 7798–802. [PubMed] [Google Scholar]
- 31. Uchida N, Kumimoto H, Nishizawa K et al . Mismatch repair and microsatellite instability in esophageal cancer cells. Int J Cancer 2001; 91: 687–91. [PubMed] [Google Scholar]
- 32. Guo M, Ren J, House MG, Qi Y, Brock MV, Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res 2006; 12: 4515–22. [DOI] [PubMed] [Google Scholar]
- 33. Uehara H, Miyamoto M, Kato K et al . Deficiency of hMLH1 and hMSH2 expression is a poor prognostic factor in esophageal squamous cell carcinoma. J Surg Oncol 2005; 92: 109–15. [DOI] [PubMed] [Google Scholar]
- 34. Aarnio M, Sankila R, Pukkala E et al . Cancer risk in mutation carriers of DNA‐mismatch‐repair genes. Int J Cancer 1999; 81: 214–8. [DOI] [PubMed] [Google Scholar]
- 35. Goecke T, Schulmann K, Engel C et al . Genotype‐phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: a report by the German HNPCC Consortium. J Clin Oncol 2006; 24: 4285–92. [DOI] [PubMed] [Google Scholar]
- 36. Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high‐frequency microsatellite instability. Cancer Res 1999; 59: 159–64. [PubMed] [Google Scholar]
- 37. Fleisher AS, Esteller M, Wang S et al . Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res 1999; 59: 1090–5. [PubMed] [Google Scholar]
- 38. Baek MJ, Kang H, Kim SE et al . Expression of hMLH1 is inactivated in the gastric adenomas with enhanced microsatellite instability. Br J Cancer 2001; 85: 1147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kitajima Y, Miyazaki K, Matsukura S, Tanaka M, Sekiguchi M. Loss of expression of DNA repair enzymes MGMT, hMLH1, and hMSH2 during tumor progression in gastric cancer. Gastric Cancer 2003; 6: 86–95. [DOI] [PubMed] [Google Scholar]
- 40. Chiaravalli AM, Furlan D, Facco C et al . Immunohistochemical pattern of hMSH2/hMLH1 in familial and sporadic colorectal, gastric, endometrial and ovarian carcinomas with instability in microsatellite sequences. Virchows Arch 2001; 438: 39–48. [DOI] [PubMed] [Google Scholar]
- 41. Aaltonen LA, Salovaara R, Kristo P et al . Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998; 338: 1481–7. [DOI] [PubMed] [Google Scholar]
- 42. Percesepe A, Borghi F, Menigatti M et al . Molecular screening for hereditary nonpolyposis colorectal cancer: a prospective, population‐based study. J Clin Oncol 2001; 19: 3944–50. [DOI] [PubMed] [Google Scholar]
- 43. Konishi M, Kikuchi‐Yanoshita R, Tanaka K et al . Molecular nature of colon tumors in hereditary nonpolyposis colon cancer, familial polyposis, and sporadic colon cancer. Gastroenterology 1996; 111: 307–17. [DOI] [PubMed] [Google Scholar]
- 44. Bubb VJ, Curtis LJ, Cunningham C et al . Microsatellite instability and the role of hMSH2 in sporadic colorectal cancer. Oncogene 1996; 12: 2641–9. [PubMed] [Google Scholar]
- 45. Chaves P, Cruz C, Lage P et al . Immunohistochemical detection of mismatch repair gene proteins as a useful tool for the identification of colorectal carcinoma with the mutator phenotype. J Pathol 2000; 191: 355–60. [DOI] [PubMed] [Google Scholar]
- 46. Cunningham JM, Christensen ER, Tester DJ et al . Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res 1998; 58: 3455–60. [PubMed] [Google Scholar]
- 47. Parc YR, Halling KC, Wang L et al . HMSH6 alterations in patients with microsatellite instability‐low colorectal cancer. Cancer Res 2000; 60: 2225–31. [PubMed] [Google Scholar]
- 48. Plaschke J, Kruger S, Pistorius S, Theissig F, Saeger HD, Schackert HK. Involvement of hMSH6 in the development of hereditary and sporadic colorectal cancer revealed by immunostaining is based on germline mutations, but rarely on somatic inactivation. Int J Cancer 2002; 97: 643–8. [DOI] [PubMed] [Google Scholar]
- 49. Jass JR, Whitehall VL, Young J, Leggett BA. Emerging concepts in colorectal neoplasia. Gastroenterology 2002; 123: 862–76. [DOI] [PubMed] [Google Scholar]
- 50. Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA 1993; 90: 7915–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tajiri T, Maki H, Sekiguchi M. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat Res 1995; 336: 257–67. [DOI] [PubMed] [Google Scholar]
- 52. Tsuzuki T, Nakatsu Y, Nakabeppu Y. Significance of error‐avoiding mechanisms for oxidative DNA damage in carcinogenesis. Cancer Sci 2007; 98: 465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fujikawa K, Kamiya H, Yakushiji H, Nakabeppu Y, Kasai H. Human MTH1 protein hydrolyzes the oxidized ribonucleotide, 2‐hydroxy‐ATP. Nucleic Acids Res 2001; 29: 449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ohtsubo T, Nishioka K, Imaiso Y et al . Identification of human MutY homolog (hMYH) as a repair enzyme for 2‐hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res 2000; 28: 1355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Klungland A, Rosewell I, Hollenbach S et al . Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA 1999; 96: 13300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Minowa O, Arai T, Hirano M et al . Mmh/Ogg1 gene inactivation results in accumulation of 8‐hydroxyguanine in mice. Proc Natl Acad Sci USA 2000; 97: 4156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sakumi K, Tominaga Y, Furuichi M et al . Ogg1 knockout‐associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res 2003; 63: 902–5. [PubMed] [Google Scholar]
- 58. Tsuzuki T, Egashira A, Igarashi H et al . Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8‐oxo‐dGTPase. Proc Natl Acad Sci USA 2001; 98: 11456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DeWeese TL, Shipman JM, Larrier NA et al . Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low‐level radiation. Proc Natl Acad Sci USA 1998; 95: 11915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xie Y, Yang H, Cunanan C et al . Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K‐ras oncogene in lung tumors. Cancer Res 2004; 64: 3096–102. [DOI] [PubMed] [Google Scholar]
- 61. Sakamoto K, Tominaga Y, Yamauchi K et al . MUTYH‐null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res 2007; 67: 6599–604. [DOI] [PubMed] [Google Scholar]
- 62. Ni TT, Marsischky GT, Kolodner RD. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8‐oxo‐guanine in S. cerevisiae . Mol Cell 1999; 4: 439–44. [DOI] [PubMed] [Google Scholar]
- 63. Haracska LYuSL, Johnson RE, Prakash L, Prakash S. Efficient and accurate replication in the presence of 7,8‐dihydro‐8‐oxoguanine by DNA polymerase eta. Nat Genet 2000; 25: 458–61. [DOI] [PubMed] [Google Scholar]
- 64. Maga G, Villani G, Crespan E et al . 8‐oxo‐guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature 2007; 447: 606–8. [DOI] [PubMed] [Google Scholar]
- 65. Sampson JR, Dolwani S, Jones S et al . Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362: 39–41. [DOI] [PubMed] [Google Scholar]
- 66. Sieber OM, Lipton L, Crabtree M et al . Multiple colorectal adenomas, classic adenomatous polyposis, and germ‐line mutations in MYH. N Engl J Med 2003; 348: 791–9. [DOI] [PubMed] [Google Scholar]
- 67. Croitoru ME, Cleary SP, Di Nicola N et al . Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96: 1631–4. [DOI] [PubMed] [Google Scholar]
- 68. Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer 2006; 95: 239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Halford SE, Rowan AJ, Lipton L et al . Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol 2003; 162: 1545–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim CJ, Cho YG, Park CH et al . Genetic alterations of the MYH gene in gastric cancer. Oncogene 2004; 23: 6820–2. [DOI] [PubMed] [Google Scholar]
- 71. Shinmura K, Kohno T, Kasai H, Koda K, Sugimura H, Yokota J. Infrequent mutations of the hOGG1 gene, that is involved in the excision of 8‐hydroxyguanine in damaged DNA, in human gastric cancer. Jpn J Cancer Res 1998; 89: 825–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Audebert M, Radicella JP, Dizdaroglu M. Effect of single mutations in the OGG1 gene found in human tumors on the substrate specificity of the Ogg1 protein. Nucl Acids Res 2000; 28: 2672–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim IJ, Ku JL, Kang HC et al . Mutational analysis of OGG1, MYH, MTH1 in FAP, HNPCC and sporadic colorectal cancer patients: R154H OGG1 polymorphism is associated with sporadic colorectal cancer patients. Hum Genet 2004; 115: 498–503. [DOI] [PubMed] [Google Scholar]
- 74. Kohno T, Shinmura K, Tosaka M et al . Genetic polymorphisms and alternative splicing of the hOGG1 gene, that is involved in the repair of 8‐hydroxyguanine in damaged DNA. Oncogene 1998; 16: 3219–25. [DOI] [PubMed] [Google Scholar]
- 75. Xing DY, Tan W, Song N, Lin DX. Ser326Cys polymorphism in hOGG1 gene and risk of esophageal cancer in a Chinese population. Int J Cancer 2001; 95: 140–3. [DOI] [PubMed] [Google Scholar]
- 76. Takezaki T, Gao CM, Wu JZ et al . hOGG1 Ser (326) Cys polymorphism and modification by environmental factors of stomach cancer risk in Chinese. Int J Cancer 2002; 99: 624–7. [DOI] [PubMed] [Google Scholar]
- 77. Kim JI, Park YJ, Kim KH et al . hOGG1 Ser326Cys polymorphism modifies the significance of the environmental risk factor for colon cancer. World J Gastroenterol 2003; 9: 956–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kimura Y, Oda S, Egashira A et al . A variant form of hMTH1, a human homologue of the E. coli mutT gene, correlates with somatic mutation in the p53 tumour suppressor gene in gastric cancer patients. J Med Genet 2004; 41: e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schafmayer C, Buch S, Egberts JH et al . Genetic investigation of DNA‐repair pathway genes PMS2, MLH1, MSH2, MSH6, MUTYH, OGG1 and MTH1 in sporadic colon cancer. Int J Cancer 2007; 121: 555–8. [DOI] [PubMed] [Google Scholar]
- 80. Morita M, Kuwano H, Ohno S et al . Multiple occurrence of carcinoma in the upper aerodigestive tract associated with esophageal cancer: reference to smoking, drinking and family history. Int J Cancer 1994; 58: 207–10. [DOI] [PubMed] [Google Scholar]
- 81. Ribic CM, Sargent DJ, Moore MJ et al . Tumor microsatellite‐instability status as a predictor of benefit from fluorouracil‐based adjuvant chemotherapy for colon cancer. N Engl J Med 2003; 349: 247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hirano S, Tominaga Y, Ichinoe A et al . Mutator phenotype of MUTYH‐null mouse embryonic stem cells. J Biol Chem 2003; 278: 38121–4. [DOI] [PubMed] [Google Scholar]