

Abstract
Hepatocellular carcinoma (HCC) is a major health care problem worldwide. The prognosis of patients with HCC is poor because even in the early stages when surgical treatment might be expected to be curative, the incidence of recurrence in patients with underlying cirrhosis is very high due to multicentric carcinogenesis. Therefore, strategies to prevent recurrence and second primary HCC are required to improve the prognosis. One of the most practical approaches to prevent the multicentric development of HCC is ‘clonal deletion’ therapy, which is defined as the removal of latent (i.e. invisible) (pre)malignant clones from the liver in a hypercarcinogenic state. Retinoids, a group of structural and functional analogs of vitamin A, exert their biological function primarily through two distinct nuclear receptors, retinoic acid receptors and retinoid X receptors (RXR), and abnormalities in the expression and function of these receptors are highly associated with the development of various cancers, including HCC. In particular, a malfunction of RXRα due to phosphorylation by the Ras–mitogen‐activated protein kinase signaling pathway is profoundly associated with the development of HCC and thus may be a critical target for HCC chemoprevention. Acyclic retinoid, which has been clinically shown to reduce the incidence of a post‐therapeutic recurrence of HCC, can inhibit Ras activity and phosphorylation of the extracellular signal‐regulated kinase and RXRα proteins. In conclusion, the inhibition of RXRα phosphorylation and the restoration of its physiological function as a master regulator for nuclear receptors may be a potentially effective strategy for HCC chemoprevention and clonal deletion. Acyclic retinoid, which targets phosphorylated RXRα, may thus play a critical role in preventing the development of multicentric HCC. (Cancer Sci 2009; 100: 369–374)
Abbreviations:
- ACR
acyclic retinoid
- AFP‐L3
lectin‐reactive α‐fetoprotein isoform 3
- Erk
extracellular signal‐regulated kinase
- HCC
hepatocellular carcinoma
- HER2
human epidermal growth factor receptor‐2
- IFN
interferon
- MAPK
mitogen‐activated protein kinase
- PIVKA‐II
protein induced by vitamin K absence or antagonist‐II
- p‐RXR
retinoid X receptor
- RA
retinoic acid
- RAR
retinoic acid receptor
- RTK
receptor tyrosine kinase
- RXR
retinoid X receptor
- VK2
vitamin K2
Hepatocellular carcinoma is the fifth most common cancer worldwide and the third most common cause of cancer mortality. HCC is unique in that it usually occurs within an established background, chronic liver disease and cirrhosis. The development of HCC is frequently associated with chronic inflammation of the liver induced by a persistent infection with hepatitis B virus or hepatitis C virus. Therefore, this cancer is a major health care problem in Eastern as well as Western countries where hepatitis virus infection is endemic.( 1 , 2 ) Patients with viral liver cirrhosis are a high‐risk group for HCC because the annual rate for this cancer in those patients is approximately 7%. Even in the early stages when surgical treatment might be expected to be curative, the incidence of recurrence in patients with underlying cirrhosis is approximately 20–25% a year. Therefore, the recurrence rate at 5 years after curative treatment may exceed 70%.( 3 , 4 , 5 , 6 ) In addition, at least one‐third of secondary tumors are primary de novo cancers.( 7 ) Based on these clinical characteristics, the prognosis of patients with HCC is poor. Thus, it is a task of pressing urgency to develop more effective strategies for the chemoprevention of HCC and, for this purpose, there is a critical need to elucidate the molecular mechanisms underlying liver carcinogenesis.
Cancer chemoprevention is defined as an approach where a natural or synthetic chemical compound works to arrest or reverse premalignant cells by using physiological pathways.( 8 ) We previously reported that, in a clinical trial, the administration of ACR, a novel synthetic retinoid (Fig. 1), reduced the incidence of post‐therapeutic HCC recurrence and improved the survival rate of patients.( 9 , 10 , 11 ) We have also revealed that a malfunction of RXRα, a nuclear retinoid receptor, due to aberrant phosphorylation is associated with carcinogenesis in the liver.( 12 , 13 , 14 ) The aim of the present paper is to review the evidence that ACR exerts its chemopreventive effects on the development of HCC by targeting p‐RXRα. In addition, the concept of ‘clonal deletion’, which is one of the most practical approaches to preventing multicentric HCC development, is reviewed and the possibility of ‘combination chemoprevention’, which uses ACR as a key drug and might be an effective strategy to prevent this malignancy by pharmacological synergism, is discussed.
Figure 1.

Chemical structures of natural and representative synthetic retinoids. Retinyl esters (mainly retinyl palmitate; R, fatty acid), stored in the liver stellate cells, are hydrolyzed to retinol, which is then transported to target cells through the circulation after binding to retinol‐binding protein. Retinoic acid (RA) is biosynthesized from retinol via the intermediate metabolite retinal by oxidization in the cells of peripheral tissues. Three well‐known isomers of RA, all‐trans RA, 9‐cis RA, and 13‐cis RA activate the retinoid receptor retinoic acid receptor (RAR), whereas only 9‐cis RA activates the other receptor, retinoid X receptor (RXR). A number of synthetic retinoids have been developed to carry out their pharmacological applications including cancer chemoprevention. Acyclic retinoid and N‐(4‐hydroxyphenyl) retinamide (4HPR) successfully prevented the development of hepatocellular carcinoma and breast cancer, respectively, in clinical trials (see review reference( 27 )).
Retinoids and their receptors
Retinoids, a group of structural and functional analogs of vitamin A, exert fundamental effects on the regulation of epithelial cell growth, differentiation, and development.( 15 , 16 ) A small portion of dietary retinoids is converted to RA, which is an active metabolite of retinoids. Retinoids exert their biological functions primarily by regulating gene expression through two distinct nuclear receptors, the RXR and the RAR, which are both composed of three subtypes (α, β, and γ) that are characterized by a modular domain structure. RXR is specific for 9‐cis RA, whereas RAR binds both 9‐cis RA and all‐trans retinoic acid (Fig. 1). Nuclear retinoid receptors are ligand‐dependent transcription factors. After ligand binding, RXR form a homodimer as well as heterodimer with RAR, which interacts with the retinoid X response element or the RAR responsive element located in the promoter region of the target genes, thereby modulating gene expression. RXR also form a heterodimer with other nuclear receptors, such as peroxisome proliferator‐activated receptor.( 17 ) Among the retinoid receptors, RXRα is thought to be one of the most important receptors with respect to regulation of fundamental cell activities, including normal cell proliferation and metabolism, and act as the master regulator of nuclear receptors.( 15 , 16 )
In addition to the binding of specific ligands, recent studies have also revealed that phosphorylation processes are crucial for regulating RAR‐ and RXR‐mediated transcriptional activity.( 18 , 19 ) For instance, the phosphorylation of RXRα at its N‐terminal domain plays a role in the activation of a subset of RA‐responsive genes and in the antiproliferative effect of RA, indicating that RXRα‘positively’ regulates the transactivation of target genes through phosphorylation.( 20 ) In contrast, there are some reports that show the phosphorylation of RXRα to ‘negatively’ modulate the function of its heterodimeric binding partners. Indeed, MAPK‐mediated phosphorylation of the omega loop of the RXRα ligand binding domain impairs the transcriptional activity of RXR–RAR( 12 , 21 ) and RXR–vitamin D3 receptor( 22 , 23 ) heterodimers. These ‘negative’ effects of RXRα via its phosphorylation might be associated with certain types of human diseases, including malignant disorders.( 12 , 24 , 25 , 26 )
Hepatocellular carcinoma and RXRα phosphorylation
Because retinoids and their receptors play an essential role in normal cell proliferation and differentiation, abnormalities in the expression and function of these molecules are highly associated with the development of various human malignancies and therefore might be critical targets for cancer chemoprevention and chemotherapy.( 27 ) HCC is no exception in this concern. In the rodent model, we found that retinol was locally deficient in the HCC but not in the adjacent normal liver tissues and this was associated with aberrant metabolism of retinol.( 28 ) The expression of RXRα was also decreased not only in HCC and adenoma, but also in glutathione S‐transferase placental form‐positive foci, a precancerous lesion of HCC, suggesting that the repression of RXRα occurs even in an early stage of liver carcinogenesis.( 29 )
In addition, we have previously shown that hepatocarcinogenesis is accompanied by the accumulation of the phosphorylated (i.e. inactivated) form of RXRα.( 30 ) Specifically, RXRα protein is anomalously phosphorylated at serine and threonine residues, and accumulated both in human HCC tissue as well as in HCC cell lines.( 12 ) Phosphorylation at serine 260 of RXRα, a consensus site of MAPK, is closely linked to its retarded degradation, low transcriptional activity, and the promotion of cancer cell growth. In addition, the abrogation of phosphorylation by MAPK‐specific inhibitors restored the degradation of RXRα in a ligand‐dependent manner.( 12 , 31 ) Furthermore, in a normal liver and in non‐proliferating hepatocyte cultures, RXRα is unphosphorylated and highly ubiquitinated, thus rendering it sensitive to proteasome‐mediated degradation. In contrast, p‐RXRα is resistant to ubiquitination and proteasome‐mediated degradation in both human HCC tissues and a human HCC cell line.( 14 ) In addition, the phosphorylation of RXRα abolishes its ability to form heterodimers with RARβ and this might be associated with uncontrolled cell growth and resistance to retinoids.( 13 ) These findings suggest that the accumulation of p‐RXRα (i.e. non‐functional RXRα) may interfere with the function of normal RXRα in a dominant‐negative manner, thereby playing a critical role in the development of HCC (Fig. 2). Therefore, the inhibition of RXRα phosphorylation and the restoration of its physiological function as a master regulator of nuclear receptors, such as heterodimeric activity with other nuclear receptors, may be an effective and important strategy for inhibiting the growth of HCC cells.
Figure 2.
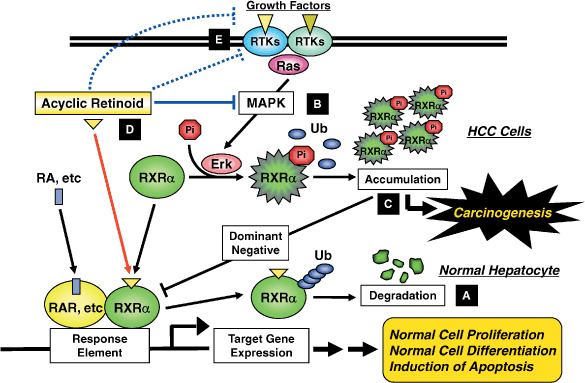
Retinoid refractoriness due to phosphorylation of retinoid X receptor (RXR) α and its restoration by acyclic retinoid (ACR) in hepatocellular carcinoma (HCC) cells. In normal hepatocytes, when the ligand binds to and activates RXRα, the receptor becomes able to heterodimerize with other nuclear receptors, such as retinoic acid receptor (RAR), and then activates the expression of target genes, which may regulate normal cell proliferation and differentiation, by binding to the specific response elements. Thereafter, RXRα dissociates from the dimer, is ubiquitinated (Ub), and is degraded by the proteasome. The whole process from ligand binding to proteasomal breakdown of RXRα is estimated to take approximately 6 h (A).( 14 ) In HCC cells, the Ras–mitogen‐activated protein kinase (MAPK) pathway is highly activated and phosphorylates RXRα at serine residues, thus impairing dimer formation and the subsequent transactivation functions of the receptor. Furthermore, phosphorylated RXRα (pi‐RXRα) escapes from ubiquitination and proteasomal degradation (B). Therefore, pi‐RXRα accumulates and interferes with the physiological function of the remaining unphosphorylated RXRα, presumably, in a dominant‐negative manner, thereby playing a critical role in the development of HCC (C). ACR is not only a ligand for RXRα but also suppresses the Ras–MAPK signaling pathway, inhibiting phosphorylation of RXRα, restoring the function of the receptor, and thus subsequently activating the transcriptional activity of the responsive element (D). ACR also directly or indirectly inhibits the ligand (i.e. specific growth factor)‐dependent receptor tyrosine kinase (RTK) activities in cancer cells (E). These effects may contribute to the inhibition of extracellular signal‐regulated kinase (Erk) and RXRα phosphorylation, thus causing inhibition of the growth of HCC cells.
Chemoprevention of HCC by ACR: Experimental study
Acyclic retinoid (NIK‐333; Kowa Pharmaceutical Co., Tokyo, Japan) is a synthetic retinoid and has an agonistic activity for both RXR and RAR.( 32 , 33 ) In experimental studies, this agent has demonstrated several beneficial effects on inhibition of HCC development. For instance, ACR inhibits chemically induced hepatocarcinogenesis in rats as well as spontaneously occurring hepatoma in mice.( 28 ) ACR also inhibits growth and induces apoptosis in human HCC‐derived cells and this might be associated with induction of apoptosis and cell differentiation in these cancer cells.( 33 , 34 , 35 , 36 , 37 ) In a human HCC cell line, ACR causes an arrest of the cell cycle in G0–G1, increases cellular levels of the p21CIP1 protein, and decreases levels of the cyclin D1 protein.( 38 ) Moreover, recent studies indicated that ACR is not only the ligand for RXRα. Indeed, in human HCC‐derived cells, ACR restores the function of RXRα by inactivating the Ras–Erk signaling system and thereby dephosphorylating RXRα, although 9‐cis RA fails to suppress phosphorylation of the Erk protein and subsequent RXRα phosphorylation.( 31 ) Both in vivo ( 39 , 40 ) and in vitro ( 41 , 42 ) studies have demonstrated that ACR reduces the development of HCC and prevents growth of cancer cells by inhibiting the activation of RTK, which play a critical role in stimulation of the Ras–MAPK signaling pathway.( 43 ) Therefore, in addition to direct inhibition of the Ras–Erk signaling system( 31 ) ACR may also cause dephosphorylation of the Erk and RXRα proteins by inactivating RTK, the upstream molecules of Ras, and thus restoring the function of RXRα. These findings suggest that ACR is a promising agent for the chemoprevention of HCC and that p‐RXRα is a useful molecular target of ACR (Fig. 2).
Chemoprevention of HCC by ACR: Clinical study
The chemopreventive effects of ACR on recurrent and secondary HCC were confirmed in patients who received anticancer treatment for an initial HCC in a double‐blind and placebo‐controlled clinical study.( 9 , 10 , 11 ) Oral administration of ACR (600 mg per day) for 12 months significantly reduced the incidence of post‐therapeutic HCC recurrence in patients who underwent potentially curative treatments.( 9 ) The survival rate was also significantly improved by the administration of this compound after a median follow up of 62 months.( 10 ) Moreover, the preventive effects of ACR lasted up to 199 weeks after randomization (or 151 weeks after completion of ACR administration).( 11 ) Therefore, administration of ACR for only 12 months confers a long‐term effect over several years, without causing any severe adverse effects of retinoids, such as dry skin, cheilitis, or conjunctivitis. However, headache or hyperlipidemia was reported in one case ACR.( 9 ) A phase II/III trial of this compound to test its effect in preventing second primary HCC is currently proceeding as a large‐scale randomized controlled study. This trial is scheduled to be completed around 2009–10 and it is expected to yield positive results.
Concept of ‘clonal deletion’
Pathologically, the high incidence of the development of second primary HCC may be explained by its characteristic mode of carcinogenesis, multicentric carcinogenesis, which is also expressed by the term ‘field cancerization’.( 44 ) Once a liver is exposed to continuous carcinogenic insults, such as hepatitis virus infection, the whole liver is regarded as a precancerous field that possesses multiple as well as independent premalignant or latent malignant clones. Therefore, the most effective strategy for HCC chemoprevention is the deletion of latent malignant clones (clonal deletion) as well as inhibition of the evolution of such clones (clonal inhibition) before they expand into a clinically detectable tumor. We therefore propose the benefits of ‘clonal deletion’ therapy for the prevention of HCC recurrence, which is defined as the removal of latent malignant (or premalignant) clones that are invisible by diagnostic imaging from the liver in a hypercarcinogenic state( 11 , 45 , 46 , 47 ) (Fig. 3).
Figure 3.

The concept of ‘clonal deletion’. Persistent inflammation caused by hepatitis B virus (HBV) or hepatitis C virus (HCV) infection transforms the liver into a ‘precancerous field’ (A). Therefore, the high incidence of hepatocellular carcinoma (HCC) as well as its recurrence in cirrhotic patients strongly suggests the presence of latent malignant clones that arise through multicentric carcinogenesis and are undetectable clinically by image analysis (invisible) (B). These multiple clones demonstrate different grades of malignancy (atypia) in the cirrhotic liver and, at some point, turn into clinical HCC (visible) (C). Even when primary HCC could be found in an early stage and surgical treatment might be expected to be curative, other clones still survive in the remaining liver and thus grow into secondary HCC again (D). Therefore, the eradication of such transformed clones, referred to as ‘clonal deletion’, may be one of the most effective strategies to prevent secondary HCC (E). Clinical experience suggests that acyclic retinoid (ACR), which inhibits phosphorylation (Pi) of retinoid X receptor (RXR) α (F), reduces the recurrence of HCC on the basis of this concept because this agent causes a decrease in the serum levels of lectin‐reactive α‐fetoprotein isoform 3 (AFP‐L3) and protein induced by vitamin K absence or antagonist‐II (PIVKA‐II), which are produced by latent malignant clones, by eradicating or inhibiting these clones. Once such clones are deleted, the preventive effect on HCC lasts several years without any continuous administration of ACR. In fact, 1‐year administration of ACR inhibited secondary HCC for the next 3 years.( 11 ) Therefore, this agent can significantly improve the survival rate of such patients.
This concept has been clinically demonstrated and implemented in a clinical trial using ACR. Indeed, in that trial, ACR significantly reduced the serum levels of AFP‐L3, which indicates the presence of latent (i.e. invisible) HCC cells in the remnant liver, after 12 months of administration.( 45 ) The administration of ACR also caused a decrease in the serum levels of protein induced by PIVKA‐II, which may also be produced by latent HCC cells.( 11 ) These results strongly suggest that ACR deleted such malignant clones producing AFP‐L3 or PIVKA‐II before they expanded to clinically detectable tumors, thereby inhibiting second primary HCC. Therefore, once such latent clones are eradicated or inhibited, it may take several years for the next cancer clone to arise clinically.( 11 ) Moreover, ACR also prevented the appearance of AFP‐L3 in patients who had been negative at entry, although there was a significant increase in the incidence of AFP‐L3‐positive patients in the placebo group and these patients had a significantly higher risk of second primary HCC.( 45 ) This is also one of the reasons for the long‐term benefit of ACR after only a 12‐month treatment with this agent.( 11 ) Therefore, we suggest that the concept of ‘clonal deletion’ seems more of a therapy rather than prevention and that ACR is a more affirmative agent to inhibit the development of HCC (Fig. 3).
Possibility of ‘combination chemoprevention’ with ACR
The combined use of two or more agents is often advantageous as it may permit lower clinical dosages, consequently decreasing the overall toxicity and thus providing the potential for synergistic effects between specific agents, including retinoids.( 24 , 48 ) Therefore, the beneficial effects, such as synergism, between ACR and other agents to inhibit the growth of HCC cells have been examined. For instance, ACR acts synergistically with IFN in suppressing growth and inducing apoptosis in human HCC cell lines and this synergism was associated with the upregulation of type 1 IFN receptor expression by ACR.( 49 ) The combination of ACR plus OSI‐461, a potent derivative of sulindac sulfone, exerts synergistic inhibition of cell growth and induction of apoptosis in HepG2 human HCC cells.( 50 ) In addition, the combination of ACR plus VK2 also synergistically induced apoptosis and inhibited the growth of HCC cells without affecting the growth of normal human hepatocytes.( 51 ) The findings that both IFN and VK2 enhance the effects of ACR seem to be of interest because these agents are expected to reduce the development and recurrence rates of HCC.( 52 , 53 )
In the above study,( 51 ) VK2 inhibited phosphorylation of the RXRα protein through the inhibition of Ras activation and Erk phosphorylation, and the inhibition of RXRα phosphorylation by VK2 was enhanced when the cells were cotreated with ACR. In addition, ACR and trastuzumab, the humanized anti‐HER2 monoclonal antibody, cooperatively inhibit the activation of HER2 and its downstream signaling pathways, subsequently inhibiting the phosphorylation of RXRα and the growth of HCC cells.( 54 ) Therefore, ACR may support the effect of the agents that target RTK, thus cooperatively or synergistically inhibiting HCC by targeting RXRα phosphorylation. These findings, together with those of previous reports,( 39 , 40 , 41 ) suggest that the combination of ACR plus a specific agent that targets RTK and the Ras–MAPK signaling pathway may be able to inhibit the phosphorylation of RXRα and it may therefore be a promising strategy to prevent the development of HCC.
Conclusion
The very high incidence of secondary HCC is mainly responsible for the poor prognosis of patients with this malignancy. This fact suggests that, in turn, the establishment of a new effective strategy to prevent the recurrence of HCC will significantly improve the outcome of these patients and thus be an urgent task worldwide. One of the most practical approaches to prevent the development of HCC is ‘clonal deletion’ and clinical trials using ACR theoretically proved the significance of this therapy in HCC chemoprevention.( 11 , 45 ) Experimental studies strongly suggest that phosphorylated RXRα is associated with HCC carcinogenesis and thus may be a critical target for HCC chemoprevention. ACR, which targets phosphorylated RXRα, may therefore play a critical role in preventing the development of HCC when it is used alone or combined with other agents.
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Science, Sports, and Culture of Japan (no. 18790457 to M.S. and no. 17015016 to H.M.).
References
- 1. El‐Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132: 2557–76. [DOI] [PubMed] [Google Scholar]
- 2. Parikh S, Hyman D. Hepatocellular cancer: a guide for the internist. Am J Med 2007; 120: 194–202. [DOI] [PubMed] [Google Scholar]
- 3. Kumada T, Nakano S, Takeda I et al . Patterns of recurrence after initial treatment in patients with small hepatocellular carcinoma. Hepatology 1997; 25: 87–92. [DOI] [PubMed] [Google Scholar]
- 4. Koda M, Murawaki Y, Mitsuda A et al . Predictive factors for intrahepatic recurrence after percutaneous ethanol injection therapy for small hepatocellular carcinoma. Cancer 2000; 88: 529–37. [PubMed] [Google Scholar]
- 5. Tsukuma H, Hiyama T, Tanaka S et al . Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 1993; 328: 1797–801. [DOI] [PubMed] [Google Scholar]
- 6. Okada S, Shimada K, Yamamoto J et al . Predictive factors for postoperative recurrence of hepatocellular carcinoma. Gastroenterology 1994; 106: 1618–24. [DOI] [PubMed] [Google Scholar]
- 7. Chen YJ, Yeh SH, Chen JT et al . Chromosomal changes and clonality relationship between primary and recurrent hepatocellular carcinoma. Gastroenterology 2000; 119: 431–40. [DOI] [PubMed] [Google Scholar]
- 8. Sporn MB, Newton DL. Chemoprevention of cancer with retinoids. Fed Proc 1979; 38: 2528–34. [PubMed] [Google Scholar]
- 9. Muto Y, Moriwaki H, Ninomiya M et al . Prevention of second primary tumors by an acyclic retinoid, polyprenoic acid, in patients with hepatocellular carcinoma. Hepatoma Prevention Study Group. N Engl J Med 1996; 334: 1561–7. [DOI] [PubMed] [Google Scholar]
- 10. Muto Y, Moriwaki H, Saito A. Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. N Engl J Med 1999; 340: 1046–7. [DOI] [PubMed] [Google Scholar]
- 11. Takai K, Okuno M, Yasuda I et al . Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. Updated analysis of the long‐term follow‐up data. Intervirology 2005; 48: 39–45. [DOI] [PubMed] [Google Scholar]
- 12. Matsushima‐Nishiwaki R, Okuno M, Adachi S et al . Phosphorylation of retinoid X receptor α at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res 2001; 61: 7675–82. [PubMed] [Google Scholar]
- 13. Yoshimura K, Muto Y, Shimizu M et al . Phosphorylated retinoid X receptor α loses its heterodimeric activity with retinoic acid receptor β. Cancer Sci 2007; 98: 1868–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adachi S, Okuno M, Matsushima‐Nishiwaki R et al . Phosphorylation of retinoid X receptor suppresses its ubiquitination in human hepatocellular carcinoma. Hepatology 2002; 35: 332–40. [DOI] [PubMed] [Google Scholar]
- 15. Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J 1996; 10: 940–54. [PubMed] [Google Scholar]
- 16. Mangelsdorf DJ, Thummel C, Beato M et al . The nuclear receptor superfamily: the second decade. Cell 1995; 83: 835–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9‐cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992; 358: 771–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rochette‐Egly C. Nuclear receptors: integration of multiple signalling pathways through phosphorylation. Cell Signal 2003; 15: 355–66. [DOI] [PubMed] [Google Scholar]
- 19. Bastien J, Rochette‐Egly C. Nuclear retinoid receptors and the transcription of retinoid‐target genes. Gene 2004; 328: 1–16. [DOI] [PubMed] [Google Scholar]
- 20. Bastien J, Adam‐Stitah S, Plassat JL, Chambon P, Rochette‐Egly C. The phosphorylation site located in the A region of retinoic X receptor α is required for the antiproliferative effect of retinoic acid (RA) and the activation of RA target genes in F9 cells. J Biol Chem 2002; 277: 28683–9. [DOI] [PubMed] [Google Scholar]
- 21. Lee HY, Suh YA, Robinson MJ et al . Stress pathway activation induces phosphorylation of retinoid X receptor. J Biol Chem 2000; 275: 32193–9. [DOI] [PubMed] [Google Scholar]
- 22. Solomon C, White JH, Kremer R. Mitogen‐activated protein kinase inhibits 1,25‐dihydroxyvitamin D3‐dependent signal transduction by phosphorylating human retinoid X receptor α. J Clin Invest 1999; 103: 1729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Macoritto M, Nguyen‐Yamamoto L, Huang DC et al . Phosphorylation of the human retinoid X receptor α at serine 260 impairs coactivator(s) recruitment and induces hormone resistance to multiple ligands. J Biol Chem 2008; 283: 4943–56. [DOI] [PubMed] [Google Scholar]
- 24. Yamazaki K, Shimizu M, Okuno M et al . Synergistic effects of RXRα and PPARγ ligands to inhibit growth in human colon cancer cells – phosphorylated RXRα is a critical target for colon cancer management. Gut 2007; 56: 1557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kanemura N, Tsurumi H, Okuno M, Matsushima‐Nishiwaki R, Shimizu M, Moriwaki H. Retinoid X receptor α is highly phosphorylated in retinoic acid‐resistant HL‐60R cells and the combination of 9‐cis retinoic acid plus MEK inhibitor induces apoptosis in the cells. Leuk Res 2008; 32: 884–92. [DOI] [PubMed] [Google Scholar]
- 26. Lattuada D, Vigano P, Mangioni S et al . Accumulation of retinoid X receptor‐α in uterine leiomyomas is associated with a delayed ligand‐dependent proteasome‐mediated degradation and an alteration of its transcriptional activity. Mol Endocrinol 2007; 21: 602–12. [DOI] [PubMed] [Google Scholar]
- 27. Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer 2001; 1: 181–93. [DOI] [PubMed] [Google Scholar]
- 28. Muto Y, Moriwaki H. Antitumor activity of vitamin A and its derivatives. J Natl Cancer Inst 1984; 73: 1389–93. [PubMed] [Google Scholar]
- 29. Ando N, Shimizu M, Okuno M et al . Expression of retinoid X receptor α is decreased in 3′‐methyl‐4‐dimethylaminoazobenzene‐induced hepatocellular carcinoma in rats. Oncol Rep 2007; 18: 879–84. [PubMed] [Google Scholar]
- 30. Matsushima‐Nishiwaki R, Shidoji Y, Nishiwaki S, Yamada T, Moriwaki H, Muto Y. Aberrant metabolism of retinoid X receptor proteins in human hepatocellular carcinoma. Mol Cell Endocrinol 1996; 121: 179–90. [DOI] [PubMed] [Google Scholar]
- 31. Matsushima‐Nishiwaki R, Okuno M, Takano Y, Kojima S, Friedman SL, Moriwaki H. Molecular mechanism for growth suppression of human hepatocellular carcinoma cells by acyclic retinoid. Carcinogenesis 2003; 24: 1353–9. [DOI] [PubMed] [Google Scholar]
- 32. Araki H, Shidoji Y, Yamada Y, Moriwaki H, Muto Y. Retinoid agonist activities of synthetic geranyl geranoic acid derivatives. Biochem Biophys Res Commun 1995; 209: 66–72. [DOI] [PubMed] [Google Scholar]
- 33. Yamada Y, Shidoji Y, Fukutomi Y et al . Positive and negative regulations of albumin gene expression by retinoids in human hepatoma cell lines. Mol Carcinog 1994; 10: 151–8. [DOI] [PubMed] [Google Scholar]
- 34. Nakamura N, Shidoji Y, Yamada Y, Hatakeyama H, Moriwaki H, Muto Y. Induction of apoptosis by acyclic retinoid in the human hepatoma‐derived cell line, HuH‐7. Biochem Biophys Res Commun 1995; 207: 382–8. [DOI] [PubMed] [Google Scholar]
- 35. Nakamura N, Shidoji Y, Moriwaki H, Muto Y. Apoptosis in human hepatoma cell line induced by 4,5‐didehydro geranylgeranoic acid (acyclic retinoid) via down‐regulation of transforming growth factor‐α. Biochem Biophys Res Commun 1996; 219: 100–4. [DOI] [PubMed] [Google Scholar]
- 36. Fukutomi Y, Omori M, Muto Y, Ninomiya M, Okuno M, Moriwaki H. Inhibitory effects of acyclic retinoid (polyprenoic acid) and its hydroxy derivative on cell growth and on secretion of α‐fetoprotein in human hepatoma‐derived cell line (PLC/PRF/5). Jpn J Cancer Res 1990; 81: 1281–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yasuda I, Shiratori Y, Adachi S et al . Acyclic retinoid induces partial differentiation, down‐regulates telomerase reverse transcriptase mRNA expression and telomerase activity, and induces apoptosis in human hepatoma‐derived cell lines. J Hepatol 2002; 36: 660–71. [DOI] [PubMed] [Google Scholar]
- 38. Suzui M, Masuda M, Lim JT, Albanese C, Pestell RG, Weinstein IB. Growth inhibition of human hepatoma cells by acyclic retinoid is associated with induction of p21 (CIP1) and inhibition of expression of cyclin D1. Cancer Res 2002; 62: 3997–4006. [PubMed] [Google Scholar]
- 39. Kagawa M, Sano T, Ishibashi N et al . An acyclic retinoid, NIK‐333, inhibits N‐diethylnitrosamine‐induced rat hepatocarcinogenesis through suppression of TGF‐α expression and cell proliferation. Carcinogenesis 2004; 25: 979–85. [DOI] [PubMed] [Google Scholar]
- 40. Sano T, Kagawa M, Okuno M et al . Prevention of rat hepatocarcinogenesis by acyclic retinoid is accompanied by reduction in emergence of both TGF‐α‐expressing oval‐like cells and activated hepatic stellate cells. Nutr Cancer 2005; 51: 197–206. [DOI] [PubMed] [Google Scholar]
- 41. Shimizu M, Suzui M, Deguchi A, Lim JT, Weinstein IB. Effects of acyclic retinoid on growth, cell cycle control, epidermal growth factor receptor signaling, and gene expression in human squamous cell carcinoma cells. Clin Cancer Res 2004; 10: 1130–40. [DOI] [PubMed] [Google Scholar]
- 42. Shao RX, Otsuka M, Kato N et al . Acyclic retinoid inhibits human hepatoma cell growth by suppressing fibroblast growth factor‐mediated signaling pathways. Gastroenterology 2005; 128: 86–95. [DOI] [PubMed] [Google Scholar]
- 43. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000; 103: 211–25. [DOI] [PubMed] [Google Scholar]
- 44. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium: clinical implications of multicentric origin. Cancer 1953; 6: 963–8. [DOI] [PubMed] [Google Scholar]
- 45. Moriwaki H, Yasuda I, Shiratori Y, Uematsu T, Okuno M, Muto Y. Deletion of serum lectin‐reactive α‐fetoprotein by acyclic retinoid: a potent biomarker in the chemoprevention of second primary hepatoma. Clin Cancer Res 1997; 3: 727–31. [PubMed] [Google Scholar]
- 46. Moriwaki H, Shimizu M, Okuno M, Nishiwaki‐Matsushima R. Chemoprevention of liver carcinogenesis with retinoids: Basic and clinical aspects. Hepatol Res 2007; 37 (Suppl 2): S299–302. [DOI] [PubMed] [Google Scholar]
- 47. Moriwaki H. Prevention of liver cancer: basic and clinical aspects. Exp Mol Med 2002; 34: 319–25. [DOI] [PubMed] [Google Scholar]
- 48. Shimizu M, Moriwaki H. Synergistic effects of PPARγ ligands and retinoids in cancer treatment. PPAR Res 2008; 2008: 181047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Obora A, Shiratori Y, Okuno M et al . Synergistic induction of apoptosis by acyclic retinoid and interferon‐β in human hepatocellular carcinoma cells. Hepatology 2002; 36: 1115–24. [DOI] [PubMed] [Google Scholar]
- 50. Shimizu M, Suzui M, Deguchi A et al . Synergistic effects of acyclic retinoid and OSI‐461 on growth inhibition and gene expression in human hepatoma cells. Clin Cancer Res 2004; 10: 6710–21. [DOI] [PubMed] [Google Scholar]
- 51. Kanamori T, Shimizu M, Okuno M et al . Synergistic growth inhibition by acyclic retinoid and vitamin K2 in human hepatocellular carcinoma cells. Cancer Sci 2007; 98: 431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kubo S, Nishiguchi S, Hirohashi K et al . Effects of long‐term postoperative interferon‐α therapy on intrahepatic recurrence after resection of hepatitis C virus‐related hepatocellular carcinoma. A randomized, controlled trial. Ann Intern Med 2001; 134: 963–7. [DOI] [PubMed] [Google Scholar]
- 53. Mizuta T, Ozaki I, Eguchi Y et al . The effect of menatetrenone, a vitamin K2 analog, on disease recurrence and survival in patients with hepatocellular carcinoma after curative treatment: a pilot study. Cancer 2006; 106: 867–72. [DOI] [PubMed] [Google Scholar]
- 54. Tatebe H, Shimizu M, Shirakami Y, Tsurumi H, Moriwaki H. Synergistic growth inhibition by 9‐cis‐retinoic acid plus trastuzumab in human hepatocellular carcinoma cells. Clin Cancer Res 2008; 14: 2806–12. [DOI] [PubMed] [Google Scholar]