

Abstract
Melanoma inhibitory activity (MIA) has been identified as a small protein secreted from malignant melanoma cells, which strongly enhances melanoma cell migration and invasion. Detailed analyses performed by our group showed interaction of MIA with extracellular matrix proteins and integrin α4β1 and α5β1 leading to cellular detachment. In this study, we identified cadherin‐7 as a new MIA‐binding protein using surface‐enhanced laser desorption/ionization‐mass spectrometry technology and co‐immunoprecipitation. Cadherin‐7 is a classical cell–cell adhesion molecule which was shown to be upregulated in malignant melanoma. We demonstrated enhanced expression of cadherin‐7 in primary tumor cells compared to metastatic cells. Upregulation of cadherin‐7 expression in metastatic cell lines but also downregulation of expression in cells derived from primary melanomas resulted in reduced cell migration. In addition, we speculate that MIA/cadherin‐7 interaction may regulate cell–cell adhesion of malignant melanoma cells influencing the migration of the cells. Interestingly, overexpression of cadherin‐7 resulted in a decreased MIA mRNA expression. In addition, MIA effects on cell migration were abrogated in cell clones overexpressing cadherin‐7. In conclusion, these findings suggest that cadherin‐7 regulates the expression and activity of MIA and the migration of melanoma cells playing a role in tumor development of malignant melanoma. (Cancer Sci 2009; 100: 261–268)
The protein melanoma inhibitory activity (MIA), which is strongly expressed in melanoma cells but not in melanocytes, represents a key molecule in melanoma progression. We have previously described the isolation of the 11‐kDa protein MIA, which is secreted from melanoma cells into the tissue culture supernatant.( 1 ) MIA inhibits adhesion of cells to the extracellular matrix leading to enhanced migration and invasion. The induction of MIA expression correlates with the tumor progression in malignant melanoma.( 2 ) Additionally, we detected enhanced MIA protein levels specifically in the serum of patients with metastatic melanomas representing MIA as a serum marker for systemic malignant melanoma.( 3 )
MIA was shown to interact with several matrix proteins such as fibronectin and laminin.( 4 ) Recently, we reported that MIA additionally directly interacts with integrin α4β1 and α5β1 leading to a modulation of integrin activity and to a reduced integrin signaling via externally regulated kinases (ERK)1/2.( 5 ) These findings suggest that MIA plays a role in tumor progression of malignant melanoma via mediating detachment of cells from extracellular matrix molecules.
It is also known that cadherins play a role in tumorigenesis of different tumor types including malignant melanoma. Loss of E‐cadherin expression and an increased N‐cadherin expression during the development and progression of malignant melanoma was observed.( 6 ) Cadherins are transmembrane proteins that mediate calcium‐dependent adhesive interactions between animal cells mainly through homophilic interactions.( 7 , 8 ) Cadherins are a large family of cell adhesion molecules that includes the classical cadherins, comprised of type I and type II cadherins, as the most important. Type I cadherins promote much stronger adhesiveness than type II which is mediated by their extracellular domains. The fact that cadherins are different in their ability to promote intercellular adhesiveness has implications for the regulation of tissue morphogenesis.( 9 )
Until now, expression patterns of cadherin‐7, a type II cadherin, has been examined in chicken central nervous system( 10 , 11 ) and in human in various tissue samples.( 12 ) Moore et al. revealed that cadherin‐7 is upregulated in malignant melanoma whereas melanocyte cell lines did not express cadherin‐7.( 13 ) Cadherin‐7 is also expressed by neural crest cells that emigrate from the neural tube and plays important roles in the regulation of morphogenesis and tissue formation during embryo development.( 14 ) The precise function of cadherin‐7 in malignant melanoma has not yet been determined.
In this study, we identified cadherin‐7 as an MIA binding partner and investigated the role of cadherin‐7 and its interaction with MIA in the progression of malignant melanoma.
Materials and Methods
Cell lines and culture condition. The melanoma cell lines Mel Ei, Mel Wei, Mel Ho, Mel Juso (derived from primary cutaneous melanomas), Mel Ju, SK‐Mel‐28, HTZ19 and HMB2 (derived from metastases of malignant melanomas) have been described previously.( 15 ) For tissue culture, the cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin (400 U/mL), streptomycin (50 µg/mL), L‐glutamine (300 µg/mL) and 10% fetal calf serum (Sigma, Deisenhofen, Germany). Normal human epidermal melanocytes derived from normal skin were cultivated in melanocyte medium MGM‐3 (Life Technologies, Eggenstein, Germany) under a humidified atmosphere of 5% CO2 at 37°C.
Expression of recombinant MIA and TANGO protein. A MIA prokaryotic expression vector with a 15 amino acid Avi‐tag peptide sequence including an FXa cleavage site was constructed by overlap extension polymerase chain reaction (PCR). The MIA cDNA construct was cloned into the vector pIVEX2.3‐MCS (Roche, Mannheim, Germany). The expression vector was used in the Rapid Translation System, a cell‐free Escherichia coli‐based protein transcription/translation system (Roche). By adding biotin, adenosine triphosphate (ATP) and the E. coli biotin protein ligase BirA during the procedure, the protein was biotinylated at the introduced Avi‐tag at the N‐terminus and called recombinant biotinylated MIA.( 5 ) Recombinant biotinylated TANGO was generated using the same technique.( 16 )
Detection of protein interactions. Protein interactions were assessed in a modified procedure as described elsewhere.( 17 ) Surface‐enhanced laser desorption/ionization‐mass spectrometry (SELDI‐MS) technology is comprised of ProteinChip arrays and mass spectrometry identifying protein interactions. First, co‐immunoprecipitation was performed incubating protein lysates of Mel Im cells with biotinylated MIA or TANGO. Then, interacting proteins were identified by SELDI‐MS technology.
In brief, 30 µL of streptavidin agarose beads (Molecular Probes, Eugene, OR, USA) were washed with phosphate‐buffered saline (PBS) followed by incubation of recombinant biotinylated MIA or TANGO in PBS overnight at 4°C in an end‐over‐end mixer, or, as a negative control, streptavidin agarose beads incubated without recombinant protein underwent the same procedure. In parallel, 5 × 106 Mel Im cells were lyzed in a buffer containing 100 mM sodium phosphate pH 7.5, 5 mM ethylene diamine tetra acetate (EDTA), 2 mM MgCl2, 3 mM 2‐β‐mercaptoethanol, 0.1% 3[(3‐Cholamidopropyl) dimethylammonio]‐propanesulfonic acid (CHAPS), 500 µM leupeptin, and 0.1 mM phenylmethylsulfonyl fluoride (PMSF). Afterwards, the cell lysate was incubated with streptavidin agarose beads for depletion of proteins that bind non‐specific to the streptavidin agarose beads. Pellets were discarded and the pre‐cleared supernatant was incubated with 50 ng recombinant MIA or TANGO and 30 µL streptavidin agarose beads for 2 h at room temperature in an over‐end‐over mixer. Bound proteins were eluted from the beads by 30 µL 50% acetonitrile/0.5% trifluoroacetic acid and gently vortexed for 5 min. Eluate (2 µL) were applied to the activated, hydrophobic surface of an H50 ProteinChip Array (Ciphergen Biosystem, Fremont, CA, USA) and dried on air. After washing with 3 µL aqua bidest, 0.5 µL sinapinic acid (saturated solution in 0.5% trifluoroacetic acid (TFA)/50% acetonitrile) was applied twice and the array was analyzed in a ProteinChip Reader (series 4000, Ciphergen) according to an automated data collection protocol.
Identification of interacting proteins. The identification of interesting proteins was performed as described.( 18 ) Briefly, the eluates were reduced to a maximal volume of 10 µL by speed‐vac and subjected to sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS‐PAGE) for separation of containing proteins followed by staining with Simply Blue Safe Stain (Enhanced Coomassie, Invitrogen). Afterwards, interesting specific gel bands were excised, destained and dried followed by rehydration and digestion with 10 µL of a trypsin solution (0.02 µg/µL; Promega) overnight at 37°C. The supernatants of in‐gel digestion were applied directly to NP20 ProteinChip arrays (Ciphergen). After addition of the matrix, peptide fragment masses were analyzed using the ProteinChip Reader, series 4000 instrument. A standard protein mix (All‐In‐1 Peptide Standard Mix; Ciphergen), including Arg8‐vasopressin (1082.2 Da), somatostatin (1637.9 Da), dynorphin (2147.5 Da), adrenocorticotrophic hormone (ACTH; 2933.5 Da), and insulin β‐chain (3495.94 Da) was used for calibration. Proteins were identified using the fragment masses searching in a publicly available database (http://prowl.rockefeller.edu/prowl‐cgi/profound.exe).
Co‐immunoprecipitation. For co‐immunoprecipitation, cell lysates dissolved in binding buffer (20 mM NaPO4, 150 mM NaCl, pH 7.5) were incubated with 0.6 µg of the anti‐Cad7 antibody (CCD‐7( 19 )) with shaking at 4°C overnight. Then, 20 µL protein G Sepharose 4 Fast Flow (Amersham Biosciences) was added for 1 h, pelleted, washed three times with binding buffer, resuspended in 20 µL of Laemmli's buffer, heated at 95°C for 5 min, and separated on a 15% SDS‐PAGE. MIA binding to cadherin‐7 were identified by western blot with anti‐MIA antibody.( 5 ) Recombinant MIA protein was used as positive control.
Transfection experiments. Mel Im and Mel Juso cell clones varying with respect to cadherin‐7 expression were established by stable transfection with a cadherin‐7 expression vector (full‐length chicken cadherin‐7 in pCMX‐PL2) into Mel Im or antisense–cadherin‐7 expression plasmids (858‐bp fragment of the cadherin‐7 coding region cloned in antisense orientation into the pCMX‐PL1) into Mel Juso and Mel Im. Plasmids were co‐transfected with pcDNA3 (Invitrogen NV, Leek, Holland), which contains the selectable marker for neomycin resistance. Controls (mock) received pCDNA3 alone. Transfections were performed using the Lipofectamine Plus method (Gibco). One day after transfection, cells were placed into selection medium containing 50 µg/mL of G418 (Sigma). After 25 days of selection, individual G418‐resistant colonies were subcloned. The amount of cadherin‐7 expression in these clones was determined by reverse transcription (RT)‐PCR and fluorescence‐activated cell sorter (FACS) analyses.
For transient transfections, 2 × 105 melanoma cells per well were seeded into six‐well plates and transfected with 0.5 µg of a chicken cadherin‐7 overexpressing plasmid using the Lipofectamine Plus method (Gibco) according to the manufacturer's instructions. The cells were used for experiments 24 h after transfection.
RNA isolation and reverse transcription. Total cellular RNA was isolated from cultured cells using the RNeasy kit (Qiagen, Hilden, Germany). cDNA were generated by RT reaction performed in 20 µL reaction volume containing 2 µg of total cellular RNA, 4 µL of 5 × first strand buffer (Invitrogen), 2 µL of 0.1 M 1,4‐dithiothreitol (DTT), 1 µL of dN6‐primer (10 mM), 1 µL of deoxynudeoside triphoshate (DTT) (10 mM), and diethyl pyrocarbonate (DEPC)‐water. The reaction mixture was incubated for 10 min at 70°C, 200 U of Superscript II reverse transcriptase (Invitrogen) were added and RNA were transcribed for 1 h at 37°C. Reverse transcriptase was inactivated at 70°C for 10 min and the RNA was degraded by digestion with 1 µL RNase A (10 mg/mL) at 37°C for 30 min.
Analysis of expression by quantitative PCR. Quantitative real‐time PCR for cadherin‐7 and MIA was performed on a LightCycler (Roche, Mannheim, Germany). cDNA template (2 µL), 0.5 µL (20 mM) of forward and reverse primers (Cad7 forward, 5′‐TCA CCA ACA AAC CCG TGG A‐3′ and Cad7 reverse, 5′‐ATC ATC AGC ATC CGT CGC T‐3′; MIA forward, 5′‐CAT GCA TGC GGT CCT ATG CCC AAG CTG‐3′ and MIA reverse 5′‐GAT AAG CTT TCA CTG GCA GTA GAA ATC‐3′) and 10 µL of SYBR Premix Ex Taq (TaKaRa Bio Inc., Foster City, CA, USA) in a total of 20 µL were applied to the following PCR program: 30 s at 95°C (initial denaturation); 20°C/s temperature transition rate up to 95°C for 15 s, 3 s at 68°C, 5 s at 72°C, 86°C acquisition mode single, and repeated for 40 times (amplification). The PCR reaction was evaluated by melting curve analysis and checking the PCR products on 2% agarose gels. β‐Actin was amplified to ensure cDNA integrity and to normalize expression.
Fluorescence‐activated cell sorting (FACS) analysis. To analyze expression of cadherin‐7 on the cell surface, cells were detached from flasks using 5 mM EDTA and resuspended in PBS. Cells (106) were fixed with 4% paraformaldehyde/PBS for 1 h at 4°C. After three washing steps with PBS, the cells were incubated for 1 h at 4°C with a 1:1000 dilution of anti‐Cad7 antibody in 1% bovine serum albumin (BSA)/PBS. Crude serum was obtained from a rabbit immunized against a small peptide corresponding to the extracellular domain of human cadherin‐7 (Sigma‐Genosys, TX, United States). The polyclonal CDH‐7 antibody was subsequently purified from the crude rabbit serum using an affinity column with immobilized GST‐CDH7 fusion protein. Then, the cells were washed again and the secondary antibody (FITC antirabbit; DakoCytomation, Denmark) was added for 30 min at 4°C. After additional washing with PBS, the cells were resuspended in a volume of 300–500 µL of PBS and FACS analysis was performed. FACS data were analyzed using the CellQuest software (Becton Dickinson).
Migration assays. For migration assays, Boyden chambers containing polycarbonate filters with 8‐mm pores (Costar, Bodenheim, Germany) were used, as described previously.( 15 ) Filters were coated with gelatine (5 mg/L) to improve cell attachment. The lower compartment was filled with fibroblast‐conditioned medium, used as a chemoattractant. Melanoma cells were harvested by trypsinization for 2 min, resuspended in DMEM without fetal calf serum at a density of 2.5 × 104 cells/mL and placed in the upper compartment of the chamber. In groups with MIA treatment, MIA was added to the cell suspension in a concentration of 200 ng/mL. After incubation at 37°C for 4 h, the filters were collected and the cells adhering to the lower surface fixed, stained and counted. Experiments were carried out in triplicate and were repeated three times with consistent results.
Immunofluorescence. Cells (105) were grown on a four‐well chamber slide for 1 day and immunofluorescence was performed using the F‐Actin Rhodamine‐Phalloidin based kit (Amersham Biosciences, Germany) according to the manufacturer's instructions. One hour before fixation, cells were treated with 100 ng/mL recombinant biotinylated MIA. After fixation, permeabilization and washing, the cells were covered with blocking solution (1% BSA/PBS) for 1 h. Thereafter, cells were incubated with a 1:20 dilution of MIA (1705) antibody (Biogenes, Berlin, Germany) for 1 h at room temperature. After washing, the cells were incubated with a 1:20 dilution of secondary antibody (FITC antirabbit, DakoCytomation, Denmark) in PBS for 30 min followed by washing again. After mounting with Hard Set Mounting Medium with DAPI (Vectashield, H‐1500) images were collected by fluorescence microscopy.
Statistical analysis. The results are expressed as the means ± standard deviation (range) or percentages. All of the calculations were performed using the GraphPad Prism software (GraphPad Software, San Diego, CA, USA).
Results
Characterization of cadherin‐7 as MIA binding partner. Previously, we described that MIA interacts with matrix proteins and integrins leading to the idea that other proteins may also interact with MIA. To identify additional MIA binding proteins, we performed SELDI technology. Total protein lysates of the melanoma cell line Mel Im and recombinant biotinylated MIA was used for co‐immunoprecipitation. Hereby, a specific protein was captured by the recombinant biotinylated MIA as detected in a subsequent electrophoretic separation of the eluted proteins. To exclude that this protein binds to the biotinylated site, we used recombinant biotinylated TANGO, an MIA‐homologous protein,( 20 ) as additional negative control supporting specific binding to MIA. Thus, we confirmed the presence of an MIA‐interacting protein. Cadherin‐7 was identified as this MIA binding protein (Fig. 1A). Phage display assays support the interaction of MIA and cadherin‐7 and revealed a binding of MIA near the transmembrane domain of cadherin‐7 (data not shown).
Figure 1.
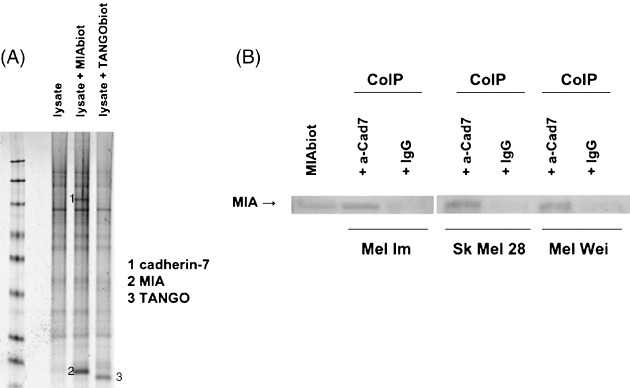
Identification of the melanoma inhibitory activity (MIA) binding partner cadherin‐7. (A) Surface‐enhanced laser desorption/ionization (SELDI) technology revealed specific binding of MIA and cadherin‐7. Co‐immunoprecipitation (CoIP) was performed using protein lysates of Mel Im incubated with biotinylated MIA or TANGO. Coomassie staining showed additional bands through interaction with MIA in comparison to the negative control and TANGO. SELDI mass spectrometry followed by digestion and analyses of gel bands identified one band as cadherin‐7. (B) Co‐immunoprecipitations confirmed the direct interaction of MIA and cadherin‐7. For immunoprecipitation an anti‐cadherin‐7 antibody (a‐Cad7) was used. Interaction was detected in three melanoma cell lines using western blot analysis with MIA antibody. Recombinant MIA was used as positive control.
The interaction was further unambiguously confirmed by co‐immunoprecipitation using total protein lysates of melanoma cell lines. Co‐immunoprecipitation revealed an interaction of cadherin‐7 with MIA. This assay was performed with three melanoma cell lines confirming the interaction of MIA and cadherin‐7 (Fig. 1B). Herewith, we identified cadherin‐7 as an MIA binding partner.
Expression of cadherin‐7 in melanoma cell lines. Next, we analyzed the expression pattern of cadherin‐7 in malignant melanoma. Expression of cadherin‐7 was detected in the melanoma cell lines using RT‐PCR (Fig. 2A). Quantitative real‐time PCR revealed cadherin‐7 expression in all melanoma cell lines. Interestingly, primary melanoma cell lines showed a high expression of cadherin‐7 in comparison to the metastatic cell lines. In normal human epidermal melanocytes (NHEM), the cadherin‐7 expression was very low or undetectable.
Figure 2.
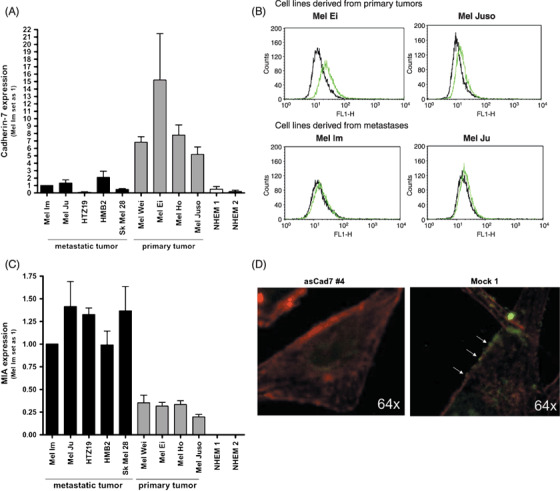
Cadherin‐7 is upregulated in malignant melanoma. (A) Quantitative polymerase chain reaction (PCR) revealed mRNA expression of cadherin‐7 in all malignant melanoma cell lines with higher expression in cell lines derived from primary tumors. The melanocytes (normal human epidermal melanocytes, NHEM) showed only a very low cadherin‐7 level. β‐Actin was used for normalization. (B) Fluorescence‐activated cell sorting experiments using primary and metastatic melanoma cell lines were performed. The cell lines were incubated with the Cad7 antibody and revealed binding to cadherin‐7 on the cell surface (gray lines). The black lines present negative controls. Higher expression of cadherin‐7 was found in the primary melanoma cell lines Mel Ei and Mel Juso in comparison to the metastatic cell lines Mel Im and Mel Ju. (C) Quantitative PCR was used to measure mRNA expression of melanoma inhibitory activity (MIA) in all malignant melanoma cell lines with higher expression in cell lines derived from metastases. The melanocytes (NHEM) showed no MIA expression. β‐Actin was used for normalization. (D) Immunofluorescence staining of MIA in Mel Juso cells expressing cadherin‐7 (Mock 1) or reduced levels of cadherin‐7 (asCad7 #4). Actin cytoskeleton was stained with rhodamine–phalloidin. The cells were treated with recombinant biotinylated MIA for 1 h, fixed and stained for MIA. The mock control cells showed a higher amount of MIA located at the cell membrane (arrows) than the asCad7 stably transfected cells.
In order to confirm differential expression of cadherin‐7 in primary melanoma and metastasis, we performed FACS analysis to determine the presentation of cadherin‐7 on the cell surface. In agreement with the mRNA data, the amount of cadherin‐7 in the primary melanoma cell lines (Mel Ei, Mel Juso) was higher in comparison to the metastatic melanomas (Mel Im, Mel Ju) (Fig. 2B). In contrast to cadherin‐7, MIA expression is induced in melanoma cells derived from primary melanoma in comparison to NHEM but expression even further increases in cell lines derived from metastases (Fig. 2C).
Next, we wanted to show that MIA co‐localizes with cadherin‐7 at the cell membrane. Therefore, we performed immunofluorescence staining of MIA in Mel Juso cells expressing cadherin‐7 (mock control) or reduced levels or cadherin‐7 (asCad clone). In Mel Juso control cells, high amounts of MIA are located at the cell membrane. In comparison, in asCad7 cell clones less MIA is found (Fig. 2D) suggesting that different amounts of MIA binding are dependent of cadherin‐7 on the cell surface and that MIA is associated with cadherin‐7 at the cell border of melanoma cells.
Cadherin‐7 expression influences the migration of melanoma cells. To analyze the function of cadherin‐7 in melanoma, we stably transfected a cell line from primary melanoma cell lines (Mel Juso) with an antisense cadherin‐7 construct to reduce cadherin‐7 expression (asCad7) and quantified changes in cadherin‐7 expression using RT‐PCR (data not shown) and FACS analysis (Fig. 3A). Proliferation assays did not reveal any changes after modulation of cadherin‐7 expression. However, Boyden chamber assay revealed decreased migration of the asCadherin‐7 transfected Mel Juso cells in comparison to the control‐transfected cells revealing that cadherin‐7 plays a supportive role in migration of melanoma cells derived from primary tumors (Fig. 3B). Overexpression of cadherin‐7 also led to reduced migration in Boyden chamber assay suggesting that very high, non‐physiological cadherin‐7 levels disturb cell motility (Fig. 3C).
Figure 3.
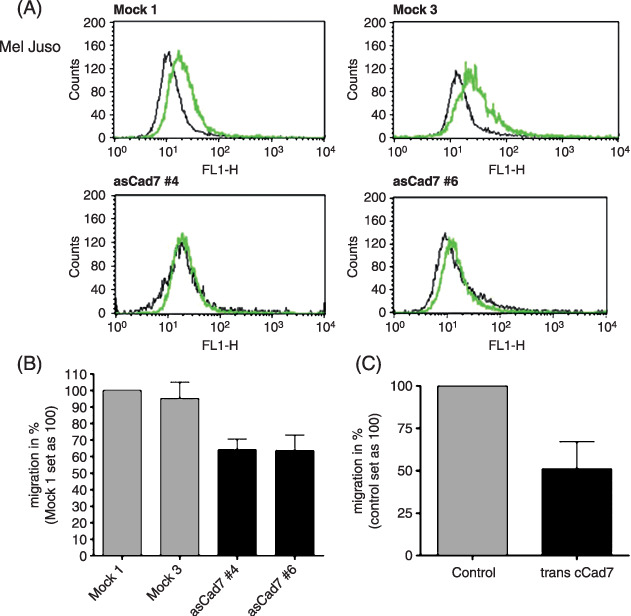
Role of cadherin‐7 in the migration of melanoma cells derived from primary tumors. (A) The primary melanoma cell line Mel Juso was stably transfected with an antisense cadherin‐7 construct and cell clones (asCad7 #4 and asCad7 #6) were analyzed by fluorescence‐activated cell sorting analysis. The gray lines present anti cadherin‐7 antibody staining. The black lines mark the negative controls. AsCad7 clones showed a reduced expression of Cad7 in comparison to the control‐transfected mock clones (Mock 1, Mock 3). (B) The Boyden chamber assay revealed a reduced migration of the asCadherin‐7 transfected Mel Juso cells compared to the mock control. (C) Compared with control cells, Mel Juso cells transiently transfected with a cadherin‐7 expressing vector showed a decreased migration capability.
Next, we stably transfected a cell line derived from a melanoma metastasis (Mel Im) with a sense cadherin‐7 expression construct to induce cadherin‐7 expression (cCad7) and investigated proliferation and migration. We confirmed changes in cadherin‐7 expression by RT‐PCR (data not shown) and FACS analysis (Fig. 4A). No changes in proliferation were observed after induction of cadherin‐7 expression in Mel Im cells. Interestingly, induced expression of cadherin‐7 in the Mel Im cell line derived from metastasis resulted in reduced migration (Fig. 4B) whereas stable antisense cadherin‐7 expression did not result in changes (data not shown). Here, it seems that melanoma cells in metastasis need to downregulate cadherin‐7 expression to ensure migratory ability. Next, we asked whether this finding is associated with MIA binding to cadherin‐7 and speculated that MIA is bound to and captured by excess of cadherin‐7 leading to reduced migratory ability of the cell clones. To test this hypothesis we treated the stably transfected cadherin‐7 cell clones with recombinant MIA and investigated migration in the Boyden chamber assay. MIA is known to block migration in this assay by inhibiting cellular attachment (for review see ( 2 )). As expected, the control cells showed decreased migration after treatment with MIA, whereas the cadherin‐7 overexpressing cell clones did not show changes in their migratory potential (Fig. 4C). Treatment of MIA in Mel Juso cells stably transfected with asCad7 construct showed no differences in migration in comparison to the control cells (data not shown).
Figure 4.
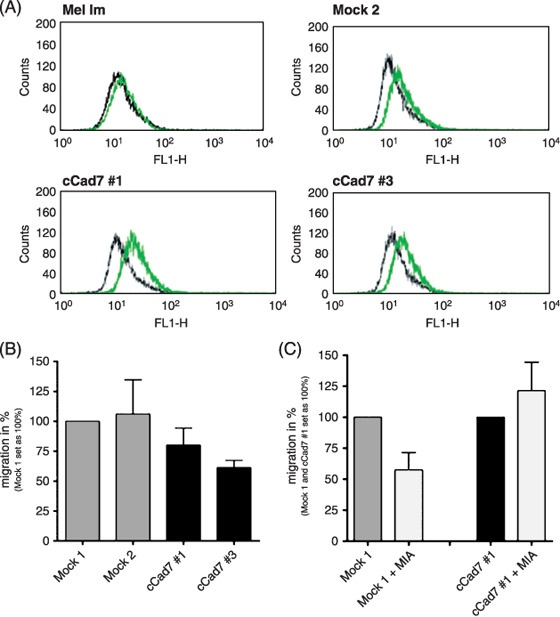
Role of cadherin‐7 in the migration of melanoma cells derived from metastases. (A) Mel Im cells were stably transfected with a cadherin‐7 overexpressing vector and cadherin‐7 expression was analyzed by fluorescence‐activated cell sorting analysis. The cCad7 cell clones (cCad7 #1, #3) showed induced cadherin‐7 expression compared to the control‐transfected mock clone and the parental Mel Im cell line. (B) The cCad7 cell clones showed a decreased migration in comparison to the control in the Boyden chamber assay. (C) The Boyden chamber assay revealed a reduced migration after treatment of the mock control cells with recombinant melanoma inhibitory activity (MIA) (200 ng/mL). In contrast, in cadherin‐7 overexpressing cells migration stayed unchanged after treatment with recombinant MIA.
Cadherin‐7 regulates the expression of MIA. As inhibition of MIA function can only partially explain the strong reduction in migration seen in Figure 4(B) we wondered whether this effect was also due to regulation of MIA expression and quantified MIA mRNA in Mel Im cells either overexpressing cadherin‐7 (Cad7 #1, #3, #4) or showing reduced cadherin‐7 expression (asCad7 #2, #7, #8) after transfection with an antisense cadherin‐7 expression construct using real‐time PCR. Overexpression of cadherin‐7 led to decreased MIA mRNA expression whereas downregulation further enhanced the MIA mRNA amount (Fig. 5A). These results were confirmed in Mel Juso cells stably transfected with asCad7‐plasmid and transiently transfected with cadherin‐7 overexpressing plasmid. Overexpression of cadherin‐7 in Mel Juso cells showed only a small reduction of MIA expression as these cells already express high levels of cadherin‐7 (Fig. 5B). This finding reveals that cadherin‐7 can influence MIA expression by so far unknown signaling pathways.
Figure 5.
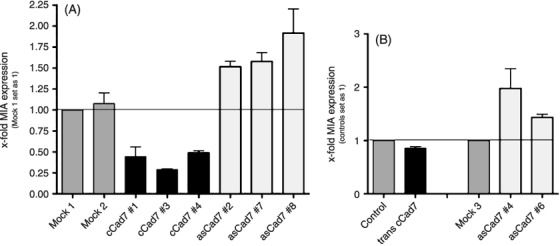
Cadherin‐7 regulates the melanoma inhibitory activity (MIA) expression. (A) MIA mRNA levels of Mel Im cell clones stably transfected with cadherin‐7 were analyzed by real‐time polymerase chain reaction (PCR) analysis and showed decreased MIA expression in comparison to the control cells. In addition, Mel Im cells were stably transfected with antisense cadherin‐7 constructs and analyzed for MIA expression. Here, induction of MIA expression was observed. (B) The amount of MIA mRNA expression was quantified by quantitative real‐time PCR in Mel Juso cells transiently transfected with cadherin‐7 construct showing a reduced MIA expression compared to control cells. Mel Juso cells stably transfected with asCad7 plasmid increased MIA mRNA levels.
Discussion
In this study, we have identified cadherin‐7 as a new MIA binding partner in malignant melanoma. In addition, we have shown that cadherin‐7 plays a role in melanoma cell migration and that the interaction of cadherin‐7 and MIA influences migration of melanoma cells.
As recent data suggest that MIA plays a role in melanoma progression and the spread of malignant melanoma,( 5 ) we aimed to search for additional MIA binding partner by SELDI technology. SELDI‐MS and co‐immunoprecipitation led to the identification of cadherin‐7, a cell adhesion molecule, as novel MIA binding protein.
It is known that cadherins are involved in processes like development, dynamic cell adhesion, detachment and migration of cells.( 21 ) We therefore assumed that cadherin‐7 is involved in the process of tumor progression of malignant melanoma potentially mediated by MIA.
Recently, Moore et al. showed that cadherin‐7 is upregulated in several malignant melanoma cell lines.( 13 ) We confirmed this finding by quantitative real‐time PCR, however, interestingly the primary melanoma cell lines showed a higher mRNA and cell surface expression of cadherin‐7 than the metastatic cell lines. This led to the hypothesis that cadherin‐7 contributes to early delamination of melanoma cells from keratinocytes as cadherin‐7 expression was shown to be important in the delamination of neural crest cells from the neural tube with a strict time‐dependent regulation.( 14 ) Many studies revealed that other cadherins, E‐ and N‐cadherin, influences early development of malignant melanoma where E‐cadherin was described as tumor suppressor. The loss of E‐cadherin was associated with a high potential to invade and metastasize.( 22 , 23 ) The demonstrated switch of E‐cadherin to N‐cadherin expression in melanoma cells has a main impact on tumor development and progression. Through the expression of N‐cadherin, the melanocytes changed their binding partners and interacted with fibroblasts instead of keratinocytes contributing to the malignant transformation of melanocytes. N‐cadherin also promotes survival and migration of melanoma cells.( 6 ) In addition, in breast cancer a switch from type I (E‐cadherin) to a type II (cadherin‐11) cadherin during tumor progression was shown.( 24 , 25 ) Also in melanoma, the switch to a type II cadherin like cadherin‐7, which forms weaker intercellular adhesions, may participate in increased migration of tumor cells. This switch may lead to weaker intercellular adhesion between the cells promoting the migration of melanoma cells.
Our studies revealed an impact of cadherin‐7 on the migration of melanoma cells. Reduction of cadherin‐7 level led to a decreased migration in cells derived from a primary tumor usually showing high cadherin‐7 expression. In contrast to the finding in cells derived from primary tumors, cells from metastasis showed only weak expression of cadherin‐7. Here, reduction of cadherin‐7 expression seems to correlate with tumor progression. In agreement, overexpression of cadherin‐7 decreased the migration of melanoma cells.
We speculated that the interaction of MIA and cadherin‐7 could have an impact on the observed effects. MIA is necessary for melanoma cell metastasis as MIA binds to fibronectin and integrins, and inhibits the cell‐matrix contacts leading to detachment of cells and cell migration.( 4 ) We could show that overexpression of cadherin‐7 in metastatic cell lines results in abrogation of MIA functional activity and in reduced migratory potential of the cells. Overexpression of cadherin‐7 compensated the migratory effects of MIA which were observed in control cells using Boyden chamber assay. This could be due to interaction between cadherin‐7 and MIA as cadherin‐7 captures MIA resulting in reduced MIA levels and thereby inhibiting the migratory effects of MIA.
In addition to modulating the amount of MIA by protein–protein interaction, we could show that cadherin‐7 regulates MIA mRNA expression. It was demonstrated in several studies that cadherins participate in the regulation of gene expression. The loss of E‐cadherin leads to upregulation of nuclear factor (NF)κB activity in malignant melanoma.( 26 ) MIA expression was previously shown to be regulated via NFκB.( 27 ) The impact of cadherin‐7 on signaling pathways has not been analyzed in detail until today. We can therefore only speculate that cadherin‐7 regulates the gene expression of MIA via modulating defined signaling pathways which are necessary for strong expression of MIA in melanoma cells from metastases.
In summary, we revealed a biphasic expression of cadherin‐7 in malignant melanoma with strong expression in primary tumors and reduced expression in metastases. This hints to the fact that cadherin‐7 promotes early tumor development potentially by supporting the delamination to surrounding cell types. Later in progression, cadherin‐7 expression seems to be dispensable or, even more, negatively influencing migration. This negative influence can be in part explained by a negative regulation of MIA expression and inhibition of MIA activity preventing the detachment and migration of cells.
Acknowledgments
We thank Sibylla Lodermeyer for excellent technical assistance; Dr Johnson (University of Munich, Germany) for the melanoma cell lines; and Professor Takeichi (Kyoto University, Japan) for providing the CCD7 antibody. This work was supported by grants from the Deutsche Krebshilfe and the Deutsche Forschungsgemeinschaft (DFC) to A. K. B. and from the Department of Defense (DOD) to V. C. S. and Teresa Johnson‐Pais.
References
- 1. Blesch A, Bosserhoff AK, Apfel R et al . Cloning of a novel malignant melanoma‐derived growth‐regulatory protein, MIA. Cancer Res 1994; 54: 5695–701. [PubMed] [Google Scholar]
- 2. Bosserhoff AK, Echtenacher B, Hein R, Buettner R. Functional role of melanoma inhibitory activity in regulating invasion and metastasis of malignant melanoma cells in vivo . Melanoma Res 2001; 11: 417–21. [DOI] [PubMed] [Google Scholar]
- 3. Bosserhoff AK, Kaufmann M, Kaluza B et al . Melanoma‐inhibiting activity, a novel serum marker for progression of malignant melanoma. Cancer Res 1997; 57: 3149–53. [PubMed] [Google Scholar]
- 4. Bosserhoff AK. Melanoma inhibitory activity (MIA): an important molecule in melanoma development and progression. Pigment Cell Res 2005; 18: 411–6. [DOI] [PubMed] [Google Scholar]
- 5. Bauer R, Humphries M, Fassler R, Winklmeier A, Craig SE, Bosserhoff AK. Regulation of integrin activity by MIA. J Biol Chem 2006; 281: 11669–77. [DOI] [PubMed] [Google Scholar]
- 6. Hsu MY, Wheelock MJ, Johnson KR, Herlyn M. Shifts in cadherin profiles between human normal melanocytes and melanomas. J Invest Dermatol Symp Proc 1996; 1: 188–94. [PubMed] [Google Scholar]
- 7. Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science 1991; 251: 1451–5. [DOI] [PubMed] [Google Scholar]
- 8. Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 1996; 84: 345–57. [DOI] [PubMed] [Google Scholar]
- 9. Chu YS, Eder O, Thomas WA et al . Prototypical type I E‐cadherin and type II cadherin‐7 mediate very distinct adhesiveness through their extracellular domains. J Biol Chem 2006; 281: 2901–10. [DOI] [PubMed] [Google Scholar]
- 10. Becker T, Redies C. Internal structure of the nucleus rotundus revealed by mapping cadherin expression in the embryonic chicken visual system. J Comp Neurol 2003; 467: 536–48. [DOI] [PubMed] [Google Scholar]
- 11. Luo J, Treubert‐Zimmermann U, Redies C. Cadherins guide migrating Purkinje cells to specific parasagittal domains during cerebellar development. Mol Cell Neurosci 2004; 25: 138–52. [DOI] [PubMed] [Google Scholar]
- 12. Kools P, Van Imschoot G, Van Roy F. Characterization of three novel human cadherin genes (CDH7, CDH19, and CDH20) clustered on chromosome 18q22‐q23 and with high homology to chicken cadherin‐7. Genomics 2000; 68: 283–95. [DOI] [PubMed] [Google Scholar]
- 13. Moore R, Champeval D, Denat L et al . Involvement of cadherins 7 and 20 in mouse embryogenesis and melanocyte transformation. Oncogene 2004; 23: 6726–35. [DOI] [PubMed] [Google Scholar]
- 14. Nakagawa S, Takeichi M. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development 1998; 125: 2963–71. [DOI] [PubMed] [Google Scholar]
- 15. Rothhammer T, Poser I, Soncin F, Bataille F, Moser M, Bosserhoff AK. Bone morphogenic proteins are overexpressed in malignant melanoma and promote cell invasion and migration. Cancer Res 2005; 65: 448–56. [PubMed] [Google Scholar]
- 16. Arndt S, Bosserhoff AK. TANGO is a tumor suppressor of malignant melanoma. Int J Cancer 2006; 119: 2812–20. [DOI] [PubMed] [Google Scholar]
- 17. Lehmann R, Melle C, Escher N, Von Eggeling F. Detection and identification of protein interactions of S100 proteins by ProteinChip technology. J Proteome Res 2005; 4: 1717–21. [DOI] [PubMed] [Google Scholar]
- 18. Melle C, Osterloh D, Ernst G, Schimmel B, Bleul A, Von Eggeling F. Identification of proteins from colorectal cancer tissue by two‐dimensional gel electrophoresis and SELDI mass spectrometry. Int J Mol Med 2005; 16: 11–7. [PubMed] [Google Scholar]
- 19. Nakagawa S, Takeichi M. Neural crest cell–cell adhesion controlled by sequential and subpopulation‐specific expression of novel cadherins. Development 1995; 121: 1321–32. [DOI] [PubMed] [Google Scholar]
- 20. Bosserhoff AK, Moser M, Buettner R. Characterization and expression pattern of the novel MIA homolog TANGO. Gene Expr Patterns 2004; 4: 473–9. [DOI] [PubMed] [Google Scholar]
- 21. Takeichi M. Morphogenetic roles of classic cadherins. Curr Opin Cell Biol 1995; 7: 619–27. [DOI] [PubMed] [Google Scholar]
- 22. Poser I, Dominguez D, De Herreros AG, Varnai A, Buettner R, Bosserhoff AK. Loss of E‐cadherin expression in melanoma cells involves up‐regulation of the transcriptional repressor Snail. J Biol Chem 2001; 276: 24661–6. [DOI] [PubMed] [Google Scholar]
- 23. Hajra KM, Fearon ER. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer 2002; 34: 255–68. [DOI] [PubMed] [Google Scholar]
- 24. Feltes CM, Kudo A, Blaschuk O, Byers SW. An alternatively spliced cadherin‐11 enhances human breast cancer cell invasion. Cancer Res 2002; 62: 6688–97. [PubMed] [Google Scholar]
- 25. Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N‐cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol 2000; 148: 779–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuphal S, Poser I, Jobin C, Hellerbrand C, Bosserhoff AK. Loss of E‐cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene 2004; 23: 8509–19. [DOI] [PubMed] [Google Scholar]
- 27. Poser I, Golob M, Buettner R, Bosserhoff AK. Upregulation of HMG1 leads to melanoma inhibitory activity expression in malignant melanoma cells and contributes to their malignancy phenotype. Mol Cell Biol 2003; 23: 2991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]