

Abstract
Estrogen receptors play a key role in breast cancer development. One of the current therapeutic strategies for the treatment of estrogen receptor (ER)‐α‐positive breast cancers relies on the blockade of ERα transcriptional activity. In the present study, we characterized Hakai, originally characterized as an E‐cadherin binding protein, as a strong blockade of ERα in breast cancer cells. We showed that Hakai inhibited the transcriptional activity of ERα by binding directly to ERα. The DNA‐binding domain of ERα was found to be responsible for its interaction with Hakai. Hakai competed with ERα coactivators, such as steroid receptor coactivator‐1 (SRC‐1) and glucocoriticord receptor interacting protein‐1 (GRIP‐1), for the modulation of ERα transactivation, while its ubiquitin‐ligase activity was not required. Further, overexpression of Hakai inhibited the proliferation and migration of breast cancer cells. Taken together, these results suggest that Hakai is a novel corepressor of ERα and may play a negative role in the development and progression of breast cancers. (Cancer Sci 2010)
Estrogen receptors (ERs) ERα and ERβ belong to the nuclear hormone receptor superfamily and mediate estrogen actions in regulating cell growth and differentiation in mammary glands. In mice with a homozygous disruption of the ERα gene, the mammary glands remain undeveloped, indicating an indispensable role of ERα in the growth of mammary glands.( 1 ) In addition, estrogen receptors are overexpressed in approximately 70% of breast cancer cells, which are referred to as estrogen receptor‐positive cells. Estrogen‐responded ERα influences cell division, cell proliferation, and DNA replication, and thus stimulates the rapid growth of cancers.( 2 ) Estrogen administration in breast cancer cells is also associated with the membrane translocation of ERα and with the rapid formation of specialized cell membrane structures, such as ruffles and pseudopodia. Loss of stress fibers in these membrane structures is associated with cancer cell transformation, metastasis, and cytoskeletal rearrangement.( 3 )
Estrogen receptor (ER)‐α consists of three functional domains: an N‐terminal region, containing a constitutive activation function (AF‐1); a central, highly conserved DNA‐binding domain (DBD); and a C‐terminal ligand‐binding domain (LBD), containing a dimerization and a ligand‐dependent activation function (AF‐2). Regulation of gene expression by ERα requires the coordinated activity of ligand binding, phosphorylation, and cofactor interactions in particular combinations, which probably results in tissue‐specific responses elicited by the receptor.( 4 ) Cofactors interacting with ERα include coactivators and corepressors. Most of the cofactors regulate ERα transcriptional activity in a ligand‐dependent manner. Ligand‐dependent coactivators include the SRC‐1 family,( 5 ) CREB binding protein (CBP) p300,( 6 ) p68,( 7 ) and andorgen receptor‐associated coregulator 70 (ARA70).( 8 ) On the other hand, coactivators, including cyclin D1( 9 ) and x‐box binding protein‐1 (XBP‐1),( 10 ) enhance ERα transcriptional activity in a ligand‐independent manner. Ligand‐dependent corepressors include nuclear receptor coreprescor (NcoR),( 11 ) silencing mediator of retinoic acid and thyroid hormone receptor (SMRT),( 12 ) short heterodimer partner (SHP),( 13 ) forkhead homolog 1 (FKHR),( 14 ) thyroid hormone receptor (TR2),( 15 ) and breast cancer 1 (BRCA1).( 16 ) BRCA1 regulates both the ligand‐dependent and ligand‐independent activity of ERα.( 17 )
Hakai, which was first characterized as a novel E‐cadherin binding protein, contains the phosphotyrosine‐binding, RING finger, and proline‐rich domains. Although it structurally and functionally related to c‐Cbl, the linear order of these domains is different in these two proteins, and sequence similarities outside of the domains are not detectable. Hakai interacts with E‐cadherin in a tyrosine phosphorylation‐dependent manner, and acts as an E3 ubiquitin‐ligase. Upon its overexpression in epithelial cells, Hakai functions in the generation of cell motility by perturbing cell–cell adhesions and enhancing hematocyte growth factor (HGF)‐induced cell scattering and endocytosis of E‐cadherin.( 18 ) Recently, Hakai has shown to affect not only cell–cell contacts, but also proliferation in both epithelial and fibroblast cells.( 19 )
In this study, we characterized Hakai as a transcriptional coregulator in breast cancer cells. Transiently transfected Hakai repressed the transactivation of ERα through direct binding to ERα and interference with the recruitment of coactivators. Finally, Hakai inhibited the proliferation and migration of ERα‐dependent breast cancer cells. Thus, we propose that Hakai is a strong corepressor of ERα, and may play an important role in the development and progression of breast cancers.
Materials and Methods
Plasmids. Plasmids for mammalian expression and in vitro translation of ERβ, androgen‐receptor (AR), retinoic acid receptor (RAR)α, and retinoid x receptor (RXR)γ were generated by cloning into pCDNA3 (Invitrogen, San Diego, CA, USA). Hakai was cloned into pCDNA3, pCDNA3‐Flag, GST4T‐1, and B42 vectors digested with EcoRI and XhoI, and into GAL4N and GFP vectors digested with BamHI. ΔN‐term Hakai (a.a.1‐148 deleted) and ΔPEST Hakai (a.a.195‐220 deleted) were constructed from pCDNA3‐Hakai by digesting with PstI/NdeI and NdeI, respectively. The shRNA sequences specifically targeting Hakai (sense: 5′‐GATCCCCACAAAGCGAAACCTGCACCTTCAAGAGAGGTGCAGGTTTGGCTTTGTTTTTTGGAAA‐3′ and antisense: 5′‐AGCTTTTCCAAAAAACAAAGCGAAACCTGCACCTCTCTTGAAGGTGCAGGTTTCGCTTTGTGGG‐3′) were custom‐designed. Dimerlized shRNA oligomers were cloned into pSUPER‐retro vector (OligoEngine, Seattle, WA, USA). pUHDrtTA2S‐M2 (TRE) was kindly provided by Dr H. Bujard (ZMBH, Heidelberg, Germany). pCDNA3‐ERα, pCDNA3‐GR, MMTV‐luc, and ERE‐luc plasmids have been previously described.( 20 ) pCDNA3HA‐ERα deletion mutants were generated by cloning the regions spanning AF1 + DBDh + LBD, AF1 + DBDh, DBDh + LBD + AF‐2, AF‐1, and LBD of ERα into the pCDNA3 vector digested with diverse restriction enzymes. pCDNA3‐SRC‐1, ‐GRIP‐1 (SRC‐2), and ‐p300 have been previously described.( 21 )
Cell culture and transient transfection. Cells were maintained in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% FBS, and transfected as previously described.( 22 ) Cells were plated in DMEM containing 5% charcoal‐stripped FBS, and transfected with the indicated amount of expression plasmids, the reporter ERE‐Luc, and the control β‐gal expression plasmid pRSV using Superfect reagent (Qiagen, Hilden, Germany) or Lipofectamine (Invitrogen Life Technologies, San Diego, CA, USA). Cells were treated with either 100 nM 17β‐estradiol or vehicle for 24 h following 24 h transfection. The levels of luciferase activity were normalized to β‐gal expression. Transfections were performed in duplicate for each construct, and all values represent the mean ± SD of at least three independent experiments.
Yeast two‐hybrid assay. Plasmid encoding LexA fusion and B42 fusions were cotransformed into S. cerevisiae EGY48 containing the lacZ reporter plasmid. Transformants were grown in the inducing medium and processed for liquid β‐galactosidase assays, as previously described.( 23 )
Glutamine‐S‐transferase (GST) pull‐down assay. Glutamine‐S‐transferase (GST) pull‐down assay was conducted as previously described.( 24 ) Immobilized GST fusion protein, GST‐Hakai, was then incubated with [35S] methionine‐labled ERα produced by in vitro translation using the TNT‐coupled transcription‐translation system (Promega, Madison, WI, USA). After eletrophoresis by SDS‐PAGE, radiolabeled proteins were visualized by autoradiography.
Coimmunoprecipitation and western blot analysis. In vivo coimmunoprecipitation assays were performed using Cos‐7 cells transfected with pCDNA‐HA‐ERα and GFP‐Hakai plasmids as previously described.( 25 ) Whole‐cell lysate was incubated with 2 μg of anti‐ERα antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C, and further incubated for another 2 h after adding 20 μL of protein A‐agarose bead slurry (Invitrogen). Bound proteins were separated by SDS‐PAGE, transferred to nitrocellulose transfer membrane (Schleicher and Schuell Bioscience, Keene, NH, USA), and subjected to western blot analysis with anti‐GFP, anti‐ERα (Santa Cruz Biotechnology), and anti‐Hakai antibodies (Zymed, San Francisco, CA, USA).
Establishment of stable cell lines. We used the Tet‐On‐inducible system to establish stable cell lines. The cloned Hakai gene and enhanced green fluorescent protein (EGFP) are induced by TRE activation through the activation of transactivator (rtTA) in the presence of doxycycline.( 26 ) Mcf‐7 cells were cotransfected with pUHDrtTA2S‐M2 (TRE) and pBI‐EGFP‐Hakai, or pBI‐EGFP as a control (Clontech, Palo Alto, CA, USA) using the Superfect reagent (Qiagen). On the next day after transfection, cells were subcultured and selected in culture media containing 800 μg/mL G418 (Geneticin; Invitrogen) for 2 weeks. Each selected clone was analyzed by fluorescence microscopy and western blot analysis. Mcf‐7/Hakai cells were treated with 300 ng/mL doxycycline (Sigma, St. Louis, MO, USA) to induce expression of HA‐tagged Hakai and EGFP.
Thymidine incorporation assay. Hakai‐stable cells and MOCK cells were cultured in 96‐well plates at a density of 4 × 103 per well in phenol red‐free DMEM containing 5% charcoal‐stripped FBS, and treated with 300 ng/mL doxycycline in the presence or absence of 10 nM 17β‐estradiol after 24 h. Cells were then pulse‐labeled with [ 3 H]thymidine (10 μCi/mL, specific activity 80 Ci/mmol; Perkin‐Elmer Life Sciences, Waltham, MA, USA) for 4 h. Cells were harvested onto a glass microfiber filter (Whatman, Sanford, ME, USA) and washed intensively with distilled water. Filters were counted with a scintillation counter for the measurement of thymidine incorporation into DNA.( 21 )
Wound‐healing assay. Hakai‐stable cells and MOCK cells were plated in 12‐well plates at a confluence of 50–60%. After incubating the plates at 37°C until cells reached 100% confluence, forming a monolayer, a scratch of the cell monolayer was created using a p20 pipet tip. Plates were washed once and replaced with the medium with or without E2 and doxycycline. Cells were observed under a microscope to ensure migration.
Results
Hakai represses the transactivation of ERα. We have independently cloned Hakai as a highly expressed protein in male germ cells, in which Hakai is localized in the nucleus (data not shown). In addition, functional domain analysis of the Hakai protein revealed the presence of a nuclear localization signal, and the expression of Hakai in mammalian cells showed its localization in the nucleus (data not shown). Thus, we investigated whether Hakai is capable of functioning as a transcriptional regulator by transiently expressing GAL4N‐fused Hakai with the reporter Gal4‐tk‐luc. Hakai showed strong repressor activity (Fig. 1a). However, the transcriptional repression by Hakai was not affected by treatment with trichostantin A (TSA), an inhibitor of histone deacetylases (HDAC), suggesting no recruitment of HDAC activity in its action.
Figure 1.
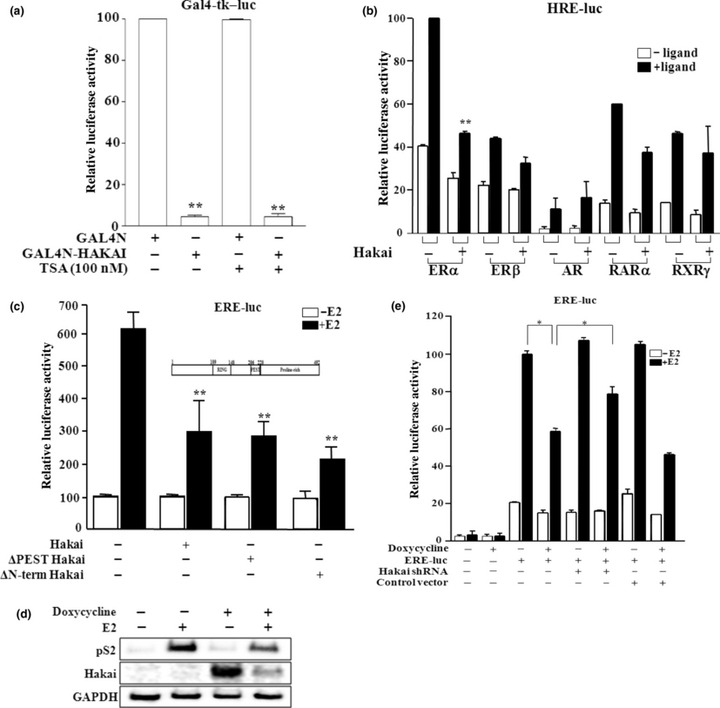
Hakai functions as a corepressor of estrogen receptor (ER)‐α. (a) GAL4N‐Hakai fusion protein functions as a transcriptional repressor, which is independent from the recruitment of histone deacetylase (HDAC) activity. Cos‐7 cells were cotransfected with GAL4N or GAL4N‐fused Hakai with the reporter Gal4‐tk‐luc. Cells were treated with or without 100 nM trichostatin A (TSA), an inhibitor of HDAC. (b) Hakai significantly represses the transactivation of ERα in Cos‐7 cells transiently transfected with the expression construct of nuclear receptors together with a corresponding reporter. (c) E3 ubiquitin‐ligase activity of Hakai is not required for the inhibition of ERα transactivation in transiently transfeced Mcf‐7 cells. Schematic diagram of Hakai with domains is shown. (d) Hakai inhibits the expression of an ERα target gene, pS2, in Mcf‐7/Hakai stable cell line, in which Hakai expression is induced by the treatment of doxycycline (300 ng/mL). (e) The expression of Hakai shRNA relieved the Hakai‐mediated inhibition of ERα transactivation in Mcf‐7/Hakai cells. Filled boxes indicate the presence of 100 nM estradiol, testosterone, or 9‐cis retinoic acid. *P < 0.05 and **P < 0.005, significant differences from the control.
Considering Hakai as an indirect transcriptional regulator, we then explored the function of Hakai as a coregulator of nuclear receptors whose action is regulated by diverse mechanisms. The results showed that transcriptional activity of ERα was significantly repressed by Hakai, while the activity of ERβ, RARα, RXRγ, or AR was not (Fig. 1b). Since Hakai is an E3 ubiquitin‐ligase, we investigated whether the ubiquitin‐ligase activity of Hakai is necessary for the Hakai‐mediated repression of ERα transactivation. We used deletion mutants of the RING finger domain and PEST sequence motif, which are important for the ubiquitin‐ligase activity and degradation of target proteins, respectively.( 18 ) As shown in Figure 1(c), both domain mutants of Hakai inhibited ERα transactivation as much as the full‐length Hakai, suggesting no involvement of Hakai ubiquitin‐ligase activity and target protein degradation.
The results let us examine the effect of Hakai on the expression of ERα target genes. We first established Mcf‐7/Hakai stable cell lines using the Tet‐On‐inducible system,( 26 ) in which Hakai expression is induced by the treatment of doxycycline. Hakai expression upon doxycycline treatment in Mcf‐7/Hakai cells caused significant down‐regulation of the expression of pS2, an endogenous ERα target gene in breast cancer cells (Fig. 1d). Interestingly, Hakai expression was markedly decreased in the presence of estrogen. We also examined the effect of Hakai shRNA on the function to ERα transcriptional activity in Mcf‐7/Hakai cells. The cells were transfected with the Hakai shRNA expression vector or empty control vector. The E2‐dependent transactivation of ERα was repressed by Hakai expression with the treatment of doxycycline, and the repressed ERα activity was significantly recovered by the expression of Hakai shRNA (Fig. 1e). Together, these results suggest that Hakai acts as a corepressor of ERα, regulating the expression of ERα target genes.
Hakai physically interacts with ERαin vitro and in vivo. Since Hakai repressed the transactivation of ERα, the physical interaction of Hakai with ERα was investigated. In a yeast‐two hybrid system, the interaction of Hakai with ERα was estrogen‐dependent (Fig. 2a). ARA70 was included as a positive control for ERα binding protein.( 8 ) To examine the in vivo interaction between Hakai and ERα, co‐immunoprecipitation assays were performed using anti‐ERα antibody in Cos‐7 cells transfected with GFP‐Hakai together with ERα expression plasmid (Fig. 2b, upper panel). The data revealed efficient co‐immunoprecipitation of ERα with Hakai. Such an in vivo interaction was further confirmed in Mcf‐7/Hakai cells by co‐immunoprecipitation of endogenous ERα with stably overexpressed Hakai (Fig. 2b, lower panel). Direct physical interaction of Hakai with ERα and the regions of each protein responsible for their interaction were assessed by GST pull‐down assays. In vitro translated deletion mutants and full‐length ERα were incubated with GST‐fused Hakai (Fig. 2c). Estrogen receptor (ER)‐α full‐length, ERαΔAF2, ERαΔLBD, and ERαΔAF‐1, which all contain the DBD‐hinge domain, showed efficient binding with GST‐Hakai. However, ERαAF‐1 and ERαLBD, ERα mutants without the DBD‐hinge domain, failed to bind with Hakai. Together, these results suggest that Hakai efficiently interacts with ERα, and that their interaction is occurring through the DBD‐hinge region of ERα.
Figure 2.
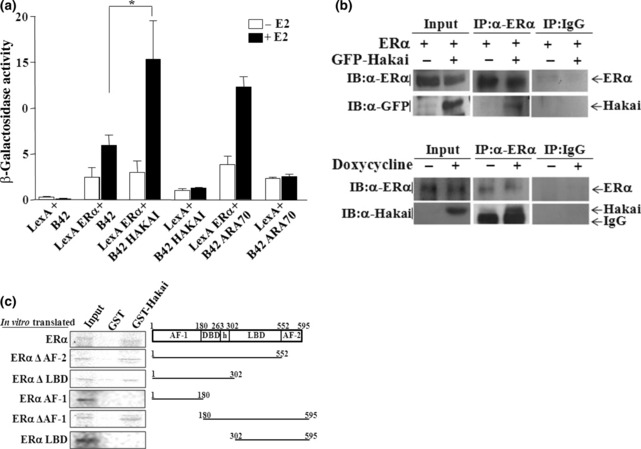
Hakai physically interacts with estrogen receptor (ER)‐α. (a) Interaction of LexA‐fused ERα with B42‐fused Hakai in yeast. All values represent the mean ± SE of at least three independent colonies. Filled boxes indicate the presence of 100 nM estradiol. *P < 0.05, significant differences from the control. (b) In vivo interaction of Hakai with ERα. Coimmunoprecipitations were carried out with ERα antibody in Cos‐7 cells transfected with GFP‐Hakai or pEGFP‐C1 (GFP only) together with ERα expression plasmid (upper panel) or in Mcf‐7/Hakai stable cells (lower panel). Inputs are shown for the expression level of each protein. (c) Direct interaction of Hakai with ERα, and definition of the ERα domain necessary for their interaction by GST full‐down assays. Schematic representation of ERα and a series of deletion mutants used in GST‐pull down assay are shown. Approximately 10% of the labeled proteins used in the binding reaction were loaded as input. Reactions were carried out with the equivalent amount of each protein as determined by Coomassie blue staining (data not shown).
The inhibitory mechanism of ERα by Hakai is through its competitive binding with ERα coactivators. To examine how Hakai represses the transactivation of ERα, we first addressed the effect of Hakai on the DNA binding affinity of ERα by electrophoretic mobility shift assays. The results showed that Hakai did not interfere with the formation of the ERα–ERE complex (data not shown). As an alternative, we then investigated whether Hakai interferes with the recruitment of coactivators by ERα for the suppression of ERα transactivation. As shown in Figure 3(a), the enhancement of ERα transactivation by SRC‐1 and GRIP‐1 (SRC‐2), known ERα coactivators, was repressed by Hakai coexpression in a dose‐dependent manner. Furthermore, SRC‐1 and GRIP‐1 were able to release the Hakai‐mediated ERα repression in a dose‐dependent manner (Fig. 3b). These findings suggest that Hakai and ERα coactivators compete for the modulation of ERα transactivation. To verify that the functional competition between ERα coactivators and Hakai for the modulation of ERα transactivation is through their physical competition for ERα binding, we performed competitive binding assays using GST–ERα. With an increase of the amount of SRC‐1 protein, the binding of ERα with Hakai was decreased in a dose‐dependent manner (Fig. 3c). Glutamine‐S‐transferase (GST)‐only protein, a negative control, failed to form a complex with either SRC‐1 or Hakai. Together, these results suggest that Hakai inhibits ERα transactivation through competitive interference with the recruitment of ERα coactivators.
Figure 3.

Inhibitory mechanism of estrogen receptor (ER)‐α by Hakai. (a) In the presence of coactivators (steroid receptor coactivator‐1 (SRC‐1), glucocoriticord receptor interacting protein‐1 (GRIP‐1)), Hakai inhibits the transactivation of ERα in a dose‐dependent manner. (b) ERα coactivators relieve the Hakai‐mediated repression of ERα transactivation. Mcf‐7 cells, endogenously expressing ERα, were cotransfected with Hakai and coactivators. The expression level of the proteins was monitored by western blot analysis (data not shown). Filled boxes indicate the presence of 100 nM estradiol. (c) SRC‐1 and Hakai binds competitively to ERα. Increased SRC‐1 inhibits the binding of Hakai to ERα. *P < 0.05 and **P < 0.005, significant differences from the control.
Hakai overexpression inhibits the proliferation and migration of breast cancer cells. Estrogen and its receptor ERα have long been recognized to play critical roles during cell proliferation and migration of breast cancer cells.( 3 ) To understand the biological role of Hakai, which acted as a corepressor of ERα, we established Mcf‐7/Hakai cell lines, which express Hakai from a tetracycline‐inducible promoter. Using these stable cell lines, we examined the effect of overexpression of Hakai on cell proliferation, which was determined by [3H]thymidine incorporation assay. As expected, Hakai overexpression showed a negative effect on the proliferation of breast cancer cells (Fig. 4a).
Figure 4.
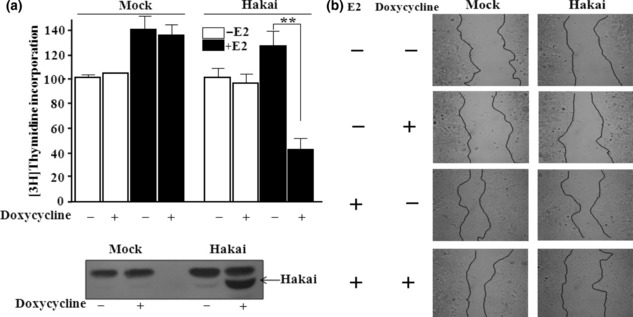
Effect of Hakai overexpression on the proliferation and migration of breast cancer cells. (a) Hakai overexpression inhibits the estrogen‐induced proliferation of Mcf‐7/Hakai stable cells. Cells were grown in the absence or presence of 300 ng/mL doxycycline and 100 nM estradiol. Cell proliferation was determined by the incorporation of [3H]thymidine during the last 4 h of culture. Data are representative of at least three independent experiments. Doxycycline‐dependent expression of Hakai in Mcf‐7/Hakai cells was examined by western blotting analysis. NS, non‐specific. **P < 0.005, significant differences from the control. (b) Hakai overexpression inhibits the estrogen‐induced migration of Mcf‐7/Hakai cells in wound‐healing assays. Uniform scratches created on the confluent cultures were sealed by culturing for 6 h in the presence or absence of 100 nM estradiol.
The wound‐healing assay is a simple method for the study of directional cell migration in vitro. We tracked the migration of Hakai‐overexpressed breast cancer cells at the leading edge of the wound. Wounded cells healed the cell damage upon estrogen addition, but Hakai overexpression delayed the healing (Fig. 4b). All together, these results support that Hakai acts as an impeder of ERα in breast cancer cells.
Discussion
In the present study, we demonstrate that Hakai acts as a corepressor of ERα and inhibits estrogen‐dependent cell proliferation and migration. Hakai modulates ERα transactivation through the competition with ERα coactivators for ERα binding. Hakai was previously characterized as an E3‐ubiqitin ligase and an E‐cadherin binding protein. However, there are evidences that suggest other functions for Hakai. First, Hakai is expressed in diverse tissues. Second, Hakai is mainly localized in the nucleus, while Hakai makes the E‐cadherin complex in the cytoplasm. Further, Hakai has strong transcriptional repressor activity when tested with GAL4N‐fused Hakai (Fig. 1a).
There are far fewer known ERα corepressors than coactivators. However, these molecules serve important roles in animal physiology by negatively regulating receptor‐dependent gene expression. sin3‐associated protein 30 (SAP30) binds to ERα in the presence of its respective antagonist, and then recruits NCoR and SMRT to certain ERα target gene promoters.( 27 ) On the other hand, SMRT/HDAC1 associated repressor protein (SHARP) binds to the steroid receptor RNA coactivator (SRA), and suppresses estrogen‐induced ERα transcriptional activity.( 28 ) SMRT/HDAC1 associated repressor protein (SHARP) also interacts directly with SMRT and HDACs to repress ERα transcriptional activity. Accordingly, SHARP has the capacity to modulate both liganded and nonliganded nuclear receptors. Corepressor activity of ERα has also been shown for nuclear receptor co‐repressor nrip1 (RIP140), repressor of ERD activity (REA), repressor of tamoxifen transcriptional activity (RTA), and DSS‐AHC critical regim on the X (DAX).( 29 ) These corepressors affect ERα activity through cooperative transcriptional regulation with other corepressors, recruitment of HDAC activity, antagonist binding, or competitive binding with coactivators. In the present study, we demonstrate that Hakai acts as a corepressor in estrogen‐activated ERα signaling through competitive binding with coactivators, such as SRC‐1 and GRIP‐1.
Like Hakai, some ubiquitin‐ligases are reported to function as transcriptional coregulators. For example, the E2 ubiquitin‐ligase BRCA1 has been shown to modulate ERα transactivation.( 16 ) BRCA1 acts as a potent inhibitor of ERα activity, in part, via its direct interaction with ERα. The N‐terminal region of BRCA1 interacts with ERα, and the C‐terminus of BRCA1 acts as a transcriptional inhibition domain. The inhibitory function for the C‐terminus of BRCA1 has been suggested by the findings that this region of BRCA1 is capable of binding to components of repression complexes, including the Rb protein, Rb‐associated proteins (RbAp46 and RbAp48), HDACs, and the carboxyl‐terminal interacting protein (CtIP).( 30 ) BRCA1‐mediated ERα repression has been also reported to occur, in part, through serine phosphorylation events in the AF‐1 domain of ERα, and also through the regulation of the relative degree of ERα acetylation versus mono‐ubiquitination.( 31 ) Another example is the ubiquitin‐ligase RING finger LIM domain‐interacting protein (RLIM), which enhances transcriptional activation mediated by ERα on target genes, while it inhibits transcriptional activity of LIM‐HDs.( 32 )
Hakai is a strong blockade of transcriptional activity (Fig. 1). The amino acid sequence shows a RING domain at its N‐terminus, which is present in many E3 ubiquitin‐ligases, and a PEST domain. In the C‐terminal sequence of Hakai, 35% of the amino acids are proline residues.( 18 ) Comparison of the repression activity of Hakai domain deletion mutants showed the activity similar to that of the full‐length (Fig. 1c). Thus, it is possible that the proline‐rich domain at the C‐terminus of Hakai is a key domain for the repression activity. The proline‐rich domain is commonly found in or near to repression domains, as evidenced in p53,( 33 ) Groucho,( 34 ) and HNF4.( 35 )
Hakai has previously been shown to regulate not only cell‐cell contacts but also cell proliferation.( 19 ) Overexpression of Hakai increased cell proliferation 2‐ to 3‐fold with MDCK stable cell lines, and transient expression of Hakai siRNA in Mcf‐7 and HEK293 cells significantly decreased cell proliferation based on the BrdU incorporation assay.( 19 ) These results contrast with our results that Hakai overexpression decreased the proliferation of Mcf‐7 breast cancer cells, although we investigated the estrogen‐dependent cell proliferation. In addition, the stable MDCK cell lines were established to constitutively overexpress Hakai, while we established stable Mcf‐7 cell lines which express Hakai in a tetracycline‐induced manner. Hakai may exert either positive or negative control of cell proliferation in different conditions. For example, Hakai may support estrogen‐independent growth of cells, while inhibiting estrogen‐dependent growth. Further studies are necessary to understand the differential functions of Hakai in estrogen‐dependent cell growth.
Acknowledgments
This study was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (A050287). We thank Dr. H. Bujard for pUHDrtTA2S‐M2.
References
- 1. Bocchinfuso WP, Hively WP, Couse JF, Varmus HE, Korach KS. A mouse mammary tumor virus‐wnt‐1 transgene induces mammary gland hyperplasia and tumorigenesis in mice lacking estrogen receptor‐α. Cancer Res 1999; 59: 1869–76. [PubMed] [Google Scholar]
- 2. Deroo BJ, Korach KS. Estrogen receptor and human disease. J Clin Invest 2006; 116: 561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giretti MS, Fu XD, De Rosa G et al. Extra‐nuclear signalling of estrogen receptor to breast cancer cytoskeletal remodeling, migration and invasion. PLoS ONE 2008; 3: e2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robyr D, Wolffe AP, Wahi W. Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol Endocrinol 2000; 14: 329–47. [DOI] [PubMed] [Google Scholar]
- 5. Yao TP, Ku G, Zhou N, Scully R, Livingston DM. The nuclear hormone receptor coactivator SRC‐1 is a specific target of p300. Proc Natl Acad Sci USA 1996; 93: 10626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chakravarti D, LaMorte VJ, Nelson MC et al. Role of CBP/p300 in nuclear receptor signaling. Nature 1996; 383: 99–103. [DOI] [PubMed] [Google Scholar]
- 7. Endoh H, Maruyama K, Masuhiro Y et al. Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor alpha. Mol Cell Biol 1999; 19: 5363–72. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Alen P, Claessens F, Schoenmakers E et al. Interaction of the putative androgen receptor‐specific coactivator ARA70/ELE1alpha with multiple steroid receptors and identification of an internally deleted ELE1beta isoform. Mol Endocrinol 1999; 13: 117–28. [DOI] [PubMed] [Google Scholar]
- 9. Neuman E, Ladha MH, Lin M et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol 1997; 17: 5338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ling L, Yan J, Zhu J et al. Ligand‐independent activation of estrogen receptor α by XBP‐1. Nucleic Acids Res 2003; 31: 5266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lavinsky RM, Jepsen K, Heinzel T et al. Diverse signaling pathways modulate nuclear receptor recruitment of N‐CoR and SMRT complexes. Proc Natl Acad Sci USA 1998; 95: 2920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4‐hydroxy‐tamoxifen. Mol Endocrinol 1997; 11: 657–66. [DOI] [PubMed] [Google Scholar]
- 13. Johansson L, Thomsen JS, Damdimopoulos AE, Spyrou G, Gustafsson JA, Treuter E. The orphan nuclear receptor SHP inhibits agonist‐dependent transcriptional activity of estrogen receptor alpha and beta. J Biol Chem 1999; 274: 345–53. [DOI] [PubMed] [Google Scholar]
- 14. Zhao HH, Herrera RE, Coronado‐Heinsohn E et al. Forkhead homologue in rhabdomyosarcoma functions as a bifunctional nuclear receptor‐interacting protein with both coactivator and corepressor functions. J Biol Chem 2001; 276: 27907–12. [DOI] [PubMed] [Google Scholar]
- 15. Hu YC, Shyr CR, Che W, Mu XM, Kim E, Chang C. Suppression of estrogen receptor‐mediated transcription and cell growth by interaction with TR2 orphan receptor. J Biol Chem 2002; 277: 33571–9. [DOI] [PubMed] [Google Scholar]
- 16. Fan S, Ma YX, Wang C et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001; 20: 77–87. [DOI] [PubMed] [Google Scholar]
- 17. Zheng L, Annab LA, Afshari CA, Lee WH, Boyer T. BRCA1 mediateds ligand‐independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci USA 2001; 98: 9587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujita Y, Krause G, Scheffner M et al. Hakai, a c‐Cbl‐like protein, ubiquitinates and induces endocytosis of the E‐cadherin complex. Nat Cell Biol 2002; 4: 222–31. [DOI] [PubMed] [Google Scholar]
- 19. Figueroa A, Kotani H, Toda Y et al. Novel roles of Hakai in cell proliferation and oncogenesis. Mol Biol Cell 2009; 20: 3533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hong CY, Suh JH, Kim K et al. Modulation of androgen receptor transactivation by the SWI3‐related gene product (SRG3) in multiple ways. Mol Cell Biol 2005; 25: 4841–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seo JH, Gong EY, Kim JB, Lee IK, Choi HS, Lee K. Sterol regulatory element‐binding protein‐1c represses the transactivation of androgen receptor and androgen‐dependent growth of prostatic cells. Mol Cancer Res 2008; 6: 314–24. [DOI] [PubMed] [Google Scholar]
- 22. Chattopadhyay S, Gong EY, Hwang M et al. The CCAAT enhancer‐binding protein‐a negatively regulates the transactivation of androgen receptor in prostate cancer cells. Mol Endocrinol 2006; 20: 984–95. [DOI] [PubMed] [Google Scholar]
- 23. Lee HJ, Chattopadhyay S, Gong EY, Ahn RS, Lee K. Antiandrogenic effects of bisphenol A and nonylphenol on the function of androgen receptor. Toxicol Sci 2003; 75: 40–6. [DOI] [PubMed] [Google Scholar]
- 24. Jeong BC, Hong CY, Chattopadhyay S et al. Androgen receptor corepressor‐19 kDa (ARR19), a leucine‐rich protein that represses the transcriptional activity of androgen receptor through recruitment of histone deacetylase. Mol Endocrinol 2004; 18: 13–25. [DOI] [PubMed] [Google Scholar]
- 25. Lee SY, Gong EY, Hong CY et al. ROS inhibit the expression of testicular steroidogenic enzyme genes via the suppression of Nur77 transactivation. Free Radic Biol Med 2009; 47: 1591–600. [DOI] [PubMed] [Google Scholar]
- 26. Solera J, Espinosa A, Geijo P et al. Treatment of human brucellosis with netilmicin and doxycycline. Clin Infect Dis 1996; 22: 441–5. [DOI] [PubMed] [Google Scholar]
- 27. Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science 2002; 295: 2465–8. [DOI] [PubMed] [Google Scholar]
- 28. Colley SM, Iyer KR, Leedman PJ. The RNA coregulator SRA, its binding proteins and nuclear receptor signaling activity. IUBMB Life 2008; 60: 159–64. [DOI] [PubMed] [Google Scholar]
- 29. Zhang H, Thomsen JS, Johansson L, Gustafsson JA, Treuter E. DAX‐1 functions as an LXXLL‐containing corepressor for activated estrogen receptors. J Biol Chem 2000; 275: 39855–9. [DOI] [PubMed] [Google Scholar]
- 30. Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C‐terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriotional repression. J Biol Chem 1998; 25: 25388–92. [DOI] [PubMed] [Google Scholar]
- 31. Xian Ma Y, Tomita Y, Fan S et al. Structural determinants of the BRCA1: estrogen receptor interaction. Oncogene 2005; 24: 1831–46. [DOI] [PubMed] [Google Scholar]
- 32. Johnsen SA, Gungor C, Prenzel T et al. Regulation of estrogen‐dependent transcription by LIM cofactors CLIM and RLIM in breast cancer. Cancer Res 2009; 69: 128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zilfou JT, Hoffman WH, Sank M, Geprge DL, Murphy M. The corepressor mSin3a interacts with the proline‐rich domain of p53 and protects p53 from proteasome‐mediated degradation. Mol Cell Biol 2001; 21: 3974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen G, Courey AJ. Groucho/TLE family proteins and transcriptional repression. Gene 2000; 249: 1–16. [DOI] [PubMed] [Google Scholar]
- 35. Ilyemere VP, Davies NH, Brownlee GG. The activation function 2 domain of hepatic nuclear factor 4 is regulated by a short C‐terminal proline‐rich repressor domain. Nucleic Acids Res 1998; 26: 2098–104. [DOI] [PMC free article] [PubMed] [Google Scholar]