

Abstract
Polycomb group protein EZH2, frequently overexpressed in malignant tumors, is the catalytic subunit of polycomb repressive complex 2 (PRC2). PRC2 interacts with HDACs in transcriptional silencing and relates to tumor suppressor loss. We examined the expression of HDAC isoforms (HDAC 1 and 2) and EZH2, and evaluated the possible use of HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) and EZH2 repressor for gallbladder carcinoma. We used 48 surgically resected gallbladders and cultures of human gallbladder epithelial cells (HGECs), gallbladder carcinoma (TGBC2TKB), and cholangiocarcinoma (HuCCT‐1 and TFK‐1) cell lines for examination. Immunohistochemically, EZH2 was overexpressed in gallbladder carcinoma, especially poorly differentiated carcinoma, but not in normal epithelium. In contrast, HDAC1/2 were expressed in both carcinoma and normal epithelium in vivo. This pattern was verified in cultured cells; EZH2 was highly expressed only in TGBC2TKB, whereas HDAC1/2 were expressed in HGECs and TGBC2TKB. Interestingly, SAHA treatment caused significant cell number decline in three carcinoma cells, and this effect was synergized with EZH2 siRNA treatment; however, HGECs were resistant to SAHA. In TGBC2TKB cells, the expression of EZH2 and HDAC1/2 were decreased by SAHA treatment, and p16 INK4a , E‐cadherin, and p21were simultaneously activated; however, no such findings were obtained in HGECs, suggesting that the effect of SAHA depends on the EZH2‐mediated tumor suppressor loss. In conclusion, this study suggests a possible mechanism by which carcinoma cells but not normal cells are sensitive to SAHA and indicates the efficacy of this new anticancer agent in combination with EZH2 repression in gallbladder carcinoma. (Cancer Sci 2009)
Gallbladder carcinoma is an aggressive and depressing disease, particularly affecting older people and women.( 1 , 2 , 3 ) It is usually difficult to detect gallbladder carcinoma in the early stage, because characteristic signs or symptoms are lacking and high risk factors have not been established in a majority of patients. Although complete surgical resection is the only curative therapy, invasion to surrounding organs or metastases is usually found in a majority of gallbladder carcinoma patients at the time of diagnosis. The current chemotherapy for patients with advanced gallbladder carcinoma is limited, and the development and exploration of new anticancer agents is necessary.( 4 , 5 )
Polycomb group proteins are epigenetic chromatin modifiers involved in cancer development.( 6 , 7 ) PcG proteins exist in at least two separate protein complexes, polycomb repressive complex 2 (PRC2) and PRC1. The PRC2 complex involves embryonic ectoderm development and EZH2.( 8 , 9 ) EZH2 is a transcriptional repressor involved in controlling cell growth and proliferation and an abnormal expression of EZH2 may be involved in the tumorigenesis process and provides proliferative advantages to eukaryotic cells through interaction with the pathways of key elements that control cell growth arrest and differentiation.( 10 , 11 ) In addition, various studies have identified EZH2 as a potential marker to distinguish aggressive from indolent or benign cancers.( 12 , 13 , 14 , 15 )
PRC2 is known to interact with HDAC proteins.( 8 , 9 ) Recent studies have disclosed that HDACs have many protein substrates involved in the regulation of gene expression, cell proliferation, and cell death. Acetylation and deacetylation of histones play an important role in the transcription regulation of cells,( 16 , 17 ) and the acetylation status of histones and non‐histone proteins is determined by HDACs.( 18 ) Inhibition of HDACs causes the accumulation of acetylated forms of these proteins with alterations of their function, and the CDK inhibitor p21 (WAF/CIP1) is one of the most common genes induced by HDAC inhibitor.( 4 ) In human cells, PRC2 interacts with class I HDAC, which includes HDAC1 and 2,( 8 , 9 , 18 ) and recent data suggest that transient interactions likely provide functional synergy between the silencing enzymes for tumor suppressor genes in vivo. The precise mechanisms of this synergy are not yet clear. Functional links between EZH2 and HDACs contribute to an emerging view that all of these types of epigenetic silencing machinery contribute to abnormal control of gene expression in malignant cells.
HDAC inhibitors are a group of recently discovered targeted anticancer agents. HDAC inhibitors induce different phenotypes in various transformed cells, including growth arrest, activation of extrinsic and/or intrinsic apoptotic pathways, autophagic cell death, mitotic cell death, and senescence.( 19 ) In comparison, normal cells are relatively more resistant to HDAC inhibitor‐induced cell death;( 20 , 21 ) however, the mechanism underlying this difference is not well understood.
Among HDAC inhibitors, SAHA is one of the most advanced in clinical fields as an anticancer agent, which interacts directly with the catalytic site of HDAC‐like protein and inhibits its enzymatic activity.( 18 ) SAHA inhibits all 11 members of the class I and II HDAC family, and causes specific modifications in the pattern of acetylation and methylation of lysines in histones H3 and H4 associated with the proximal promoter of the p21 gene.( 18 ) Although considerable progress has been made in elucidating the role of HDACs and the effects of HDAC inhibitors, these areas are still in the early stages of discovery.
Several previous studies reported abnormal promoter methylation of important genes including p16 INK4a and E‐cadherin in gallbladder carcinoma,( 22 , 23 ) however, there have been few studies addressing alteration of epigenetic chromatin modification by PcG and HDACs in gallbladder carcinoma.( 24 , 25 ) In this study using human gallbladders and cultures of gallbladder epithelial cells and the gallbladder carcinoma cell line, we assessed the expression pattern of EZH2 and class I HDACs (HDAC 1 and 2). We considered the possible mechanism by which carcinoma cells are sensitive to SAHA and the possible efficacy of this new anticancer agent in therapeutic treatment in gallbladder carcinoma.
Materials and Methods
Human tissue studies.
Preparation of tissue specimens. Forty‐eight surgically resected gallbladders were used, 10 with cholecystolithiasis and chronic cholecystitis, which were used as a control, and 38 with gallbladder carcinoma (Table 1). Gallbladder carcinoma consisted of 9 well differentiated, 12 moderately differentiated, and 10 poorly differentiated adenocarcinomas, and 7 papillary adenocarcinomas. All surgically resected gallbladders were fixed in 10% formalin. After embedding in paraffin, approximately 20 serial thin sections were cut from each block, and used for histological and immunohistochemical analyses. This study was approved by the Ethics Committee of Kanazawa University (Kanazawa, Japan).
Table 1.
Main clinicopathological features of cases examined in this study
| Gallbladder | Cases (n) | Age (years) | Gender |
|---|---|---|---|
| Mean ± SD (range) | Male:female | ||
| Cholecystolithiasis without BilIN‐1/2 | 10 | 61.0 ± 14.6 (38–81) | 7:3 |
| Gallbladder cancer | 38 | 77.1 ± 8.3 (57–90) | 16:22 |
| Papillary adenocarcinoma | 7 | 77.4 ± 3.4 (60–90) | 3:4 |
| Well differentiated adenocarcinoma | 9 | 73.2 ± 3.2 (57–88) | 5:4 |
| Moderately differentiated adenocarcinoma | 12 | 79.3 ± 1.5 (72–85) | 5:7 |
| Poorly differentiated adenocarcinoma | 10 | 78.9 ± 3.6 (65–90) | 3:7 |
BilIN‐1/2, biliary intraepithelial neoplasm‐1/2.
Immunohistochemistry. The expression of EZH2, HDAC1, and HDAC2 were assessed immunohistochemically using monoclonal antibodies: anti‐EZH2 (clone 11, 1:100; BD Transduction Laboratories, San Jose, CA, USA); anti‐HDAC1 (clone 3E1, 1:250; Sigma, St Louis, MO, USA); and anti‐HDAC2 (ab12169, 1:500; Abcam, Cambridge, UK), as described previously.( 6 , 24 ) As a negative control, similar dilution of the control mouse IgG (Dako, Glostrup, Denmark) was applied.
The expressions of EZH2, HDAC1, and HDAC2 were evaluated semiquantitatively according to the percentage of positive cells: 1 (negative, 0–10% epithelial cells in the lesion were positive); 2 (focal, 10–50% positive); and 3 (extensive, 50–100% positive). In addition, the percentage of cells positive for EZH2 was semiquantitatively calculated in high‐power fields of two or three foci containing approximately 100 contiguous epithelial cells, and the mean was regarded as the labeling index in each case.
Cell culture study.
Reagents. SAHA was purchased from Cayman Chemical (Ann Arbor, MI, USA). Media, FBS, and antibiotic–antimycotic were purchased from Gibco (Rockville, MD, USA).
Primary culture of HGECs. Human gallbladder epithelial cells were established from a surgically resected gallbladder with cholecystolithiasis as described,( 24 , 26 ) and grown as monolayers. Epithelial nature of these cells was confirmed by the expression of CK7. HGECs were incubated in culture medium as described, and HGECs between passages four and eight were used.
Culture of carcinoma cell line. The human gallbladder carcinoma cell line TGBC2TKB (RCB1130) was provided by the Riken Cell Bank (Tsukuba, Japan). Two human cholangiocarcinoma cell lines, HuCCT‐1 and TFK‐1, were provided by Cell Resource Center for Biochemical Research, Tohoku University, Sendai, Japan. TGBC2TKB cells were maintained in DMEM containing 5% FBS, and HuCCT‐1 and TFK‐1 in RPMI‐1640 with 10% FBS in a standard condition.
Assay for relative cell number. Carcinoma cells (TGBC2TKB, HuCCT‐1, and TFK‐1) and HGECs were seeded in a 96‐well plate (3–5 × 103 cells per well). After several treatments, these cells were cultured for 48 h and the relative cell number was assessed using Cell Proliferation Reagent WST‐1 according to the manufacturer’s protocol (Roche, Mannheim, Germany).
Assay for apoptosis. Apoptosis was detected using the ssDNA apoptosis ELISA kit (Millipore, Billerica, MA, USA) according to the manufacturer’s protocol.
Western blot analysis. Western blotting was carried out using a standard method.( 6 , 24 ) The membrane was incubated with primary antibodies against EZH2 (1:500), HDAC1 (1:500), HDAC2 (1:500), p21 (Cip1/WAF1, clone 70, dilution 1:100; BD Transduction Laboratories), p16INK4a (clone JC8, dilution 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), E‐cadherin (clone 36/E‐cadherin, dilution 1:100; BD Transduction Laboratories), and β‐actin (1:5000; Abcam).
RT‐PCR. RT‐PCR was carried out using a standard method.( 6 , 24 ) The annealing temperature was 54°C for EZH2, HDAC1, and HDAC2, and 56°C for G3PDH. The number of PCR cycles was 27 for EZH2, and 35 for HDAC1 and HDAC2. Primer sequences were as follows: EZH2, 5′‐gtggagagattatttctcaagatg‐3′ and 5′‐ccgacatacttcagggcatcagcc‐3′; HDAC1, 5′‐agccaagagagtcaaaacaga‐3′ and 5′‐ggtccattcaggccaact‐3′; HDAC2, 5′‐gagcaatgcggagaaatgtt‐3′ and 5′‐tgctctgcttgttacatggc‐3′; and G3PDH, 5′‐gaacgggaagctcactggcatggc‐3′ and 5′‐tgaggtccaccaccctgttgctg‐3′.
Quantitative real‐time PCR. Quantitative real‐time PCR was carried out to measure HDAC1, HDAC2, and G3PDH mRNA according to a standard protocol using the SYBR Green PCR Master Mix (Toyobo, Tokyo, Japan) and an ABI Prism 7700 sequence detection system (Applied Biosystems, Tokyo, Japan). Fold difference in comparison with the control was calculated. Each experiment was carried out in triplicate, and the mean was adopted in each experiment.
Knockdown of EZH2 by siRNA. Validated siRNA for EZH2 and negative control siRNA were purchased from Santa Cruz Biotechnology and Qiagen (Hilden, Germany), respectively. One day before transfection, cells were plated in 12‐well plates (3–5 × 104 cells), then transiently transfected with either EZH2 or control siRNA (100 nM), using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol.
ChIP assay. We examined the effect of EZH2 knockdown and SAHA on the interaction and chromatin modification of the p16INK4a and E‐cadherin promoter in TGBC2TKB cells. ChIP assay was carried out using an EZ ChIP kit (Upstate, Lake placid, NY, USA) according to the manufacturer’s protocol. Antibodies specific for trimethyl histone H3‐Lys27 (TM‐H3K27; Upstate) and acetyl H3‐Lys27 (Ac‐H3K27; Upstate), EZH2 (described above) and normal mouse or normal rabbit IgG as a control were used for immunoprecipitation. PCR was carried out using the following pairs of primers:( 27 , 28 ) p16INK4a, 5′‐ggctagttttatgaccaagacg‐3′ and 5′‐gaggstggtggtgttaaagagg‐3′; E‐cadherin, 5′‐tagagggtcaccgcgtctat‐3′ and 5′‐tcacaggtgctttgcagttc‐3′; and GAPDH, 5′‐ggtagggagttcgagaccag‐3′ and 5′‐tcaacgcagttcagttaggc‐3′.
Statistical analysis. Data are expressed as the mean ± SD. For comparisons of multiple groups, we used Welch’s t‐test. Differences and χ2‐test with values of P < 0.05 were considered significant.
Results
Human tissue studies.
Immunohistochemical expression of EZH2. EZH2 was expressed in the nuclei of non‐neoplastic epithelial cells and carcinoma cells, when detectable (Fig. 1). EZH2 expression was clear and frequent in carcinoma, but faint and infrequent in non‐neoplastic epithelium of chronic cholecystitis. EZH2 expression was more extensive in gallbladder carcinoma than in chronic cholecystitis (Fig. 2a) (P < 0.05). In gallbladder carcinoma, EZH2 expression was higher in poorly differentiated adenocarcinoma than in other histological types of gallbladder carcinoma, including well and moderately differentiated adenocarcinoma and papillary adenocarcinoma (Fig. 2b).
Figure 1.
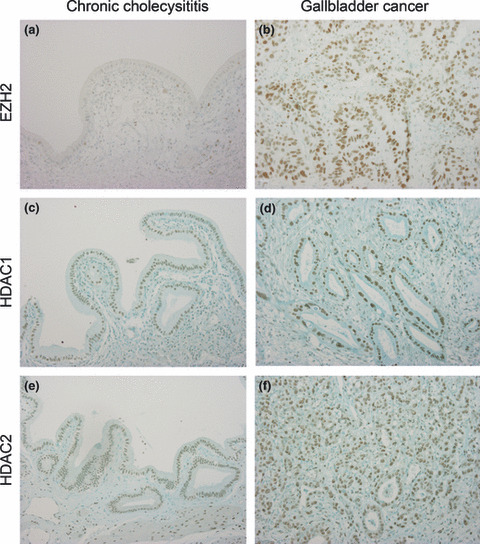
Immunohistochemical expression of polycomb group protein EZH2 (a,b), histone deacetylase (HDAC) 1 (c,d), and HDAC2 (e,f) in chronic cholecystitis (left) and gallbladder carcinoma (right). EZH2 is expressed in the nuclei of gallbladder epithelium and carcinoma. EZH2 is overexpressed in gallbladder carcinoma in comparison with chronic cholecystitis. HDAC1 and HDAC2 are expressed in the nuclei of gallbladder epithelium and carcinoma, and also in the stromal cells of chronic cholecystitis and gallbladder carcinoma. Nuclear staining of HDAC1 and HDAC2 is slightly stronger in gallbladder carcinoma than in non‐neoplastic gallbladder epithelium. Original magnification, ×200.
Figure 2.
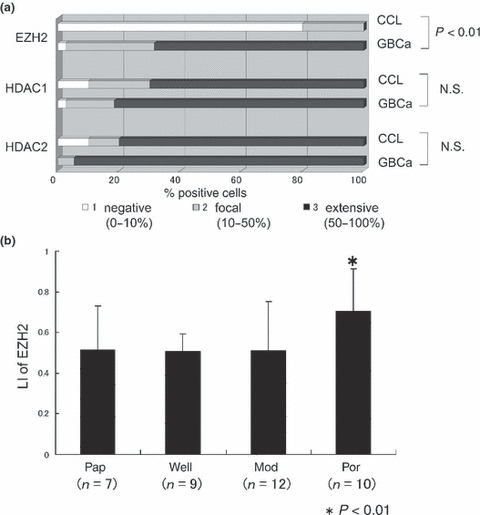
(a) Frequency and degree of immunohistochemical expression of polycomb group protein EZH2, histone deacetylase (HDAC) 1, and HDAC2 in chronic cholecystitis (CCL) and gallbladder cancer (GBCa). EZH2 expression is extensive in GBCa in comparison with CCL (P < 0.05), whereas HDAC1 and HDAC2 is expressed similarly in CCL and GBCa. N.S., not significant. (b) Labeling index (LI) of the expression of EZH2 in gallbladder carcinoma. Poorly differentiated adenocarcinomas (Por) have a higher labeling index of EZH2 (LI, 70.5 ± 20.7%) than other histological types of carcinoma (LI of papillary adenocarcinoma [Pap], 51.4 ± 21.5%; LI of well differentiated adenocarcinoma [Well], 50.6 ± 8.8%; LI of moderately differentiated adenocarcinoma [Mod], 51.2 ± 23.8%). n, number of cases. *P < 0.05.
Immunohistochemical expressions of HDAC1 and HDAC2. HDAC1 and HDAC2 were expressed in the nuclei of non‐neoplastic epithelial cells and carcinoma cells in both chronic cholecystitis and gallbladder carcinoma (Fig. 1). There were no differences in the extent of HDAC1 and HDAC2 expressions between chronic cholecystitis and gallbladder carcinoma (Fig. 2a), although nuclear staining was slightly weaker in non‐neoplastic epithelial cells than in carcinoma cells. Interestingly, HDAC1 and HDAC2 were also expressed in non‐epithelial cells such as fibroblasts and smooth muscle cells in the stroma of the gallbladder (Fig. 1).
Cell culture studies.
Expression patterns of EZH2 and HDACs in cultured cells. The expression of EZH2 and HDACs was examined in cultured normal gallbladder epithelial cell (HGECs) and gallbladder carcinoma cell (TGBC2TKB) by RT‐PCR and Western blotting. RT‐PCR showed EZH2 mRNA expression in carcinoma cells, but not in HGECs (Fig. 3a). HDAC1 and 2 were expressed in both HGECs and TGBC2TKB cells, and real‐time PCR showed that their expression levels were two‐ or threefold higher in TGBC2TKB cells (P < 0.01) (Fig. 3b). Similarly, Western blotting showed HDAC1 and 2 in both HGECs and TGBC2TKB cells, and its expression levels were slightly higher in TGBC2TKB cells (Fig. 3c).
Figure 3.
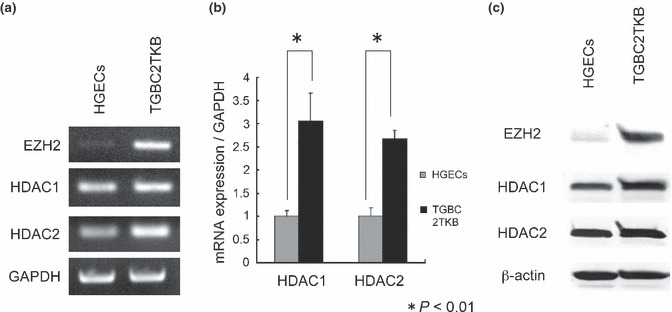
Expressions of polycomb group protein EZH2, histone deacetylase (HDAC)1, and HDAC2 in cultured cells. (a) RT‐PCR: EZH2 mRNA expression is clear in carcinoma cells (TGBC2TKB) but negligible in normal human gallbladder epithelial cells (HGECs), and HDAC 1 and 2 mRNA are clearly expressed in carcinoma cells and normal epithelial cells. (b) Real‐time PCR: Expressions of HDAC 1 and 2 mRNA are higher in carcinoma cells (TGBC2TKB) than in normal HGECs (P < 0.01). (c) Protein expression is evaluated in normal HGECs and gallbladder carcinoma cells (TGBC2TKB) by Western blot analysis. EZH2 expression is evident in carcinoma cells but is very weak in normal HGECs. HDAC 1 and 2 expressions are clear in both of these cells, but the expression level is a little lower in normal epithelial cells than in carcinoma cells.
Effect on cell number of cultured cells by SAHA and EZH2 siRNA. To evaluate the effect of SAHA and EZH2 treatment on the proliferation of cultured cells, EZH2 siRNA or control siRNA was first transfected in HGECs, TGBC2TKB, HuCCT‐1, and TFK‐1 cells, then these cultured cells were treated with 1.0 μM SAHA for 48 h. Relative cell number was calculated by WST‐1 assay (Fig. 4a). It was found that EZH2 siRNA and SAHA treatment induced a relative reduction of TGBC2TKB, HuCCT‐1, and TFK‐1 cells (P < 0.01), and furthermore, the combination of these treatments induced a synergistic effect in the relative reduction of cell numbers (P < 0.01), suggesting that TGBC2TKB, HuCCT‐1, and TFK‐1 cells are sensitive to EZH2 siRNA and SAHA treatment. However, these treatments had no effect on the relative number of HGECs. Considering the expression status of EZH2 and HDACs in HGECs, SAHA treatment had no effect on the number of cultured HGECs, suggesting that HGECs were also resistant to SAHA treatment in spite of their HDAC expression.
Figure 4.
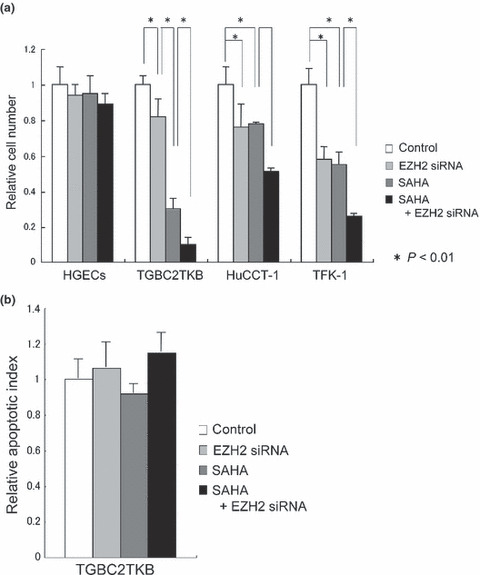
Effect of suberoylanilide hydroxamic acid (SAHA) and polycomb group protein EZH2 siRNA on cell proliferation and apoptosis of cultured cells. After transfection of control siRNA and EZH2 siRNA, cultured cells were treated with SAHA (1.0 μM for 48 h). (a) Relative cell numbers of normal human gallbladder epithelial cells (HGECs) are not reduced by SAHA and/or EZH2 siRNA treatment. In contrast, the relative cell numbers of carcinoma cells (TGBC2TKB) are reduced by SAHA and EZH2 siRNA treatment (P < 0.01), and this effect is synergized (P < 0.01). Relative cell number was evaluated by WST‐1 assay. HuCCT‐1 and TFK‐1 are cholangiocarcinoma cell lines. (b) Apoptosis of carcinoma cells (TGBC2TKB) is not significantly increased by SAHA and EZH2 siRNA treatment (P > 0.01). Apoptosis was detected using an ssDNA apoptosis ELISA kit.
Effect on apoptosis of cultured cells by SAHA and EZH2 siRNA. To evaluate the effect of SAHA and EZH2 treatment on apoptosis of cultured cells, EZH2 siRNA or control siRNA was first transfected in TGBC2TKB cells, then these cultured cells were treated with 1.0 μM SAHA for 48 h. Apoptosis was detected using the ssDNA apoptosis ELISA kit (Fig. 4b). It was found that combined treatment of SAHA and EZH2 siRNA tended to induce a slight increase of apoptosis in TGBC2TKB cells, but there was no significant difference. This finding suggests that apoptosis might not be related to the synergistic effect of combined treatment of SAHA and EZH2 siRNA on relative reduction of cell numbers and that other processes, probably cell cycle arrest, may be responsible for the synergistic effect.
Effect of SAHA on HDAC expression in cultured cells. To understand the mechanism of the different effects of SAHA in the two types of cultured cells, the expression levels of EZH2 and HDACs in cultured cells treated by EZH2 siRNA and/or SAHA were assayed by Western blotting (Fig. 5a). EZH2 siRNA or control siRNA was transfected in HGECs and TGBC2TKB cells, then treated with SAHA (1.0 μM for 48 h). The EZH2 level increased in TGBC2TKB cells was considerably reduced by EZH2 siRNA, and more by SAHA, and also by SAHA + EZH2 siRNA. HDACs levels were repressed by SAHA treatment, and this repression was seen in both HGECs and TGBC2TKB cells almost equally. This indicates that the reduction of HDACs alone is not the cause of the SAHA treatment effect, because the repression of HDACs in HGECs does not reflect the resistance to SAHA treatment indicated in the WST‐1 assay. However, SAHA repressed the EZH2 level in TGBC2TKB cells.
Figure 5.
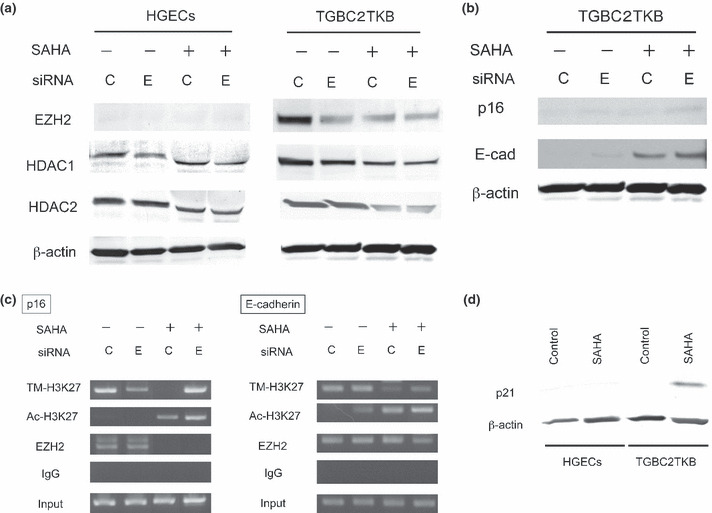
(a) Effect on polycomb group protein EZH2, histone deacetylase (HDAC) 1, and HDAC2 protein expression in cultured cells by suberoylanilide hydroxamic acid (SAHA) and EZH2 siRNA treatment. Western blotting: The increased expression of EZH2 in carcinoma cells (TGBC2TKB) was reduced by SAHA (1.0 μM for 48 h) and EZH2 siRNA treatment, whereas EZH2 was not expressed in normal human gallbladder epithelial cells (HGECs) before or after treatment. Expression of HDAC 1 in normal cells and carcinoma cells was repressed by SAHA treatment and also EZH2 siRNA, and HDAC2 expression was repressed in normal cells and carcinoma cells by SAHA treatment, but not clearly by EZH2 siRNA treatment. C, control siRNA; E, EZH2 siRNA. (b) Effect on p16INK4a (p16) and E‐cadherin (E‐cad) protein expression in cultured cells by SAHA and EZH2 siRNA treatment. Western blotting: The expression of p16INK4a and E‐cadherin was increased by SAHA (1.0 μM for 48 h) and EZH2 siRNA treatment in TGBC2TKB cells. C, control siRNA; E, EZH2 siRNA. (c) ChIP assays were carried out using antibodies against trimethylated histone H3Lys27 (TM‐H3K27), acetylated histone H3Lys27 (Ac‐H3K27), EZH2, and IgG (control) in TGBC2TKB cells treated with SAHA (1.0 μM for 48 h) and/or EZH2 siRNA. PCR was carried out using primers for each amplicon targeting human p16INK4a (left) and E‐cadherin (right) promoter. EZH2 binding to the p16INK4a promoter was decreased by SAHA, whereas Ac‐H3K27 levels increased at the p16INK4a promoter by SAHA. TM‐H3K27 levels decreased at the p16INK4a promoter by SAHA. EZH2 binding to the E‐cadherin promoter was decreased by combined SAHA and EZH2 siRNA treatment. Ac‐H3K27 levels increased at the E‐cadherin promoter by SAHA and/or EZH2 siRNA. TM‐H3K27 levels decreased at the E‐cadherin promoter by SAHA. (d) p21 protein expression in cultured cells by SAHA treatment. Western blotting: normal HGECs and TGBC2TKB carcinoma cells were treated with SAHA (1.0 μM for 6 h). p21 was detectable (activated) in carcinoma cells but not in normal epithelial cells.
Effect on p16 and E‐cadherin expression and chromatin modification in TGBC2TKB cells. The expression levels of p16INK4a and E‐cadherin in TGBC2TKB cells treated by EZH2 siRNA and/or SAHA (described above) were assayed by Western blotting (Fig. 5b). The protein expression level of p16INK4a and E‐cadherin was increased by EZH2 siRNA, more by SAHA, and also by SAHA and EZH2 siRNA in TGBC2TKB cells.
ChIP assays showed that the p16INK4a and E‐cadherin promoter was bound by EZH2 directly in TGBC2TKB cells (Fig. 5c). EZH2 binding to the p16INK4a promoter was decreased by SAHA treatment, whereas acetylated‐H3K27 levels increased at the p16INK4a promoter by SAHA. Trimethylated‐H3K27 levels decreased at the p16INK4a promoter by SAHA. The reason remains unknown why trimethylated‐H3K27 levels recovered at the p16INK4a promoter by combined SAHA and EZH2 siRNA treatment. EZH2 binding to the E‐cadherin promoter was decreased by combined SAHA and EZH2 siRNA treatment. Acetylated‐H3K27 levels increased at the E‐cadherin promoter by SAHA and/or EZH2 siRNA. Trimethylated‐H3K27 levels decreased at the E‐cadherin promoter by SAHA. These findings suggest that EZH2 binds to the p16INK4a and E‐cadherin promoter and that SAHA and/or EZH2 siRNA treatment affect trimetylated and acetylated levels at the p16INK4a and E‐cadherin promoter and increase the expression levels of p16INK4a and E‐cadherin.
SAHA induces p21 expression in cultured TGBC2TKB cells but not in HGECs. We then examined whether tumor suppressor genes are activated by SAHA treatment by evaluating p21 activation in TGBC2TKB cells and HGECs by SAHA treatment (1.0 μM for 6 h). It was found that p21 was expressed in TGBC2TKB cells but not in HGECs (Fig. 5d), suggesting that HDACs are responsible for p21 repression in TGBC2TKB cells, and that the suppressed p21 gene is re‐activated by the repression of HDACs induced by SAHA. In contrast, in HGECs, HDACs were not responsible for p21 gene repression and HDACs inhibition did not activate p21 expression.
Discussion
Chromatin alterations are causally related to the development and progression of malignant tumors. The most well‐characterized alteration is CpG DNA hypermethylation, which contributes to tumor suppressor loss through epigenetic silencing,( 29 ) and epigenetic modification of histones is also implicated in oncogenesis.( 30 ) A common biological function of PRC2 is transcriptional silencing of differentiation genes, and frequent targets of PRC2 are transcription factors and signaling components with key roles in cell fate decisions in a wide variety of organisms. PRC2 contains a conserved catalytic subunit, EZH2, which contains the signature domain providing the methylation active site,( 31 ) and EZH2 levels are abnormally elevated in malignant tissues, with the highest EZH2 levels correlating with advanced stages and poor prognosis.( 10 , 12 , 15 ) This study showed that EZH2 was overexpressed in gallbladder carcinoma, particularly poorly differentiated carcinoma, but its expression was faint and infrequent in non‐neoplastic epithelium. Culture studies also showed EZH2 mRNA and protein expression in gallbladder carcinoma cells, but not in normal cells. Furthermore, downregulation of EZH2 by siRNA induces significant growth inhibitory effect in gallbladder carcinoma cells and cholangiocarcinoma cells to various degrees. EZH2 siRNA also increased the expression of E‐cadherin, which is supposed to control invasiveness of carcinoma, by chromatin modification. Taken together, EZH2 may be related to factors controlling cell proliferation, differentiation, and invasiveness in gallbladder carcinoma as reported in other types of cancers.( 10 , 11 , 12 , 13 , 14 , 15 , 27 , 28 )
PRC2 is known to interact with HDAC proteins.( 8 , 9 ) Recent studies disclosed that HDACs have many protein substrates involved in the regulation of gene expression, cell proliferation, and cell death. Acetylation and deacetylation of histones play an important role in transcription regulation of eukaryotic cells.( 16 , 17 ) Recently, several reports showed that HDACs are strongly expressed in cancerous tissue, and the expression of class I HDACs is an independent prognostic marker in various cancers, such as ovarian, colorectal and cervical cancer.( 32 , 33 , 34 ) The present study showed that HDAC1/2 were expressed in the nuclei of non‐neoplastic epithelial cells and carcinoma cells in the gallbladder, although nuclear staining was slightly weaker in the former than in the latter. This pattern was verified in cultured normal epithelial and carcinoma cells, indicating that HDAC1/2 expression level is a little higher in cancerous tissue; however, its expression is not specific to carcinoma cells in the gallbladder.
Both epigenetic changes and genetic alterations to DNA sequence in the malignant cell genome might contribute to disease progression. Once the DNA sequence is changed by mutation, it is difficult to restore the gene; however, epigenetic changes can potentially be reserved with inhibitors that block the relevant chromatin‐modifying enzymes. Epigenetic silencing of the tumor suppressor gene in carcinoma has inspired potential therapeutic strategies that use inhibitors of epigenetic enzymes. Many inhibitors target either DNA methyltransferases or HDACs.( 16 ) HDAC inhibitors have been found to have profound anticancer effects in clinical trials.( 5 ) Among HDAC inhibitors, SAHA is one of the most advanced in clinical fields as an anticancer agent. SAHA has shown significant anticancer activity in tumor‐bearing animals and in phases I and II clinical trials with little evidence of adverse effects on normal cells.( 5 )
It was found in this study that SAHA treatment reduced the number of gallbladder carcinoma cells and cholangiocarcinoma cells, suggesting that carcinoma cells are sensitive to SAHA treatment. However, this treatment had no effect on the number of normal epithelial cells in spite of their HDAC expression. The reason why carcinoma cells are more sensitive to SAHA compared to normal cells remains speculative. High expression levels of EZH2 might be related to high sensitivity to SAHA in carcinoma cells, as discussed below. SAHA significantly repressed the EZH2 level in carcinoma cells. These differences in sensitivity to SAHA‐induced cell reduction of carcinoma cells compared with normal cells appeared not to be caused by a difference in the expression levels of HDACs, because normal cells and the normal epithelium of the gallbladder has relatively weak but significant expression levels of HDAC1/2, and moreover, it was found that the expression level of HDACs was significantly repressed in normal cells and also carcinoma cells by SAHA treatment.
Histone deacetylase inhibitors are known to alter gene expression followed by the expression of the proteins responsible for composing the transcription factor complex to which HDACs are recruited.( 8 , 9 ) We therefore examined whether tumor suppressor genes are activated by SAHA treatment by evaluating p21 activation in gallbladder carcinoma cells and normal cells by SAHA treatment. It was found that p21 was expressed in carcinoma cells but not in normal cells, indicating that HDACs forcibly repress the p21 gene in carcinoma cells, that inhibition of HDACs by SAHA treatment re‐activated the p21 gene, and that HDACs are not responsible for p21 gene repression in normal cells. One hypothesis is that p21 is not forcibly repressed in normal epithelial cells, and HDACs do not act as a p21 gene suppressor; therefore, HDAC inhibition does not result in activation of the p21 gene in normal cells.
Almost all carcinomas have multiple alterations in the expression and/or structure of proteins that regulate cell proliferation and death. The multiple alterations of tumor suppressor genes in cancer cells might explain why transformed cells are more sensitive than normal cells to the HDAC inhibitor. EZH2 are reported to mediate tumor suppressor genes, such as p16 INK4A and E‐cadherin.( 28 , 35 , 36 ) In this context, the effect of HDAC inhibitor might be linked to EZH2 expression. In the link between EZH2 and HDACs it has been reported that PRC2‐mediated transcriptional silencing is impeded by the HDAC inhibitor, trichostatin A (TSA).( 12 , 15 ) EZH2 is reported to have a SET domain, which is responsible for histone methylation, and this activity was abrogated in the presence of TSA.( 12 , 15 , 16 ) Interestingly, the present study showed that SAHA decreased EZH2 expression itself in carcinoma cells, in addition to HDAC repression. Similar downregulation of EZH2 by HDAC inhibitor has been reported by Fiskus et al. ( 37 ) Whether the mechanism by which HDAC inhibitors such as SAHA deplete EZH2 level is transcriptional, post‐transcriptional, or increased protein degradation remains to be addressed in future study. This double‐repression effect might be an important mechanism in the anticancer effect of SAHA and the reason why normal cells are resistant to SAHA treatment. Normal cells express HDAC1/2 in gallbladder epithelial cells, but not EZH2. Whereas gallbladder carcinoma cells, which have both HDAC and EZH2 expression, work together as transcriptional repressors of tumor suppressor genes, and SAHA repressed these epigenetic enzymes together and re‐activates tumor suppressor genes. From this point of view, the effect of SAHA on gallbladder carcinoma might be associated with EZH2 expression, rather than HDAC expression.
As epigenetic enzymes often synergize in vivo, there is also great interest in testing combined inhibitor treatments that target more than one epigenetic enzyme, suggesting that combination of an HDAC inhibitor and other anticancer agents might be very attractive therapeutic strategies. It was found in this study that the combination of SAHA and EZH2 siRNA decreased cell numbers more than either single treatment. It was disclosed that cell cycle arrest, not apoptosis, might be related to the synergistic effect of the combined treatment. Furthermore, this study showed that SAHA and/or EZH2 siRNA treatment affect trimetylated and acetylated levels at the p16INK4a and E‐cadherin promoter and the combined treatment increased the expression levels of p16INK4a and E‐cadherin. Although the mechanism how the addition of EZH2 siRNA enhanced antitumor activity of SAHA remains to be clarified, EZH2 downregulation might affect other members of PRC2( 37 ) and HDAC activity, which result in the enhanced antitumor effect of SAHA. Similar to HDACs, EZH2 histone methyltransferase has emerged as a key target in potential epigenetic strategies; however, specific inhibitors of EZH2 histone methyltransferase have not been described. The most encouraging inhibitory agent of PRC2 reported so far is deazaneplanocin A, which can deplete PRC2 subunits in breast cancer cell lines and reactivate PRC2‐silenced genes.( 38 ) However, as this type of inhibitor might affect many processes that require methyl transfer, there are concerns about its specificity in potential therapy. The availability of histone methyltransferase inhibitors specific for EZH2, EZH2 inhibitor, and HDAC inhibitor should expand the repertoire of new possibilities in combined epigenetic therapy.
In conclusion, we showed that EZH2 is overexpressed in carcinoma cells, and HDAC1/2 were clearly expressed in both carcinoma cells and normal epithelial cells of the gallbladder. This expression pattern was also followed in cultured cells. SAHA treatment reduced the expression levels of HDAC1/2 and EZH2 in cultured carcinoma cells along with their reduced cell numbers, and this effect was synergized when treated with EZH2 siRNA. Because gallbladder carcinoma shows increased expressions of EZH2 and HDAC1/2, therapeutic SAHA treatment with EZH2 repressors, such as EZH2‐specific methyltransferase inhibitor, is a promising therapeutic approach in gallbladder carcinoma.
Abbreviations
- HDAC
histone deacetylase
- HGEC
human gallbladder epithelial cell
- PcG
polycomb group
- PRC
polycomb repressive complex
- SAHA
suberoylanilide hydroxamic acid
References
- 1. Albores‐saavedra J, Scoazec JC, Wittekind C et al. World Health Organization Classification of tumors, Pathology and Genetics, Tumor of the Digestive System; Carcinoma of the gallbladder and extrahepatic bile ducts. Lyon: International Agency for Research on Cancer, 2000. [Google Scholar]
- 2. Yamato T, Sasaki M, Watanabe Y, Nakanuma Y. Expression of MUC1 and MUC2 mucin core proteins and their messenger RNA in gall bladder carcinoma: an immunohistochemical and in situ hybridization study. J Pathol 1999; 188: 30–7. [DOI] [PubMed] [Google Scholar]
- 3. Sasaki M, Yamato T, Nakanuma Y, Ho SB, Kim YS. Expression of MUC2, MUC5AC and MUC6 apomucins in carcinoma, dysplasia and non‐dysplastic epithelia of the gallbladder. Pathol Int 1999; 49: 38–44. [DOI] [PubMed] [Google Scholar]
- 4. Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21 (WAF1) involves changes in promoter‐associated proteins, including HDAC1. Proc Natl Acad Sci USA 2004; 101: 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 2007; 25: 84–90. [DOI] [PubMed] [Google Scholar]
- 6. Sasaki M, Yamaguchi J, Itatsu K, Ikeda H, Nakanuma Y. Over‐expression of polycomb group protein EZH2 relates to decreased expression of p16 INK4a in cholangiocarcinogenesis in hepatolithiasis. J Pathol 2008; 215: 175–83. [DOI] [PubMed] [Google Scholar]
- 7. Valk‐Lingbeek ME, Bruggeman SW, Van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell 2004; 118: 409–18. [DOI] [PubMed] [Google Scholar]
- 8. Kuzmichev A, Nishioka K, Erdjument‐Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 2002; 16: 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb‐group protein EED involves histone deacetylation. Nat Genet 1999; 23: 474–478. [DOI] [PubMed] [Google Scholar]
- 10. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB‐E2F pathway, essential for proliferation and amplified in cancer. EMBO J 2003; 22: 5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 2005; 24: 6269–6280. [DOI] [PubMed] [Google Scholar]
- 12. Varambally S, Dhanasekaran SM, Zhou M et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419: 624–629. [DOI] [PubMed] [Google Scholar]
- 13. Rhodes DR, Sanda MG, Otte AP, Chinnaiyan AM, Rubin MA. Multiplex biomarker approach for determining risk of prostate‐specific antigen‐defined recurrence of prostate cancer. J Natl Can Inst 2003; 95: 661–668. [DOI] [PubMed] [Google Scholar]
- 14. Collett K, Eide GE, Arnes J et al. Expression of enhancer of zeste homologue 2 is significantly associated with increased tumor cell proliferation and is a marker of aggressive breast cancer. Clin Cancer Res 2006; 12: 1168–1174. [DOI] [PubMed] [Google Scholar]
- 15. Kleer CG, Cao Q, Varambally S et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 2003; 100: 11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lehrmann H, Pritchard LL, Harel‐Bellan A. Histone acetyltransferases and deacetylases in the control of cell proliferation and differentiation. Advances in Cancer Research 2002; 86: 41–65. [DOI] [PubMed] [Google Scholar]
- 17. Mai A, Massa S, Rotili D et al. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Medic Res Rev 2005; 25: 261–309. [DOI] [PubMed] [Google Scholar]
- 18. Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol 2005; 23: 3971–3993. [DOI] [PubMed] [Google Scholar]
- 19. Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26: 5541–5552. [DOI] [PubMed] [Google Scholar]
- 20. Ungerstedt JS, Sowa Y, Xu WS et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA 2005; 102: 673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burgess A, Ruefli A, Beamish H et al. Histone deacetylase inhibitors specifically kill nonproliferating tumour cells. Oncogene 2004; 23: 6693–6701. [DOI] [PubMed] [Google Scholar]
- 22. Garcia P, Manterola C, Araya JC et al. Promoter methylation profile in preneoplastic and neoplastic gallbladder lesion. Mol Carcinog 2009; 48: 79–89. [DOI] [PubMed] [Google Scholar]
- 23. Takahashi T, Shicapurkar N, Riquelme E et al. Aberrant promoter hypermethylation of multiple genes in gallbladder carcinoma and chronic cholecystitis. Clin Cancer Res 2004; 10: 6126–6133. [DOI] [PubMed] [Google Scholar]
- 24. Yamaguchi J, Sasaki M, Harada K et al. Papillary hyperplasia of the gallbladder in pancreatobiliary maljunction represents a senescence‐related lesion induced by lysolecithin. Lab Invest 2009; 89: 1018–1031. [DOI] [PubMed] [Google Scholar]
- 25. Xu LN, Wang X, Zou SQ. Effect of histone deacetylase inhibitor on proliferation of biliary tract cancer cell lines. World J Gastroenterol 2008; 14: 2578–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harada K, Ohba K, Ozaki S et al. Peptide antibiotic human beta‐defensin‐1 and ‐2 contribute to antimicrobial defense of the intrahepatic biliary tree. Hepatology 2004; 40: 925–932. [DOI] [PubMed] [Google Scholar]
- 27. Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes Dev 2007; 21: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cao Q, Yu J, Dhanasekaran SM et al. Repression of E‐cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008; 27: 7274–7284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Egger G, Liang GN, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429: 457–463. [DOI] [PubMed] [Google Scholar]
- 30. Sparmann A, Van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 2006; 6: 846–856. [DOI] [PubMed] [Google Scholar]
- 31. Rea S, Eisenhaber F, O’Carroll N et al. Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature 2000; 406: 593–599. [DOI] [PubMed] [Google Scholar]
- 32. Weichert W, Denkert C, Noske A, Darb‐Esfahani S, Dietel M, Kalloger SE. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia 2008; 10: 1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weichert W, Roske A, Niesporek S et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo . Clin Cancer Res 2008; 14: 1669–1677. [DOI] [PubMed] [Google Scholar]
- 34. Lin ZH, Bazzaro M, Wang MC, Chan KC, Peng SW, Roden RBS. Combination of proteasome and HDAC inhibitors for uterine cervical cancer treatment. Clin Cancer Res 2009; 15: 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujii S, Ito K, Ito Y, Ochiai A. Enhancer of zeste homologue 2 (EZH2) down‐regulates RUNX3 by increasing histone H3 methylation. J Biol Chem 2008; 283: 17324–17332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bracken AP, Kleine‐Kohlbrecher D, Dietrich N et al. The Polycomb group proteins bind throughout the INK4A‐ARF locus and are disassociated in senescent cells. Genes Dev 2007; 21: 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fiskus W, Pranpat M, Balasis M et al. Histone deacetylase inhibitors deplete enhancer of zeste 2 and associated polycomb repressive complex 2 proteins in human acute leukemia cells. Mol Cancer Ther 2006; 5: 3096–3104. [DOI] [PubMed] [Google Scholar]
- 38. Tan J, Yang XJ, Zhuang L et al. Pharmacologic disruption of polycomb‐repressive complex 2‐mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007; 21: 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]