

Abstract
The adaptor protein ASC (also called TMS1) links certain NLR proteins (e.g., NLRC4, NLRP3) and caspases. It is involved in the chemosensitivity of tumor cells and inflammation. Here, we found that ASC activation using NLRC4 mimicry or an autoinflammatory disease‐associated NLRP3 mutant induced necrosis in COLO205 colon adenocarcinoma cells, but induced caspase‐8‐dependent apoptosis in NUGC‐4 stomach cancer cells. As the Fas ligand induced caspase‐8‐dependent apoptosis in COLO205 cells, caspase‐8 was intact in this cell line. ASC‐mediated necrosis was preceded by lysosomal leakage, and diminished by inhibitors for vacuolar H+‐ATPase, cathepsins, and calpains but not by inhibitors for caspase‐8, or aspartic proteases, suggesting that lysosomes and certain proteases were involved in this process. Finally, growing tumors of transplanted human cancer cells in nude mice were eradicated by the activation of endogenous ASC in the tumor cells, irrespective of the form of cell death. Thus, ASC mediates distinct forms of cell death in different cell types, and is a promising target for cancer therapy. (Cancer Sci 2010)
The adaptor protein ASC was originally identified as a protein that forms large aggregates in HL‐60 human leukemia cells treated with chemotherapeutic agents,( 1 ) and as the product of a gene that is silenced in cancer cells by DNA methylation.( 2 ) ASC interacts with NLRC4 (also called CARD12, IPAF, or CLAN), a caspase recruitment domain (CARD)‐containing member of the NLR family, through a homophilic interaction between the CARDs, and mediates intracellular signals for apoptosis and nuclear factor‐κB activation.( 3 , 4 , 5 ) In addition, the genes for ASC and NLRC4 are targets of p53 and are required for the apoptosis induced by p53 and chemotherapeutic drugs.( 6 , 7 ) Thus, the NLRC4–ASC axis seems to be important for tumor suppression and the chemosensitivity of cancer cells. However, the potential of ASC as a molecular target for cancer therapy has not been evaluated.
Another line of research revealed that ASC functions as an adaptor protein that recruits caspase‐1 to several pyrin domain‐containing members of the NLR family, such as NLRP3 (also called PYPAF1, cryopyrin, or NALP3).( 8 ) NLRP3 serves as a sensor for cell‐invading pathogens( 9 , 10 ) or various inflammatory substances, such as uric acid crystals( 11 ) and asbestos.( 12 ) Thus, ASC plays an essential role in the caspase‐1‐dependent maturation of pro‐inflammatory cytokines such as interleukin (IL)‐1β and IL‐18 in response to these inflammatory stimuli. In addition, disease‐associated NLRP3 mutations that are responsible for familial autoinflammatory syndromes cause aberrant ASC‐dependent caspase‐1 activation and IL‐1β secretion.( 13 ) NLRC4 also functions as a sensor for certain cell‐invading bacteria such as Salmonella typhimurium, and ASC is required for the NLRC4‐dependent caspase‐1 activation and IL‐1β secretion.( 14 ) In mouse macrophages, the caspase‐1 activation induced by S. typhimurium causes a unique form of cell death called pyroptosis.( 15 )
We previously showed that the activation of ASC using NLRC4 mimicry induces caspase‐8‐dependent apoptosis in human cancer cell lines.( 5 ) However, recent studies show that the exogenous expression of disease‐associated NLRP3 mutants or the activation of endogenous wild‐type NLRP3 by bacterial infection induces necrosis in an ASC‐dependent manner in human monocytic leukemia THP‐1 cells.( 16 ) This necrosis (called pyronecrosis) is inhibited by the cathepsin B inhibitor CA‐074Me;( 16 , 17 ) however, the precise mechanism of this necrosis and the reason ASC induces different forms of cell death (apoptosis or necrosis) in different contexts are unknown.
In this study, we investigated the types of cell death induced by the activation of ASC using NLRC4 mimicry or NLRP3 mutants in two cancer cell lines, and showed that the cell type, rather than the means of activating ASC, determines whether cells die by apoptosis or necrosis. We also showed for the first time that the activation of ASC in tumors results in tumor eradication, irrespective of the type of cell death.
Materials and Methods
Reagents. Soluble mouse Fas ligand was prepared as described previously.( 18 ) Muramyl dipeptide (MDP; Sigma, St Louis, MO, USA), Z‐IETD‐fluoromethylketone (Z‐IETD‐FMK; R&D Systems, Minneapolis, MN, USA), CA‐074Me, cathepsin L inhibitor IV, cathepsin S inhibitor, calpain inhibitor XI (Merck Calbiochem, Tokyo, Japan), pepstatin A (Peptide Institute, Minoh, Osaka, Japan) and bafilomycin A (Fermentek, Jerusalem, Israel) were purchased.
To generate an anti‐human ASC mAb, lymph node cells from a BALB/c mouse immunized with human ASC (amino acids 113–195) were fused with P3U1 mouse myeloma cells. Hybridoma clones producing mAb that detected human ASC expressed in HEK293 cells by Western blotting were selected. The mAb purified from the hybridoma clone 71 was used in this study. Anti‐caspase‐8 mAb (MBL, Nagoya, Japan), anti‐cathepsin B polyclonal Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐β‐actin mAb (Sigma), and anti‐GAPDH mAb (Millipore, Billerica, MA, USA) were purchased.
Plasmids. The expression plasmids for Flag‐C12N2 (pEF‐Flag‐C12N2) and NLRP3 (pEF‐PYPAF1) were described previously.( 4 , 19 ) The expression plasmid for the NLRP3‐Y570C mutant in the pEF‐BOS vector was provided by Dr. Naotomo Kambe (Chiba University, Chiba, Japan).( 17 ) To generate lentiviral vectors expressing NLRP3 or NLRP3‐Y570C, the GFP cDNA in pLVCT‐tTR‐KRAB (Addgene, Cambridge, MA) was replaced by the corresponding cDNA.
Human cell lines. The NUGC‐4 stomach cancer and COLO205 colon adenocarcinoma cell lines were obtained from the Cell Resource Center for Biomedical Research (Tohoku University, Sendai, Japan). The NUC12N2 cell line was described previously.( 5 ) To generate the CLC12N2 cell line, COLO205 cells were cotransfected with pEF‐FLAG‐C12N2 and a vector carrying the puromycin‐resistance gene. Among the puromycin‐resistant clones, those that underwent cell death upon stimulation with MDP were established as stable cell lines.
Mice. Six‐week‐old female BALB/c and KSN nu/nu mice were purchased from SLC (Shizuoka, Japan). The Kanazawa University Committee on Animal Welfare approved all animal protocols.
Apoptosis and necrosis assay. The proportions of apoptotic and necrotic cells were determined by flow cytometry after staining the cells with propidium iodide (Sigma) and Cy5–annexin V (Biovision, Mountain View, CA, USA), as described previously.( 5 ) Cell viability was assessed by WST‐1 assay as described previously.( 20 ) When cells were transduced with lentiviral vectors, the proportions of apoptotic and necrotic cells were determined in situ as described below. Hoechst 33342 (ABD Bioquest, Sunnyvale, CA, USA) and propidium iodide (both 2.5 μg/mL) were added to the culture wells at the end of transduction. After an additional 30‐min incubation, the apoptotic cells (showing nuclear condensation and/or fragmentation revealed by Hoechst 33342 staining) and necrotic cells (stained by propidium iodide without nuclear condensation) were counted under a fluorescence microscope (Keyence, Osaka, Japan). More than 500 cells in five fields were examined.
Acridine orange staining. Cells were stained with acridine orange (1 μg/mL; Wako Pure Chemical Industries, Osaka, Japan) for 30 min. Acridine orange absorbs light at approximately 490 nm and emits red fluorescence (600–650 nm) under acidic conditions, especially in lysosomes. The red fluorescence of the acridine orange was analyzed by flow cytometry.
Transfection of genes and siRNAs. NUC12N2 cells were transfected with siRNA (100 nM) using RNAiMAX (Invitrogen, Tokyo, Japan). CLC12N2 cells were transfected with siRNA (20 nM) using the Neon Transfection System (Invitrogen). The ASC‐targeting siRNA (Stealth Select HSS147065) and the caspase‐8‐targeting siRNA (Stealth siRNA HSS141460) were purchased from Invitrogen. For viral transduction, the target cells were seeded into a 96‐well plate, and the viral supernatant was added to the cells together with 10 μg/mL polybrene. The culture plate was centrifuged at 1200g for 60 min at room temperature to improve the transduction efficiency.
Cancer therapy models. KSN nu/nu mice were injected s.c. in the dorsal region with 2.5 × 106 tumor cells. MDP (200 μg) was given to the mice i.v. on days 5 and 8 after the tumor injection. The long and short diameters of the tumors were measured at 3‐day intervals using a hand‐held caliper. The tumor size was calculated as follows. Tumor size (mm2) = long diameter (mm) × short diameter (mm). For the peritoneally disseminated tumor model, 2.5 × 106 NUC12N2 cells were injected i.p. into KSN nu/nu mice. Three and 10 days after the tumor inoculation, MDP (200 μg) was given to the mice i.p. Forty‐nine days after the tumor inoculation, the mice were killed and the development of ascites and peritoneal nodules was examined.
Results
Necrotic or apoptotic cell death induced by ASC, depending on the cell type. To investigate the functions of ASC in cells, we previously established an experimental system in which a chimeric protein (C12N2) consisting of the CARD from NLRC4 and the nucleotide‐binding oligomerization domain (NOD) and leucine‐rich repeats from NOD2 (also called NLRC2), a sensor for MDP, was expressed in cells.( 4 ) In this system, the simple addition of MDP to the culture medium induced ASC‐dependent responses in the cells. Using this system, we previously showed that the activation of ASC induces caspase‐8‐dependent apoptosis in stomach, colon, and lung cancer cell lines.( 5 )
Here, we generated another stable C12N2 transfectant cell line, CLC12N2, from the COLO205 colon adenocarcinoma cell line. Unexpectedly, the MDP‐induced cell death was associated with cell swelling in CLC12N2 cells (Fig. 1a). MDP did not induce death in the parental cell line. Flow cytometry analyses after fluorochrome‐conjugated annexin V and propidium iodide staining indicated that MDP induced rapid plasma membrane permeabilization without the preceding externalization of phosphatidylserine on the cell surface, showing that the CLC12N2 cells underwent necrosis (Figs 1b and S1). In contrast, consistent with our previous data,( 5 ) when NUC12N2 cells, a stable C12N2 transfectant derived from the NUGC‐4 stomach cancer cell line, were treated with MDP, typical apoptosis was induced, as revealed by the extensive blebbing of the dying cells (Fig. 1a) and the externalization of phosphatidylserines preceding permeabilization of the plasma membrane (Figs 1b and S1). The MDP‐induced necrosis in CLC12N2 cells was more rapid than the apoptosis in NUC12N2 cells (Fig. 1c). The reduction of ASC expression by RNAi abrogated the MDP‐induced apoptosis in NUC12N2 cells and necrosis in CLC12N2 cells (Fig. 1d), confirming that both types of cell death were mediated by ASC.
Figure 1.
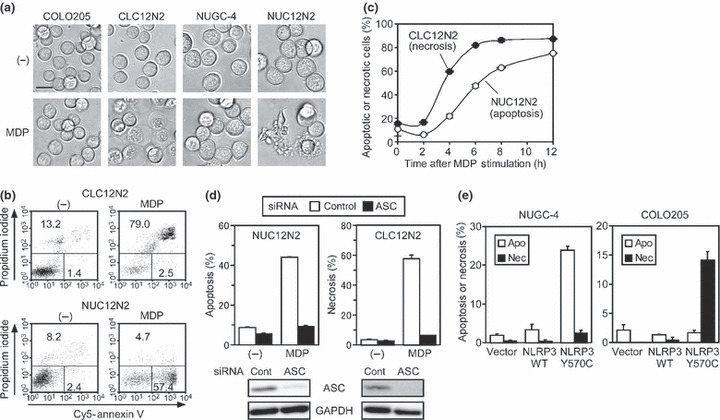
Activation of ASC induces apoptosis and necrosis, depending on the cell type. (a) Cells were cultured without (−) or with muramyl dipeptide (MDP; 100 ng/mL) for 8 h (NUGC‐4 and NUC12N2 cells) or 6 h (COLO205 and CLC12N2 cells). Morphological changes were observed under a microscope. Scale line = 20 μm. (b) Cells were cultured without (−) or with MDP (100 ng/mL) for 8 h (NUC12N2 cells) or 6 h (CLC12N2 cells), stained with Cy5–annexin V and propidium iodide, and analyzed by flow cytometry. The percentages of early apoptotic cells (propidium iodide−, Cy5–annexin V+) or necrotic cells (propidium iodide+) are indicated. (c) CLC12N2 and NUC12N2 cells were cultured with MDP (100 ng/mL) for the indicated period, and analyzed as described in (b). The percentages of apoptotic NUC12N2 cells and necrotic CLC12N2 cells were plotted. (d) Cells were transfected with control (cont) or ASC‐targeting siRNA. The cells were then incubated without (−) or with MDP (100 ng/mL) for the last 16 h (NUC12N2) or 8 h (CLC12N2) of a 72‐h culture, and analyzed as described in (b). The expression levels of ASC and GAPDH at the end of the 72‐h culture period without MDP were examined by Western blot analysis (lower panels). (e) NUGC‐4 and COLO205 cells were transduced with a lentiviral vector for wild‐type (WT) or Y570C mutant NLRP3. After culture for 12 h, the cells were stained with Hoechst 33342 and propidium iodide. The percentages of apoptotic cells (Apo) and necrotic cells (Nec) are indicated. Experiments c–e were done in duplicate, and error bars represent the range of the duplicate data.
It was recently reported that disease‐associated mutants of NLRP3 induce ASC‐dependent necrotic cell death in the THP‐1 human monocytic leukemia cell line,( 16 ) so we investigated whether such an NLRP3 mutant (Y570C) induced necrosis or apoptosis in COLO205 and NUGC‐4. The viral transduction of mutant but not wild‐type NLRP3 induced significant necrosis and apoptosis in COLO205 and NUGC‐4 cells, respectively (Figs 1e and S2). These results indicate that the activation of ASC induces either necrosis or apoptosis, depending on the cell type rather than on the upstream molecular machinery used to activate ASC.
Caspase‐8 in CLC12N2 cells intact but not required for ASC‐mediated necrosis. We previously showed that caspase‐8 is the initiator caspase for ASC‐mediated apoptosis.( 5 ) Therefore, we investigated the expression and function of caspase‐8 in NUC12N2 and CLC12N2 cells. Western blot analyses revealed that the expression levels of caspase‐8 and ASC in NUC12N2, CLC12N2, and their parental cell lines were comparable (Fig. 2a). Consistent with our previous finding that the ASC‐mediated apoptosis in NUC12N2 cells is caspase‐8 dependent,( 5 ) the generation of the large subunit (p20) of mature caspase‐8 was observed in this cell line upon MDP stimulation (Fig. 2b). In contrast, the generation of p20 was not observed in CLC12N2 cells, even 4 h after MDP stimulation (Fig. 2b), when most of the cells showed a necrotic morphology (Fig. 1c).
Figure 2.
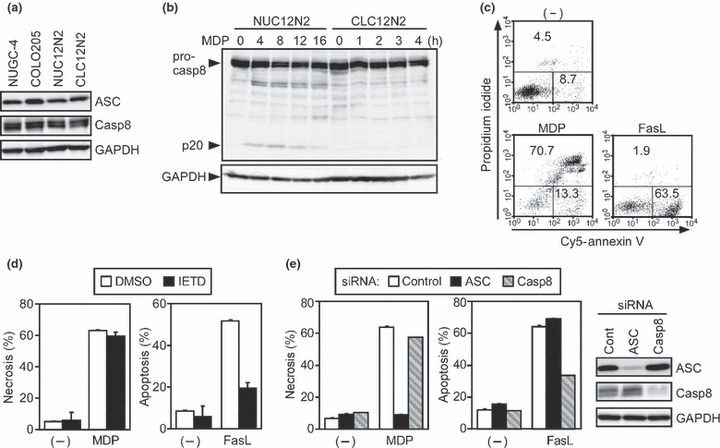
Caspase‐8 is not required for ASC‐mediated necrosis. (a) The expression levels of ASC, caspase‐8 (Casp8), and GAPDH in NUGC‐4, COLO205, NUC12N2, and CLC12N2 cells were examined by Western blotting. (b) NUC12N2 and CLC12N2 cells were cultured with muramyl dipeptide (MDP; 100 ng/mL) for the indicated period. Pro‐caspase‐8 and the large subunit of mature caspase‐8 (p20) were detected by Western blot analysis using an anti‐caspase‐8 Ab. (c) CLC12N2 cells were treated with MDP (100 ng/mL) or Fas ligand (FasL; 1000 U/mL), or left untreated (−) for 8 h. (d) CLC12N2 cells were cultured in the presence or absence (DMSO) of 40 μM Z‐IETD‐fluoromethylketone (IETD) for 7 h and treated without (−) or with MDP (100 ng/mL) for the last 6 h or FasL (1000 U/mL) for the last 4 h of the 7‐h culture. (e) CLC12N2 cells were transfected with control, ASC‐targeting, or caspase‐8‐targeting siRNA. Cells were cultured with MDP (100 ng/mL) or Fas ligand (1000 U/mL), or left untreated (−) for the last 6 h of a 48‐h culture. The expression levels of ASC, caspase‐8, and GAPDH at the end of the 48‐h culture without stimuli were examined by Western blotting (right panels). (c–e) The cultured cells were analyzed as described in Figure 1b. Experiments were done in duplicate, and error bars in (d,e) represent the range of the duplicate data.
It has also been reported that apoptotic stimuli can induce necrotic cell death in certain types of cells, especially when the caspase activity is inhibited,( 21 ) so we next investigated whether caspase‐8 function was intact in CLC12N2 cells. Intriguingly, Fas ligand induced typical apoptosis in CLC12N2 cells, whereas MDP induced necrosis under the same conditions (Fig. 2c). Importantly, the Fas ligand‐induced apoptosis of CLC12N2 cells was inhibited by the caspase‐8 inhibitor Z‐IETD‐FMK, whereas this inhibitor neither enhanced nor inhibited the MDP‐induced necrosis of the same cell line (Fig. 2d). Consistent with this finding, the reduction of caspase‐8 expression by RNAi inhibited the Fas ligand‐induced apoptosis but not the MDP‐induced necrosis of CLC12N2 cells, but the reduction of ASC did the opposite (Fig. 2e). These results indicate that the caspase‐8 function in CLC12N2 cells is intact, but is not involved in ASC‐mediated necrosis.
Inhibitors for cathepsin B, L, and S, and calpains abrogate ASC‐mediated necrosis. The necrosis of THP‐1 cells induced by disease‐associated NLRP3 mutants is inhibited by a cathepsin B inhibitor, CA‐074Me.( 16 , 17 ) Therefore, we next addressed that cathepsin B was responsible for the ASC‐mediated necrotic cell death in our experimental system. Cathepsin B protein was expressed in COLO205 and CLC12N2 cells but not in NUGC‐4 or NUC12N2 cells (Fig. 3a). Furthermore, MDP‐induced necrosis of CLC12N2 cells was reduced dose‐dependently by CA‐074Me but not by Z‐IETD‐FMK (Fig. 3b). This necrosis was also diminished by inhibitors for cathepsins L and S, and calpains (calpain‐1 and calpain‐2), but not by pepstatin A, an inhibitor of aspartic proteases (Fig. 3c). However, the knockdown of cathepsin B expression by RNAi failed to inhibit the MDP‐induced necrosis of CLC12N2 cells (Fig. S3). These results suggest that a protease cascade that includes cathepsins and calpains is redundantly involved in ASC‐mediated necrosis.
Figure 3.
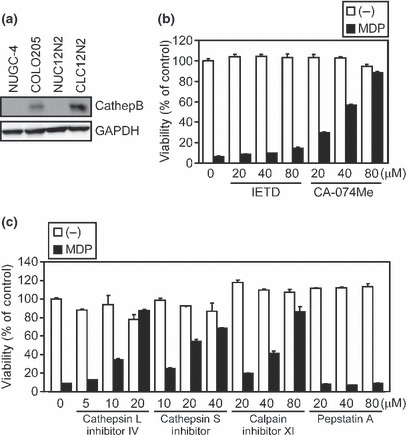
Necrotic cell death is prevented by cathepsin B inhibitor CA‐074Me. (a) The expression levels of cathepsin B (CathepB) and GAPDH in NUGC‐4, COLO205, NUC12N2, and CLC12N2 cell lines were examined by Western blotting. (b,c) The CLC12N2 cells were pretreated with the indicated inhibitors for 1 h, then stimulated with muramyl dipeptide (MDP; 100 ng/mL) for 8 h. The cell viability was assessed by WST‐1 assay. Experiments were done in duplicate, and error bars represent the range of the duplicate data. (−), no treatment; IETD, Z‐IETD‐fluoromethylketone.
Lysosomes involved in ASC‐mediated necrosis. It was previously shown that, during the course of THP‐1 cell death induced by disease‐associated NLRP3 mutants, “small‐scale lysosomal leakage (SSLL)” was observed as a reduction of red fluorescence in cells stained with acridine orange.( 17 ) Therefore, we stimulated acridine orange‐stained CLC12N2 cells with MDP and monitored the red fluorescence by flow cytometry. Consistent with the previous report, a portion of the CLC12N2 cells showed a slight reduction in red fluorescence, corresponding to SSLL, 3 h after the addition of MDP (Fig. 4a). Four hours after the MDP addition, corresponding to necrosis, a large population of the cells showed a dramatic reduction in red fluorescence (Fig. 4a). Thus, SSLL appeared to precede necrosis. Furthermore, CA‐074Me inhibited the MDP‐induced SSLL (Fig. 4b). These observations were strikingly similar to those reported for THP‐1 cells expressing the NLRP3 mutants, suggesting that the activation of ASC induces a similar form of cell death in COLO205 and THP‐1 cells. In addition, we found that the vacuolar ATPase (v‐ATPase) inhibitor bafilomycin A1 abrogated the MDP‐induced SSLL (Fig. 4b) and necrosis (Fig. 4c) in CLC12N2 cells. Collectively, these results suggest that the v‐ATPase‐dependent acidification of lysosomes and the subsequent activation of cathepsins are involved in the SSLL and necrosis of MDP‐treated CLC12N2 cells.
Figure 4.
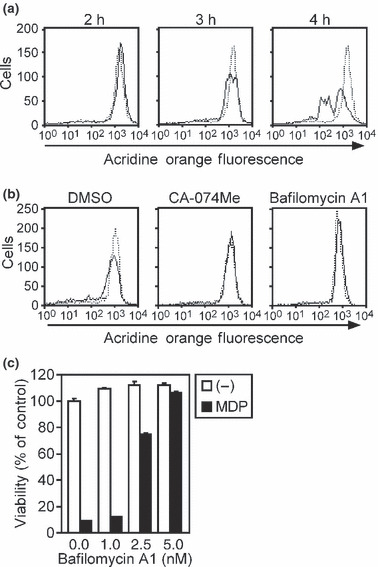
Lysosomal leakage is involved in ASC‐mediated necrosis. (a) CLC12N2 cells stimulated with muramyl dipeptide (MDP; 100 ng/mL) for the indicated period were stained with acridine orange, and the red fluorescence was measured by flow cytometry (solid line). The dotted line indicates unstimulated cells. (b) CLC12N2 cells were pretreated with or without bafilomycin A1 (10 nM) or CA‐074Me (80 μM) for 1 h, then cultured in the presence or absence of MDP (100 ng/mL) for 3 h. The cells were stained with acridine orange and analyzed as described in (a). The dotted line indicates unstimulated cells. (c) CLC12N2 cells were pretreated with the indicated concentrations of bafilomycin A1 for 1 h, then stimulated without (−) or with MDP (100 ng/mL) for 8 h. The cell viability was assessed by WST‐1 assay. Experiments were done in duplicate, and error bars represent the range of the duplicate data.
Potential molecular target for cancer therapy. Finally, we investigated whether the activation of ASC could lead to tumor eradication in vivo. For this purpose, NUC12N2 or CLC12N2 cells or their parental lines were transplanted intradermally into the back of nude mice. After tumor nodules larger than 5 mm in diameter were established, 200 μg MDP was injected i.v. Remarkably, both the NUC12N2 and CLC12N2 tumors were completely eradicated after MDP injection, whereas the tumors derived from the parental cell lines kept growing, irrespective of treatment with PBS or MDP (Fig. 5a). The injection of as little as 50 μg MDP induced the almost complete eradication of the NUC12N2 tumors (Fig. S4). Because NUGC‐4 is a stomach cancer cell line, we also tested whether MDP treatment was effective for peritoneally disseminated tumor cells. Mice that received a PBS injection 3 days after the intraperitoneal injection of NUC12N2 tumor cells developed ascites and multiple peritoneal nodules. In contrast, the four MDP‐injected mice developed no ascites, although tumor nodules were observed in one of them (Fig. 5b). These results indicate that tumor cells can be eradicated in vivo by activating ASC in them, irrespective of the type of cell death induced by the activation of ASC.
Figure 5.
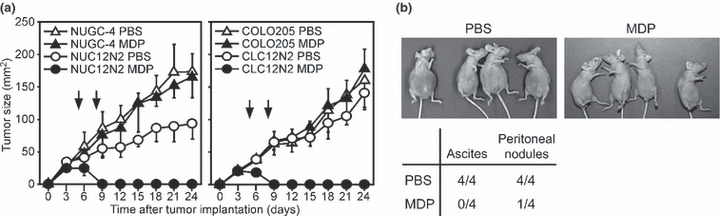
ASC activation eradicates tumor cells in vivo. (a) NUGC‐4, NUC12N2, COLO205, and CLC12N2 cells (2.5 × 106) were transplanted intradermally into the back of nude mice (n = 4) on day 0. Muramyl dipeptide (MDP; 200 μg) or control PBS was injected i.v. into mice on days 5 and 8 (vertical arrows). Error bars indicate SD. (b) NUC12N2 cells (2.5 × 106) were injected i.p. into nude mice (n = 4) on day 0. On days 3 and 10, MDP (200 μg) or PBS was injected i.p. The upper panels show mice in a supine position on day 49. The accumulation of ascites was evident in all the mice receiving PBS but not in those receiving MDP. The lower table shows the ratio of mice with ascite accumulation and peritoneal tumor nodules on day 49.
Discussion
We previously showed that the activation of ASC using NLRC4 mimicry induces caspase‐8‐dependent apoptosis in various human cancer cell lines, including the NUC12N2 stomach cancer cell line.( 5 ) However, recent reports have indicated that the activation of ASC by disease‐associated mutants of NLRP3 or Shigella flexneri infection, which triggers the NLRP3–ASC axis, induces a type of necrotic cell death called pyronecrosis in human monocytic leukemia THP‐1 cells.( 16 ) Therefore, we initially speculated that ASC leads to apoptosis or necrosis depending on its upstream effectors, that is, NLRC4 or NLRP3. Here, however, we show that ASC activation, irrespective of its upstream effectors, that is, NLRC4 mimicry or an NLRP3 mutant, induced caspase‐8‐independent necrosis in the COLO205 colon adenocarcinoma cell line. In contrast, both NLRC4 mimicry and the NLRP3 mutant induced apoptosis in NUGC‐4 cells. These results clearly indicated that the form of ASC‐mediated cell death is determined by cell type rather than by upstream effector proteins.
The molecular mechanisms that determine the form of ASC‐mediated cell death are currently unclear. Tumor necrosis factor (TNF), a well‐known death factor that induces caspase‐8‐dependent apoptosis, causes necrotic cell death in some cell types, especially when caspase activity is inhibited. Recently, RIP3 was found to be a molecular switch for TNF‐induced necrosis.( 22 , 23 , 24 ) However, the function of caspase‐8 and its pro‐apoptotic downstream effectors were intact in the CLC12N2 cells, given that Fas ligand induced caspase‐8‐dependent apoptosis in this cell line. In addition, the caspase‐8 inhibitor Z‐IETD‐FMK neither enhanced nor inhibited the ASC‐mediated necrosis of CLC12N2 cells. Furthermore, the pan‐caspase inhibitor Z‐VAD‐FMK inhibited the ASC‐mediated apoptosis of NUC12N2 cells, but did not change the cell‐death type from apoptosis to necrosis (data not shown). These results suggest that ASC‐mediated necrosis is different from TNF‐induced necrosis. Nevertheless, it would be interesting to examine whether RIP3 is also required for ASC‐mediated necrosis.
The ASC‐mediated necrosis of COLO205 cells in our system was accompanied by SSLL and inhibited by the CA‐074Me cathepsin B inhibitor, which is consistent with a previous report on the ASC‐mediated necrosis of THP‐1 cells. However, the reduction of cathepsin B expression using RNAi did not suppress the ASC‐mediated necrosis in COLO205 cells. Because inhibitors for cathepsins L and S also inhibited the ASC‐mediated necrosis, multiple cathepsins seem to be redundantly involved in this necrosis, and an inhibitor for one cathepsin might cross‐inhibit other cathepsins. Consistent with the involvement of these lysosomal proteases, which show optimal activity at acidic pH, the v‐ATPase inhibitor bafilomycin A1 inhibited the ASC‐mediated SSLL and necrosis. Thus, it is likely that lysosomal acidification followed by cathepsin activation resulted in the SSLL and necrosis. We found that a calpain inhibitor also blocked the ASC‐mediated necrosis. Because calpains are reported to be involved in the activation of cathepsins and lysosomal rupture in ischemic neuronal cell death,( 25 ) they may act upstream of cathepsins. A caveat is that the calpain inhibitor used in the present study weakly inhibits cathepsin B, so the observed reduction of necrosis by this inhibitor might have been due to the cross‐inhibition of cathepsin B.
Although it has been shown that ASC is involved in the chemosensitivity of cancer cells,( 6 ) the potential of ASC as a molecular target for cancer therapy has not been directly examined. In this study, we showed that the activation of ASC in tumor cells, irrespective of the induced cell death type, caused the complete eradication of intradermal solid tumors or of peritoneal disseminated tumor cells. Thus, ASC can be a molecular target for cancer therapy. An interesting question is whether apoptosis or necrosis is the preferable type of cell death for tumor eradication, with respect to cancer therapy. Both the NUC12N2 (apoptosis‐type) and CLC12N2 (necrosis‐type) tumors were equally and efficiently eradicated when ASC was activated by MDP. However, because we used immune‐incompetent mice as the host animals for human cancer cells, we could not investigate the immunological aspects of ASC‐mediated cancer therapy. It would be interesting to compare the apoptosis‐type and necrosis‐type tumor cells in terms of the potency of the antitumor immunity developed after ASC‐targeted therapy, using combinations of immune‐competent mice and syngeneic tumor cell lines.
Supporting information
Fig. S1. Representative flow cytometry profiles of cells shown in Fig. 1c.
Fig. S2. The Y570C mutant of NLRP3 induced apoptosis and necrosis in NUGC‐4 and COLO205 cells, respectively.
Fig. S3. Reduction of cathepsin B expression by siRNA did not inhibit muramyl dipeptide (MDP)‐induced necrosis of CLC12N2 cells.
Fig. S4. Dose‐dependent suppression of the growth of NUC12N2 tumors by treatment with muramyl dipeptide (MDP).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
This work was supported in part by Grants‐in‐Aid for Scientific Research on Priority Areas (Cancer) from the Ministry of Education, Culture, Sports, Science and Technology, Government of Japan, and a grant from the Novartis Foundation (Japan) for the Promotion of Science.
References
- 1. Masumoto J, Taniguchi S, Ayukawa K et al. ASC, a novel 22‐kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL‐60 cells. J Biol Chem 1999; 274: 33835–8. [DOI] [PubMed] [Google Scholar]
- 2. Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation‐induced gene silencing in human breast cancers. Cancer Res 2000; 60: 6236–42. [PubMed] [Google Scholar]
- 3. Masumoto J, Dowds TA, Schaner P et al. ASC is an activating adaptor for NF‐kappa B and caspase‐8‐dependent apoptosis. Biochem Biophys Res Commun 2003; 303: 69–73. [DOI] [PubMed] [Google Scholar]
- 4. Hasegawa M, Imamura R, Kinoshita T et al. ASC‐mediated NF‐kappa B activation leading to IL‐8 production requires caspase‐8 and is inhibited by CLARP. J Biol Chem 2005; 280: 15122–30. [DOI] [PubMed] [Google Scholar]
- 5. Hasegawa M, Kawase K, Inohara N et al. Mechanism of ASC‐mediated apoptosis: bid‐dependent apoptosis in type II cells. Oncogene 2007; 26: 1748–56. [DOI] [PubMed] [Google Scholar]
- 6. Ohtsuka T, Ryu H, Minamishima YA et al. ASC is a Bax adaptor and regulates the p53‐Bax mitochondrial apoptosis pathway. Nat Cell Biol 2004; 6: 121–8. [DOI] [PubMed] [Google Scholar]
- 7. Sadasivam S, Gupta S, Radha V, Batta K, Kundu TK, Swarup G. Caspase‐1 activator Ipaf is a p53‐inducible gene involved in apoptosis. Oncogene 2005; 24: 627–36. [DOI] [PubMed] [Google Scholar]
- 8. Wang L, Manji GA, Grenier JM et al. PYPAF7, a novel PYRIN‐containing Apaf1‐like protein that regulates activation of NF‐kappa B and caspase‐1‐dependent cytokine processing. J Biol Chem 2002; 277: 29874–80. [DOI] [PubMed] [Google Scholar]
- 9. Kanneganti TD, Ozoren N, Body‐Malapel M et al. Bacterial RNA and small antiviral compounds activate caspase‐1 through cryopyrin/Nalp3. Nature 2006; 440: 233–6. [DOI] [PubMed] [Google Scholar]
- 10. Mariathasan S, Weiss DS, Newton K et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440: 228–32. [DOI] [PubMed] [Google Scholar]
- 11. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–41. [DOI] [PubMed] [Google Scholar]
- 12. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008; 320: 674–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dowds TA, Masumoto J, Zhu L, Inohara N, Nunez G. Cryopyrin‐induced interleukin 1beta secretion in monocytic cells: enhanced activity of disease‐associated mutants and requirement for ASC. J Biol Chem 2004; 279: 21924–8. [DOI] [PubMed] [Google Scholar]
- 14. Mariathasan S, Newton K, Monack DM et al. Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 2004; 430: 213–8. [DOI] [PubMed] [Google Scholar]
- 15. Cookson BT, Brennan MA. Pro‐inflammatory programmed cell death. Trends Microbiol 2001; 9: 113–4. [DOI] [PubMed] [Google Scholar]
- 16. Willingham SB, Bergstralh DT, O’Connor W et al. Microbial pathogen‐induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2007; 2: 147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujisawa A, Kambe N, Saito M et al. Disease‐associated mutations in CIAS1 induce cathepsin B‐dependent rapid cell death of human THP‐1 monocytic cells. Blood 2007; 109: 2903–11. [DOI] [PubMed] [Google Scholar]
- 18. Suda T, Tanaka M, Miwa K, Nagata S. Apoptosis of mouse naive T cells induced by recombinant soluble Fas ligand and activation‐induced resistance to Fas ligand. J Immunol 1996; 157: 3918–24. [PubMed] [Google Scholar]
- 19. Wang Y, Hasegawa M, Imamura R et al. PYNOD, a novel Apaf‐1/CED4‐like protein is an inhibitor of ASC and caspase‐1. Int Immunol 2004; 16: 777–86. [DOI] [PubMed] [Google Scholar]
- 20. Fukui M, Imamura R, Umemura M, Kawabe T, Suda T. Pathogen‐associated molecular patterns sensitize macrophages to Fas ligand‐induced apoptosis and IL‐1beta release. J Immunol 2003; 171: 1868–74. [DOI] [PubMed] [Google Scholar]
- 21. Kroemer G, Galluzzi L, Vandenabeele P et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho YS, Challa S, Moquin D et al. Phosphorylation‐driven assembly of the RIP1‐RIP3 complex regulates programmed necrosis and virus‐induced inflammation. Cell 2009; 137: 1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He S, Wang L, Miao L et al. Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell 2009; 137: 1100–11. [DOI] [PubMed] [Google Scholar]
- 24. Zhang DW, Shao J, Lin J et al. RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science 2009; 325: 332–6. [DOI] [PubMed] [Google Scholar]
- 25. Yamashima T. Ca2+‐dependent proteases in ischemic neuronal death: a conserved ‘calpain‐cathepsin cascade’ from nematodes to primates. Cell Calcium 2004; 36: 285–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative flow cytometry profiles of cells shown in Fig. 1c.
Fig. S2. The Y570C mutant of NLRP3 induced apoptosis and necrosis in NUGC‐4 and COLO205 cells, respectively.
Fig. S3. Reduction of cathepsin B expression by siRNA did not inhibit muramyl dipeptide (MDP)‐induced necrosis of CLC12N2 cells.
Fig. S4. Dose‐dependent suppression of the growth of NUC12N2 tumors by treatment with muramyl dipeptide (MDP).
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item