

Abstract
Epidermal cells are the first cells to be exposed to environmental genotoxic agents such as ultraviolet and ionizing radiations, which induce DNA double strand breaks (DSB) and activate DNA damage response (DDR) to maintain genomic integrity. Defective DDR can result in genomic instability (GIN) which is considered to be a central aspect of any carcinogenic process. P53‐binding protein 1 (53BP1) belongs to a family of evolutionarily conserved DDR proteins. Because 53BP1 molecules localize at the sites of DSB and rapidly form nuclear foci, the presence of 53BP1 nuclear foci can be considered as a cytological marker for endogenous DSB reflecting GIN. The levels of GIN were analyzed by immunofluorescence studies of 53BP1 in 56 skin tumors that included 20 seborrheic keratosis, eight actinic keratosis, nine Bowen's disease, nine squamous cell carcinoma, and 10 basal cell carcinoma. This study demonstrated a number of nuclear 53BP1 foci in human skin tumorigenesis, suggesting a constitutive activation of DDR in skin cancer cells. Because actinic keratosis showed a high DDR type of 53BP1 immunoreactivity, GIN seems to be induced at the precancerous stage. Furthermore, invasive cancers exhibited a high level of intense, abnormal 53BP1 nuclear staining with nuclear accumulation of p53, suggesting a disruption of DDR leading to a high level of GIN in cancer cells. The results of this study suggest that GIN has a crucial role in the progression of skin carcinogenesis. The detection of 53BP1 expression by immunofluorescence can be a useful histological marker to estimate the malignant potential of human skin tumors. (Cancer Sci 2008; 99: 946–951)
Abbreviations:
- UV
ultraviolet
- IR
ionizing radiations
- DSB
DNA double strand breaks
- DDR
DNA damage response
- GIN
genomic instability
- 53BP1
P53‐binding protein 1
- SK
seborrheic keratosis
- AK
actinic keratosis
- BD
Bowen's disease
- SCC
squamous cell carcinoma
- BCC
basal cell carcinoma
- DAPI‐I
4′, 6‐diamidino‐2‐phenylindole dihydrochloride
- CIS
carcinoma in situ
The skin is the primary barrier for humans against the external environment. Therefore, epidermal cells are the first cells to be exposed to physical and chemical genotoxic agents such as UV and IR. IR effectively induces DSB in normal cells and activates DDR pathways to maintain genomic integrity. DDR genes, such as p35, are frequently mutated in human cancer. Thus, defective DDR can result in GIN which is generally considered to be a central aspect of any carcinogenic process.( 1 , 2 ) It has been shown that gamma irradiation induced skin tumors in mice. Most of the tumor‐bearing mice showed a loss of the wild‐type p53 allele. Since no skin tumor was found in wild‐type p53 mice, this suggested a requirement of p53 loss in irradiation‐induced skin carcinogenesis.( 3 ) The incidence of skin cancer was reported to be elevated in atomic bomb survivors, which also suggested a radiation etiology in human skin carcinogenesis.( 4 )
53BP1 belongs to a family of evolutionarily conserved DDR proteins with C‐terminal BRCT (BRCA1 C‐terminus) domains.( 5 , 6 ) The 53BP1 is a nuclear protein that rapidly localizes at the sites of DSB, and co‐operatively activates p53 with other kinases.( 7 , 8 , 9 , 10 , 11 , 12 ) Subsequently, activated p53 plays a critical role in cellular responses to genomic injury, such as cell cycle arrest, DNA repair, and apoptosis.( 13 , 14 ) It has been well documented in vitro with immunofluorescence that 53BP1 exhibits diffuse nuclear staining in untreated primary cells. However, after exposure to IR, 53BP1 localizes at the sites of DSB and forms discrete nuclear foci.( 7 , 8 , 15 , 16 ) We have recently demonstrated that immunofluorescence analysis for 53BP1 specifically detected the 53BP1 nuclear foci at the sites of DSB induced by IR in formalin‐fixed paraffin‐embedded mouse intestine.( 17 ) Because one manifestation of GIN is the induction of endogenous DSB,( 18 ) the level of 53BP1‐focus formation can be considered as a cytological marker for GIN.
Human cancers develop through a multistep process that involves the accumulation of genetic mutations.( 19 ) It is well established that any DNA damages can lead to GIN and subsequently induce DDR. Thus, measurement of GIN, a hallmark feature of solid tumors that is implicated in both initiation and progression of cancers, may serve as a valuable molecular marker of malignant potential. Our recent study of thyroid tumors from patients demonstrated a few nuclear 53BP1 foci in follicular adenoma but conspicuously more nuclear 53BP1 foci in thyroid cancers. This suggested a constitutive activation of DDR in thyroid tumors and increased GIN with progression of cancer.( 17 ) Furthermore, anaplastic thyroid cancers prominently exhibited an abnormal and intense nuclear staining of 53BP1, which was also observed in mouse colonic crypts as a delayed response to a lethally high dose of IR, suggesting increased GIN with progression to high‐grade cancer.( 17 ) Thus, we propose that immunofluorescence analysis of 53BP1 expression can be a useful tool to estimate the level of GIN and, simultaneously, the malignant potential of human thyroid tumors. The present study analyzed the presence of GIN by immunofluorescence of 53BP1 expression in a series of skin tissues from patients to evaluate the significance of GIN during skin carcinogenesis. Similar to thyroid tumorigenesis, GIN was shown to be induced in skin cells at a precancerous stage and increased significantly with progression to cancer.
Materials and Methods
Skin tumor tissues. A total of 56 archival skin tissue samples, which were obtained from surgically excised specimens, were selected for this study from the archives of our department (Table 1). Histologically, the 56 primary skin tumors comprised the following: 20 SK arising from 10 sun‐exposed (including seven faces and three necks) and 10 non‐exposed sites (including four abdomens, three chests, two axillae, and one back); eight AK; nine BD; nine SCC; and 10 BCC. All samples were formalin‐fixed paraffin‐embedded tissues, from which sections were prepared for immunofluorescence studies. For normal controls, 14 samples of non‐exposed (including five abdomens, five backs, two ramps, one chest, and one axilla) and eight samples of sun‐exposed (including five faces, one neck, one hand, and one leg) normal epidermal cells surrounding tumor sections were evaluated.
Table 1.
Summary of subjects used in this study
| Histological type | n | Mean age (range) | M/F | Site (nonexposed/ sun‐exposed) |
|---|---|---|---|---|
| Normal epidermis | ||||
| non‐exposed | 14 | 60.8 (31–80) | 11/3 | 14/0 |
| sun‐exposed | 8 | 79.9 (66–88) | 3/5 | 0/8 |
| Seborrheic keratosis | ||||
| non‐exposed | 10 | 65.6 (58–90) | 10/0 | 10/0 |
| sun‐exposed | 10 | 77.9 (53–91) | 3/7 | 0/10 |
| Actinic keratosis | 8 | 79.5 (66–91) | 3/5 | 0/8 |
| Bowen's disease | 9 | 76.5 (66–88) | 3/6 | 1/8 |
| Squamous cell carcinoma | 9 | 82.3 (74–96) | 3/6 | 0/8 |
| Basal cell carcinoma | 10 | 73.3 (53–89) | 5/5 | 3/7 |
Immunofluorescence. After antigen retrieval with microwave treatment in citrate buffer, deparaffinized sections were preincubated with 10% normal goat serum. Tissue sections were then reacted with anti‐53BP1 rabbit polyclonal antibody (Bethyl Laboratories, Montgomery, TX, USA) at a 1:200 dilution. The slides were subsequently incubated with Alexa Fluor 488‐conjugated goat anti‐rabbit antibody (Invitrogen, Carlsbad, CA, USA). Specimens were counterstained with DAPI‐I (Vysis, Downers Grove, IL, USA), and were visualized and photographed using a fluorescence microscope (Zeiss Axioplan2; Carl Zeiss Japan, Tokyo, Japan) equipped with a charge coupled device (CCD) camera, and then analyzed with IPLab/MAC image software (Scanalytics, Fairfax, VA, USA). Signals were analyzed in 10 viewing areas per case at a 1000‐fold magnification.
Evaluation of immunofluorescence results. As described in our previous report,( 17 ) the pattern of 53BP1 immunoreactivity was classified into four types: (1) stable type: faint and diffuse nuclear staining; (2) low DDR type: one or two discrete nuclear foci; (3) high DDR type: three or more discrete nuclear foci; and (4) abnormal type: intense heterogeneous nuclear staining, occasionally, with several small foci. The percentage of epidermal or tumor cells expressing each type of 53BP1 immunoreactivity in each viewing area was graded into the following four groups: (1) negative: 0 to less than 5%; (2) low: 5% to less than 30%; (3) medium: 30% to 60%; and (4) high: more than 60%. The type of 53BP1 expression pattern in each case was determined by the predominant expression pattern.
Statistical analysis. The Mann–Whitney test was used to assess differences in the type of 53BP1 expression between non‐exposed and exposed epidermal cells. Spearman's correlation coefficient by rank test was used to assess correlation between histological type of skin tumors and type of 53BP1 expression. A P‐value of less than 0.05 was considered statistically significant.
Double‐labeled immunofluorescence. To assess the colocalization of 53BP1‐foci formation and p53 expression, double‐labeled immunofluorescence was performed. We also carried out a double‐labeled immunofluorescence of 53BP1 and Ki‐67 expression to clarify the association of type of 53BP1 expression and cycling tumor cells. In double staining, tissues were incubated with a mixture of rabbit anti‐53BP1 antibody and mouse anti‐p53 monoclonal antibody (DO7; Dako, Glostrup, Denmark) at a 1:200 dilution or mouse anti‐Ki‐67 monoclonal antibody (MIB‐1; Dako) at a 1:50 dilution, and subsequently incubated with a mixture of Alexa Fluor 488‐conjugated goat anti‐rabbit antibody and Alexa Fluor 546‐conjugated goat anti‐mouse antibody. Specimens were counterstained with DAPI‐I (Vysis), and were visualized and photographed using a fluorescence microscope (Zeiss Axioplan2) equipped with a CCD camera, and then analyzed with IPLab/MAC image software (Scanalytics). Signals were analyzed at a 1000‐fold magnification.
Results
53BP1 expression in normal epidermis surrounding tumors. Of the 14 controls consisting of nonexposed normal epidermis, 13 cases (92.9%) expressed only stable type cells (Fig. 1a), while one case (7.1%) showed stable type keratinocytes but also included a small number (up to 10%) of low DDR type in the basal layer. Of the other eight controls consisting of sun‐exposed normal epidermis, two cases (20%) expressed only stable type cells, while five cases (62.5%) showed stable type in more than 70% of keratinocytes but also had up to 30% of low DDR type (Fig. 1b) in the basal layer. One other case (12.5%) also showed stable type in more than 70% of keratinocytes, but included up to 30% of high DDR type in the basal layer.
Figure 1.
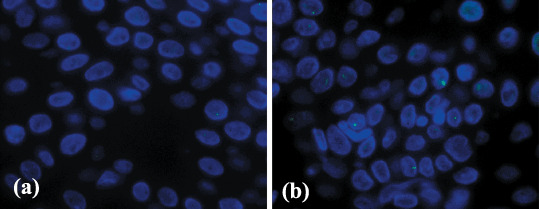
Immunofluorescence of p53‐binding protein 1 (53BP1) expression in the normal epidermis surrounding tumor sections. (a) The non‐exposed epidermis showed a stable type staining and rarely one nuclear focus. (b) Sun‐exposed epidermis occasionally showed one discrete 53BP1 nuclear focus at the basal layer.
53BP1 expression in skin tumors. The results of the immunofluorescence of staining patterns for 53BP1 in each histological type of skin tumors are presented in Table 2. Similar to the pattern observed for normal epidermal cells, all 10 of the non‐exposed SK cases expressed only the stable type (Fig. 2a). Of the 10 sun‐exposed SK cases, four (40%) expressed only the stable type, while six (60%) showed stable type in more than 70% of tumor cells, but also up to 30% of low DDR type (Fig. 2b) mainly in the basal layer.
Table 2.
Results for type of p53‐binding protein 1 (53BP1) expression in skin tumors by immunofluorescence
| n | Stable | Low DDR | High DDR | Mixed DDR and abnormal | Abnormal | |
|---|---|---|---|---|---|---|
| Epidermis | ||||||
| non‐exposed | 14 | 13 (92.9%) | 1 (7.1%) | 0 | 0 | 0 |
| sun‐exposed | 8 | 2 (25.0%) | 5 (62.5%) | 1 (12.5%) | 0 | 0 |
| SK | ||||||
| non‐exposed | 10 | 10 | 0 | 0 | 0 | 0 |
| sun‐exposed | 10 | 4 (40.0%) | 6 (60.0%) | 0 | 0 | 0 |
| AK | 8 | 1 (12.5%) | 3 (37.5%) | 4 (50.0%) | 0 | 0 |
| BD | 9 | 1 (11.1%) | 2 (22.2%) | 4 (44.4%) | 2 (22.2%) | 0 |
| SCC | 9 | 0 | 0 | 0 | 4 (44.4%) | 5 (55.6%) |
| BCC | 10 | 0 | 0 | 0 | 1 (10.0%) | 9 (90.0%) |
DDR, DNA damage response; SK, seborrheic keratosis; AK, actinic keratosis; BD, Bowen's disease; SCC, squamous cell carcinoma; BCC, basal cell carcinoma.
Figure 2.
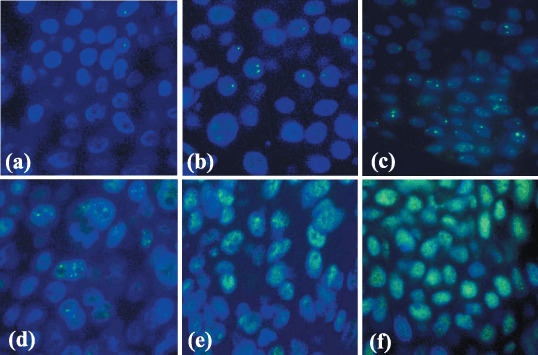
Immunofluorescence of p53‐binding protein 1 (53BP1) expression in human skin tumors. (a) Seborrheic keratosis (SK) in the non‐exposed skin expressed stable type staining with rarely one nuclear focus (stable type), (b) whereas SK in the sun‐exposed skin expressed an occasional one or two nuclear foci (low DNA damage response (DRR) type). (c) Actinic keratosis showed three or more discrete nuclear foci in dysplastic cells (high DRR type). (d) Bowen's disease showed several discrete nuclear foci mixed with intense and heterogeneous nuclear staining (mixed DRR and abnormal type). (e) Squamous cell carcinoma as well as (f) basal cell carcinoma exhibited intense and heterogeneous nuclear staining (abnormal type).
In contrast to the normal epidermis and SK, of the eight AK cases, four cases (50%) and three cases (37.5%) showed high DDR (Fig. 2c) and low DDR types, respectively, in dysplastic cells, while only one case (12.5%) expressed the stable type. In the nine BD cases, two cases (22.2%) showed low DDR type, four cases (44.4%) were of high DDR type, and two cases (22.2%) were of mixed high DDR and abnormal type (Fig. 2d), while only one case (11.1%) expressed the stable type. Of the nine SCC cases, four (44.4%) were of mixed low DDR and abnormal type and five (55.6%) were of the abnormal type (Fig. 2e). Finally, of the 10 BCC cases, one case (10%) was of mixed low DDR and abnormal type, and nine cases (90%) were of the abnormal type (Fig. 2f).
The Mann–Whitney test revealed that DDR expression of 53BP1 was significantly higher in the sun‐exposed epidermis than in the non‐exposed epidermis (P = 0.001). Furthermore, Spearman's analysis revealed that the histological type of skin tumors was significantly correlated with type of 53BP1 expression (P < 0.001).
Double‐labeled immunofluorescence of 53BP1 and p53 expressions ( Fig. 3 )/Ki‐67 expressions ( Fig. 4 ). The normal epidermis and SK expressed the stable or a low DDR type of 53BP1 immunoreactivity and only a few Ki‐67 nuclear stainings at the basal layer but no p53 nuclear staining. Discrete nuclear foci in a high DDR type of 53BP1 immunoreactivity were observed and were colocalized to dysplastic cells exhibiting p53 nuclear staining at the basal layer in AK. In BD, p53 nuclear staining was sparsely found in cancer cells which were distributed throughout the epidermal layer, and discrete nuclear foci of high DDR type of 53BP1 immunostaining were colocalized to p53‐positive cancer cells. In SCC and BCC, high levels of abnormal type of 53BP1 and strong p53 immunoreactivity were observed in nuclei of cancer cells, and cells expressing the abnormal type of 53BP1 immunoreactivity were randomly distributed in lesions. Intense 53BP1 staining was not always colocalized with p53 overexpression in cancer cells. Furthermore, double staining of 53BP1 and Ki‐67 demonstrated that discrete nuclear foci of 53BP1 immunostaining were not colocalized to Ki‐67‐positive dysplastic/cancer cells in AK/BD; whereas abnormal type of 53BP1 staining frequently expressed Ki‐67 nuclear staining.
Figure 3.
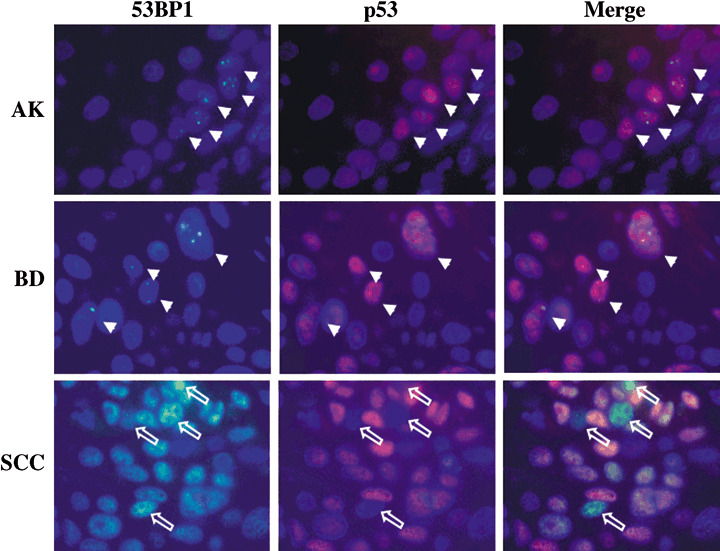
Double‐labeled immunofluorescence for p53‐binding protein 1 (53BP1) and p53 expression. Actinic keratosis (AK) showed colocalization of discrete 53BP1 nuclear foci and p53 nuclear staining in dysplastic cells at the basal layer, suggesting an activation of DNA damage response (DDR). Bowen's disease (BD) also showed colocalization of discrete 53BP1 nuclear foci and p53 nuclear staining in dysplastic cells including dispersed plump cells. Squamous cell carcinoma (SCC) exhibited intense and heterogeneous nuclear staining of both 53BP1 and p53 immunoreactivity; however, intense 53BP1 staining was not always colocalized with p53 overexpression, suggesting a disruption of the DDR pathway. Arrows indicate colocalization of 53BP1 nuclear foci and p53 staining in both AK and BD. Open arrows indicate cancer cells showing intense 53BP1 staining with no p53 staining in SCC.
Figure 4.
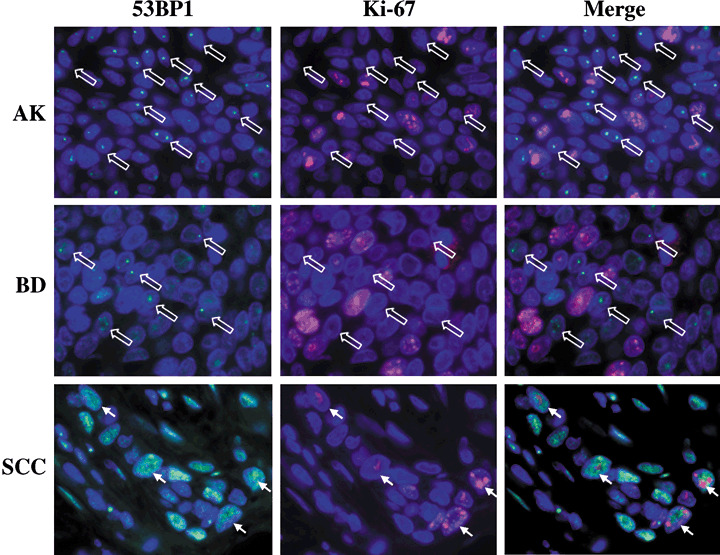
Double‐labeled immunofluorescence for p53‐binding protein 1 (53BP1) and Ki‐67 expression. Actinic keratosis (AK) showed no colocalization of discrete 53BP1 nuclear foci and Ki‐67 nuclear staining at the basal layer. Bowen's disease (BD) also showed independent discrete 53BP1 nuclear foci from Ki‐67 nuclear staining in cancer cells. Squamous cell carcinoma (SCC) occasionally exhibited intense and heterogeneous nuclear staining of both 53BP1 and Ki‐67 immunoreactivity, suggesting a proliferating ability through a disruption of the DDR pathway. Open arrows indicate cells showing 53BP1 nuclear foci with no Ki‐67 staining in both AK and BD. Arrows indicate colocalization of intense 53BP1 staining and Ki‐67 staining in SCC.
Discussion
Development of SCC of the skin is viewed as a multistep process, while BCCs are believed to develop de novo.( 20 ) In skin carcinogenesis, AK is a well established precancerous skin lesion and it has been suggested that ~10% of these sun‐induced lesions will develop into SCC.( 21 ) BD, also known as CIS, represents a preinvasive stage of SCC. The present study demonstrated apparent differences in 53BP1 staining patterns during human skin tumorigenesis, as in the following: SK/benign tumor, AK/precancerous lesion, BD/CIS, and SCC or BCC/invasive cancer. The number of discrete immunoreactive nuclear foci in DDR type of 53BP1 in the epidermis seems to increase in precancerous lesions. Furthermore, the abnormal type of 53BP1 immunoreactivity was restricted to malignancies including both CIS and invasive cancers. Similar results, which showed the differences in 53BP1 expression patterns during carcinogenesis, were also obtained in our recent study on thyroid tumors resected from patients.( 17 ) Therefore, we submit that immunofluorescence analysis of 53BP1 expression can be a useful tool to estimate the level of GIN and, simultaneously, the malignant potential of human tumors such as skin and thyroid tumors.
Interestingly, the low DDR type of 53BP1 immunoreactivity was very rare in the non‐exposed epidermis but frequently found in sun‐exposed skin. Similarly, sun‐exposed SK/benign skin tumors also frequently showed a low DDR type of 53BP1 immunoreactivity, but none of the nonexposed SK cases expressed the DDR type of 53BP1 immunoreactivity. Because the skin is the primary barrier for humans against the external environment, sun‐exposed epidermis is continuously exposed to a low level of physical and chemical genotoxic agents such as UV and IR. Thus, a low DDR type of 53BP1 immunoreactivity in the sun‐exposed epidermis may represent a minor genotoxic injury induced by external environmental factors.
P53 is activated by DDR‐associated molecules, and is essential to control GIN and to suppress tumorigenesis.( 22 ) The p53 mutation is the most prominent aberration in skin cancers, and it is now established that ~50% of all skin cancers show p53 mutations.( 23 ) Double‐labeled immunofluorescence was carried out to evaluate the association between the type of 53BP1 expression and p53 nuclear staining. In this study, although no p53 staining was observed in the epidermis and SK even at the sun‐exposed sites which showed a low DDR type of 53BP1 immunoreactivity, AK showed both p53 nuclear staining and a high DDR type of 53BP1 immunoreactivity in dysplastic cells at the basal layer in the epidermis. The colocalization of p53 nuclear staining and 53 BP 1 nuclear foci was also observed in BD. P53 mutations have been found in precanceous epidermis,( 24 , 25 , 26 ) and can be detected by immunohistochemistry. Indeed, 70% of immunohistochemical detections were shown to have an underlying mutation in the p53 gene.( 25 , 27 ) Because of the loss of p53 function based on gene mutation, environmental genotoxic agents may easily induce DSB in precancerous lesion/CIS in skin. Alternatively, because this study also demonstrated no colocalization of DDR type 53BP1 expression and Ki‐67 nuclear staining as a marker for cycling cells in AK and BD, activated DDR induced by genotoxic factors may activate p53 function to induce cell‐cycle arrest in injured cells. Furthermore, a high level of the abnormal type of 53BP1 immunoreactivity was restricted to invasive cancer and was significantly associated with p53 overexpression reflecting mutation. However, cancer cells exhibiting an abnormal type of 53BP1 staining were colocalized with Ki‐67 staining but not always colocalized with p53 staining. Thus, cancer cells expressing strong p53 immunoreactivity were cycling cells, suggesting a loss of p53 function and a disruption of the DDR pathway; whereas AK and BD cells exhibiting both 53BP1 nuclear foci and weak p53 staining were non‐cycling cells, suggesting a p53 function through the activated DDR pathway. Taken together, these findings may indicate that GIN may have already occurred at the precancerous stage during skin tumorigenesis, and that a loss of control of GIN based on p53 mutations may allow further accumulation of other genomic alterations to progress to invasive cancer through acceleration of cell proliferating.
In summary, this study demonstrated a number of nuclear 53BP1 foci in skin tumors resected from patients, which were similar to those found in irradiated cells, suggesting a constitutive activation of DDR in skin cancer cells. Because AK showed a high DDR type of 53BP1 immunoreactivity which colocalized with p53 nuclear staining as well as BD, GIN seems to be induced at the precancerous stage. Furthermore, invasive cancers exhibited the abnormal type of 53BP1 immunoreactivity with an underlying mutation in the p53 gene and an increased proliferation, suggesting disrupted DDR subsequently leading a high level of GIN in cancer cells. This study proposed that GIN has a crucial role in the progression of skin carcinogenesis. Immunofluorescence analysis of 53BP1 expression of various tumor tissues should be performed to clarify the significance of 53BP1 staining pattern as a common histological marker to estimate the malignant potential of human tumors.
Supporting information
Table S1. Odds ratio (OR) and 95% confidence interval (CI) for the incidence of DNA damage response expression of 53BP1 in both normal epidermis and seborrheic keratosis samples.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported in part through Nagasaki University Global Center of Excellence program ‘Global Strategic Center for Radiation Health Risk Control’ and by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. (No. 18590334).
Brief statements: Defective DNA damage response (DDR) can result in genomic instability (GIN) which is considered to be a central aspect of any carcinogenic process. P53‐binding protein 1 (53BP1) belongs to a family of evolutionarily conserved DDR proteins. This study demonstrated a number of nuclear 53BP1 foci in human skin tumorigenesis, suggesting a constitutive activation of DDR in skin cancer cells. The results of this study suggest that GIN has a crucial role in the progression of skin carcinogenesis.
References
- 1. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396: 643–9. [DOI] [PubMed] [Google Scholar]
- 2. Coleman WB, Tsongalis GJ. The role of genomic instability in human carcinogenesis. Anticancer Res 1999; 19: 4645–64. [PubMed] [Google Scholar]
- 3. Miyazawa T, Sato H, Hatakeyama K, Kitagawa T, Kominami R. Allelic losses in mouse skin tumors induced by gamma‐irradiation of p53 heterozygotes. Jpn J Cancer Res 2002; 93: 994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sadamori N, Mine M, Hori M. Skin cancer among atom bomb survivors. Lancet 1989; 1: 1267. [DOI] [PubMed] [Google Scholar]
- 5. Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage‐responsive cell cycle checkpoint proteins. FASEB J 1997; 11: 68–76. [PubMed] [Google Scholar]
- 6. Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes Dev 2002; 16: 583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem 2003; 278: 19579–82. [DOI] [PubMed] [Google Scholar]
- 8. Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double‐strand breaks. J Cell Biol 2000; 151: 1381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage‐signaling pathways. J Cell Biol 2001; 153: 613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 2001; 21: 1719–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xia Z, Morales JC, Dunphy WG, Carpenter PB. Negative cell cycle regulation and DNA damage‐inducible phosphorylation of the BRCT protein 53BP1. J Biol Chem 2001; 276: 2708–18. [DOI] [PubMed] [Google Scholar]
- 12. Shiloh Y, Kastan MBATM. genome stability, neuronal development, and cancer cross paths. Adv Cancer Res 2001; 83: 209–54. [DOI] [PubMed] [Google Scholar]
- 13. Xu Y, Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev 1996; 10: 2401–10. [DOI] [PubMed] [Google Scholar]
- 14. Xu Y, Yang EM, Brugarolas J, Jacks T, Baltimore D. Involvement of p53 and p21 in cellular defects and tumorigenesis in Atm‐/‐ mice. Mol Cell Biol 1998; 18: 4385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mochan TA, Venere M, DiTullio RA Jr, Halazonetis TD. 53BP1 and NFBD1/MDC1‐Nbs1 function in parallel interacting pathways activating ataxia‐telangiectasia mutated (ATM) in response to DNA damage. Cancer Res 2003; 63: 8586–91. [PubMed] [Google Scholar]
- 16. Rakhorst HA, Tra WM, Posthumus‐Van Sluijs ST et al . Quantitative analysis of radiation‐induced DNA break repair in a cultured oral mucosal model. Tissue Eng 2006; 12: 3395–403. [DOI] [PubMed] [Google Scholar]
- 17. Nakashima M, Suzuki K, Meirmanov S et al . Foci formation of p53‐binding protein 1 in thyroid tumors: activation of genomic instability during thyroid carcinogenesis. Int J Cancer 2008; 122: 1082–8. [DOI] [PubMed] [Google Scholar]
- 18. Suzuki K, Yokoyama S, Waseda S, Kodama S, Watanabe M. Delayed reactivation of p53 in the progeny of cells surviving ionizing radiation. Cancer Res 2003; 63: 936–41. [PubMed] [Google Scholar]
- 19. Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer 2002; 2: 331–41. [DOI] [PubMed] [Google Scholar]
- 20. Boukamp P. Non‐melanoma skin cancer: what drives tumor development and progression? Carcinogenesis 2005; 26: 1657–67. [DOI] [PubMed] [Google Scholar]
- 21. Johnson TM, Rowe DE, Nelson BR, Swanson NA. Squamous cell carcinoma of the skin (excluding lip and oral mucosa). J Am Acad Dermatol 1992; 26: 467–84. [DOI] [PubMed] [Google Scholar]
- 22. DiTullio RA Jr, Mochan TA, Venere M et al . 53BP1 functions in an ATM‐dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol 2002; 4: 998–1002. [DOI] [PubMed] [Google Scholar]
- 23. Giglia‐Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat 2003; 21: 217–28. [DOI] [PubMed] [Google Scholar]
- 24. Nakazawa H, English D, Randell PL et al . UV and skin cancer: specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc Natl Acad Sci USA 1994; 91: 360–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jonason AS, Kunala S, Price GJ et al . Frequent clones of p53‐mutated keratinocytes in normal human skin. Proc Natl Acad Sci USA 1996; 93: 14025–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ling G, Persson A, Berne B, Uhlen M, Lundeberg J, Ponten F. Persistent p53 mutations in single cells from normal human skin. Am J Pathol 2001; 159: 1247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ren ZP, Ponten F, Nister M, Ponten J. Two distinct p53 immunohistochemical patterns in human squamous‐cell skin cancer, precursors and normal epidermis. Int J Cancer 1996; 69: 174–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Odds ratio (OR) and 95% confidence interval (CI) for the incidence of DNA damage response expression of 53BP1 in both normal epidermis and seborrheic keratosis samples.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item