

Abstract
Malignant mesothelioma (MM) is a tumor with poor prognosis associated with asbestos exposure. While it remains to be clarified how asbestos fibers confer genetic/epigenetic alterations and induce cellular transformation in normal mesothelial cells, the understanding of key molecular mechanisms of MM cell development, proliferation, and invasion has progressed. MM shows frequent genetic inactivation of tumor suppressor genes of p16 INK4a /p14 ARF and neurofibromatosis type 2 (NF2) which encodes Merlin, and epigenetic inactivation of RASSF1A. However, no frequent mutations of well‐known oncogenes such as K‐RAS and PIK3CA have been identified. Activation of multiple receptor tyrosine kinases including the epidermal growth factor receptor (EGFR) family and MET, and subsequent deregulations of mitogen‐activated protein kinase (MAPK) and phosphatidylinositol‐3‐kinase (PI3K)–AKT signaling cascades are frequently observed in most MM cells. The tumor suppressive function of Merlin in MM cells is also being investigated by dissecting its possible downstream signaling cascade called the Hippo pathway. Further comprehensive delineation of dysregulated signaling cascades in MM cells will lead to identification of key addiction pathways for cell survival and proliferation of MM cells, which strongly promote establishment of a new molecular target therapy for MM. (Cancer Sci 2009)
Malignant mesothelioma (MM) is an aggressive neoplasm which arises primarily in the pleural or peritoneal cavity.( 1 , 2 ) In most patients with MM, a clinically overt tumor is diagnosed within 30–40 years after exposure to asbestos, indicating a long latency for tumor development.( 3 ) About 80% of MM develops in the pleura, 20% in the peritonea, and less than 1% in the pericardium. Pathologically, epithelial type accounts for 60%, sarcomatous type for 20%, and biphasic type with both components ranges around 20%. Since patients with MM are usually diagnosed at advanced stages and MM is largely unresponsive to conventional therapy, the prognosis of patients with MM is very poor. The median survival of patients with malignant pleural mesothelioma (MPM) is 9 to 12 months after diagnosis, regardless of the recent advancement of chemotherapeutical modalities combining cisplatin and antifolate such as pemetrexed or raltitrexed.( 4 , 5 )
Since MM is a relatively rare malignancy and early preneoplastic lesions are difficult to identify clinically, the understanding of molecular pathogenesis including sequential accumulation of genetic/epigenetic alterations for MM development has lagged behind other common malignancies. While several distinct cell signaling pathways including p16INK4a/p14ARF are altered in most MM similar to other common tumors, the existence of NF2 mutation suggests that mesothelial cells need rather unique dysregulation of cell regulatory mechanisms for malignant transformation. Despite the difficulties in approaching early lesions and characterizing normal mesothelial cells, many research approaches now have been revealing the underlying mechanisms and key events in MM development, utilizing in vitro systems of cell lines and mouse models of MM. Such novel knowledge is expected to be applied to the development of new diagnostic tools and molecular target therapies.( 6 , 7 )
Genetic and cellular damage induced by asbestos
A clear link has been established between asbestos exposure and MM development.( 1 ) There are two subgroups of asbestos: (i) the amphiboles, a group of rod‐like fibers including amosite (brown asbestos), crocidolite (blue asbestos), anthophyllite, actinolite, and tremolite; and (ii) the serpentine group, consisting of chrysotile (white asbestos). The association between amphibole asbestos exposure and MM development is well accepted. In particular, crocidolite is considered to be the most oncogenic type of asbestos. Crysotile is the most common type of asbestos, accounting for ∼90% of the world’s asbestos production. It is still controversial whether chryotile causes MM.
It remains unclear whether asbestos fibers act directly on mesothelial cells or indirectly cause mesothelioma.( 1 ) Compared to other cell types, human mesothelial cells are very susceptible to asbestos cytotoxicity, which raises a paradoxical issue of how asbestos causes MM if human mesothelial cells exposed to asbestos die.( 8 ) There are several possible mechanisms as to how asbestos fibers cause MPM.( 1 , 9 ) First, long and thin asbestos fibers can be inhaled deeply into the lungs and can penetrate them. Asbestos fibers repeatedly scratch the mesothelial surface and cause prolonged cycles of damage, repair, and local inflammation, thus inducing pleural irritation. Second, asbestos fibers can also physically interfere with the mitotic process of the cell cycle via disrupting the mitotic spindle, which may result in chromosomal structural abnormalities and aneuploidy of mesothelial cells. Third, asbestos fibers generate highly reactive oxygen species (ROS) and reactive nitrogen species (RNS), which lead to DNA damage and strand breaks. Finally, asbestos can induce cytokines and growth factors in exposed mesothelial cells and nearby macrophages. Those include tumor necrosis factor‐α (TNF‐α), transforming growth factor‐β (TGF‐β), and platelet‐derived growth factor (PDGF). Transcription factors such as nuclear factor kappa B (NF‐κB) and activator protein‐1 (AP‐1) were shown to be induced, which also play a critical role in the promotion and progression of mesotheliomas. In addition, crocidolite asbestos was shown to induce autophosphorylation of epidermal growth factor receptor (EGFR) in rat mesothelial cells.( 10 )
p16INK4a/p14ARF inactivation in MM
The most frequently inactivated tumor suppressor gene (TSG) in human MM is p16 INK4a /p14 ARF , while only 20–25% of MMs show a mutation of p53, which is the most frequently inactivated TSG in human malignancies. Immunohistochemical analysis showed frequent down‐regulation of p16INK4a in primary MM.( 11 ) With the fluorescence in situ hybridization (FISH) analysis of primary MM samples or MM cells cultured for less than 5 days, 50–70% showed homozygous deletions of the p16 INK4a /p14 ARF locus.( 12 ) Using established MPM cell lines, ∼90% cell lines had a homozygous deletion of the p16 INK4a /p14 ARF gene locus.( 13 , 14 ) The p16 INK4a gene product controls the cell cycle via the cyclin‐dependent kinase 4 (CDK4)/cyclin D‐RB pathway, while the p14 ARF gene product regulates p53 through inactivation of human homolog of mouse double minute 2 (HDM2), which is an upstream regulator of p53. Thus, the homozygous deletion of p16 INK4a /p14 ARF indicates the inactivation of two major tumor suppressing pathways of RB and p53 in the cell.
A recent study showed that mice deficient for Arf, but not p16 INK4a , were susceptible to accelerated asbestos‐induced MM, indicating that Arf inactivation has a significant role in driving MM pathogenesis.( 15 ) MMs arising in Arf (+/−) consistently exhibited biallelic inactivation of Arf. Furthermore, in the developed tumors in Arf (+/−) mice, FAS‐associated factor 1 (Faf1) was down‐regulated, which was implicated in leading to TNF‐α/NF‐κB signaling activation.( 15 )
NF2 inactivation and Hippo signaling pathway dysregulation in MM
The neurofibromatosis type 2 (NF2) gene responsible for NF2 familial cancer syndrome was shown to be the target gene of 22q12 loss in MM.( 16 , 17 ) The characteristic tumors of this syndrome are vestibular schwannoma and meningioma,( 18 ) while there has been no report that the NF2 patient has a higher MM susceptibility. The NF2 gene is inactivated by homozygous deletion, nonsense mutation, or missense mutation in 40–50% of MMs.( 16 , 17 ) However, it remains unclear whether MM tumors without an NF2 mutation express functional Merlin, a translation product of NF2, and whether the other molecules of Merlin‐associated signaling cascades are altered. One recent study suggested that splicing variants of NF2 also affect the protein product in MM with wild‐type NF2,( 19 ) while another suggested that upregulation of microRNA such as hsa‐miR‐885‐3p might target NF2.( 20 ) In an animal model, Nf2 (+/−) knockout mice were shown to develop MPMs in the earlier stage and more frequently after asbestos exposure.( 21 ) Remarkably, similar to human MM, tumors from Nf2 (+/−) mice showed frequent homozygous deletions of the p16 Ink4a /p19 Arf locus and adjacent p15 Ink4b tumor suppressor gene.( 21 ) In addition, mesotheliomas were shown to develop at higher incidence in Nf2:Ink4a/Arf conditional knockout mice, with median survival times of approximately 30 weeks.( 22 )
Merlin is a membrane‐cytoskeleton‐associated protein with a FERM (four‐point‐one, ezrin, radixin, and moesin) domain, and is known to interact with 34 proteins, including CD44, ezrin radixin moesin (ERM) proteins, Na+/H+ exchanger regulatory factor (NHERF), and p21‐activated kinase 1 (PAK1).( 23 ) With phosphorylation at serine 518 with PAK1 and dephosphorylation with myosin phosphatase targeting subunit 1‐protein phosphatase 1δ (MYPT‐1‐PP1δ), which is inhibited by 17‐kDa protein kinase C potentiated inhibitor (CPI‐17),( 24 ) Merlin switches between an open and closed form; the latter is dephosphorylated and active as a tumor suppressor (Fig. 1).
Figure 1.
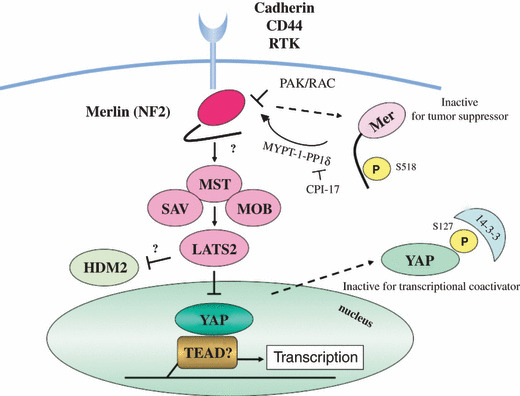
Neurofibromatosis type 2 (NF2) gene product, Merlin, and mammalian Hippo signaling cascade. Signals from the extracellular environment which are transduced via cell–cell contact (cadherin), cell–matrix contact (CD44), or growth factors (receptor tyrosine kinases, RTKs), affect the activity of Merlin. One of the downstream cascades regulated by activated Merlin is called the Hpo (Hippo) cascade, which suppresses the activity of YAP transcriptional coactivator. When Merlin is inactivated in malignant mesothelioma (MM) cells, transcription of growth promoting genes is induced with YAP and transcription factor TEAD.
The mammalian Hippo cascade, which was initially identified via genetic studies in Drosophila,( 25 ) is one of the possible downstream signaling cascades of Merlin (Fig. 1). The downstream molecules in this pathway include WW45 (also called Salvador), MST (Drosophila Hippo), LATS, and YAP.( 26 ) Noticeably, amplification of chromosomal 11q22 region including YAP was identified in a subset of MM specimens ( 14 ) and YAP was shown to play a positive role in cell proliferation and survival in MM cells.( 27 ) Exogenous NF2 transduction enhanced YAP phosphorylation and translocation from the nucleus to cytoplasm in MM cells, thus inactivating transcriptional coactivator activity of YAP.( 27 ) While genetically engineered mice of YAP also showed an increase in organ size,( 28 ) the genetic alterations of components of the Hippo pathway have also been reported in other human malignancies (Table 1). Interestingly, YAP is also known to act both for positive cell proliferation and apoptosis, which is thought to be dependent on different cell contexts.( 29 ) Recent findings indicate that one of the key contexts is distinct expression of transcription factors such as TEAD, p73, or RUNX.( 30 ) Thus, to determine the precise effects caused by NF2 inactivation in MM cells, it is necessary to detect cooperative transcription factors and target genes of YAP in MM cells. In addition, a recent study suggested that loss of Merlin function activated integrin‐dependent mTORC1 signaling in MM cells and that Merlin‐null MM cell lines were sensitive to the growth inhibitory effect of rapamycin.( 31 )
Table 1.
Genetic alterations of neurofibromatosis type 2 (NF2) and Hippo pathway components in human malignancies
| Genes | Human malignancy | Types of alteration | References |
|---|---|---|---|
| NF2 (Merlin) | Neurofibromatosis type 2 Sporadic schwannoma,meningioma | HD, nonsense, missense | Baser( 18 ) |
| Mesothelioma | HD, nonsense, missense | Sekido et al. ( 16 ) Bianchi et al. ( 17 ) | |
| MST | Soft tissue sarcoma | Hypermethylation | Seidel et al. ( 78 ) |
| WW45 (Sav) | Renal cancer | HD | Tapon et al. ( 79 ) |
| LATS2 | Leukemia | Down‐regulation | Kawahara et al. ( 80 ) |
| Lung cancer | Missense, deletion | Strazisar et al. ( 81 ) | |
| YAP | Hepatocellular carcinoma | Overexpression | Xu et al. ( 82 ) |
| Colon, lung adenocarcinomas | Overexpression | Steinhardt et al. ( 83 ) | |
| Mesothelioma | Amplification | Yokoyama et al. ( 27 ) | |
| TAZ | Breast cancer | Overexpression | Chan et al. ( 84 ) |
HD, homozygous deletion.
Activation of PI3K/AKT and MAPK signaling cascades in MM
Constitutive activation of receptor tyrosine kinases (RTKs) plays a central role in cell survival and proliferation of malignant cells, which often result from gene amplification or activating mutation that does not require their ligand binding for activation. In MM, activation of RTK family members has been investigated, and frequent expression and phosphorylation (thus indicating activation) of MET or EGFR family members were identified in both primary tumors and cell lines of MM (Fig. 2).( 32 , 33 ) Although frequent mutation of any RTK genes has not yet been shown in MM,( 33 ) the downstream signaling cascades including the mitogen‐activated protein kinase (MAPK) and phosphatidylinositol‐3‐kinase (PI3K)‐AKT cascades are activated in most MMs.( 34 ) MMs with positive AKT phosphorylation status have also been shown to be phosphorylated (activated) mTOR (mammalian target of rapamycin), a downstream molecule of the AKT pathway.( 34 ) PTEN homozygous deletion was detected in a small subset of MM cell lines, which was also responsible for AKT activation.( 34 , 35 ) In addition, while K‐RAS mutation has not been reported in MM, one study showed activated mutation of N‐RAS in three of 38 MMs.( 36 )
Figure 2.
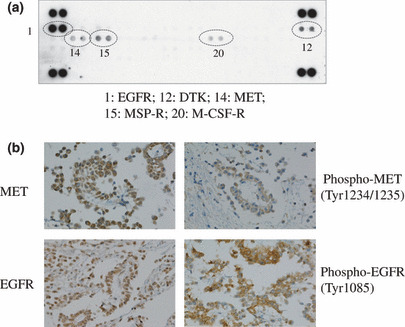
Co‐activation of multiple receptor tyrosine kinases (RTKs) in a primary malignant pleural mesothelioma (MPM) specimen. (a) A phospho‐RTK array (R&D Systems, Minneapolis, MN, USA) was performed using protein lysate extracted from surgically resected human MPM specimen (MMY‐39). Positive duplicated spots for individual RTK were numbered and indicated below the array. Five of 42 RTKs were scored positive. Two dots at each corner indicate positive controls. (b) Expression of MET and epidermal growth factor receptor (EGFR) and their phosphorylation (phospho‐MET and phospho‐EGFR) were detected with immunohistochemical analysis. DTK, developmental tyrosine kinase; MSP‐R, macrophage‐stimulating protein receptor; M‐CSF‐R, macrophage‐colony stimulating factor receptor.
Based on the observations of frequent activation of RTKs in MMs, small molecule inhibitors of RTKs were applied to clinical studies, but clear effectiveness was not observed.( 37 , 38 ) This less than expected responsiveness reflects the intrinsic resistance of MM cells against RTK inhibitors. As acquired secondary resistance, effectiveness of inhibitors against constitutive activation of a specific RTK is thought to be abrogated via either a new genetic change in the RTK gene or activation of other RTK. Indeed, phosphorylation of multiple RTKs was observed in most MM cell lines.( 39 ) Among them, MET and EGFR family members were the most frequently activated in 42 RTKs studied (Fig. 2). Expectedly, simultaneous treatment of MET and EGFR inhibitors induced stronger inhibition of cell growth than each.( 39 )
MM cell lines produce many other growth factors and cytokines, which are also other mechanisms to constitutively activate signaling cascades of cell survival and/or proliferation.( 1 ) PDGF, TGF‐β, and insulin‐like growth factor (IGF) have been well studied. Factors involving angiogenic pathways were also demonstrated to be expressed in MM cells, including vascular endothelial growth factors (VEGFs), fibroblast growth factors (FGFs), and interleukin (IL)‐8.( 40 ) In addition, the PI3K/MEK5/fos‐related antigen 1 pathway was also shown to be activated by HGF, a ligand for MET, for mesothelioma cell proliferation.( 41 )
Genome‐wide approaches for searching new target genes in MM
Since a relatively small number of cancer‐associated genes have been identified in MM, other as yet unidentified genes may well be responsible for its development. Allelotyping and karyotype analyses revealed nonrandom chromosomal abnormalities in MM cells,( 42 , 43 , 44 , 45 ) while subsequent studies revealed p16 INK4a /p14 ARF and NF2 as target genes for 9p and 22q loss, respectively. However, there still seem to be many chromosomal regions with gain, loss, or fusion, which may lead a gene or genes located in the target region to be constitutively active or inactive. For instance, a recent study with array‐based comparative genomic hybridization (CGH) analysis identified high copy gain at 1p32 in a subset of MMs, which includes the JUN protooncogene.( 14 ) JUN encodes a component of the transcription factor AP‐1 complex, which is formed via heterodimerization with FOS. Since crocidolite and chrysotile asbestos were previously shown to induce JUN and FOS expression in rat pleural mesothelial cells ( 46 ) and the JUN amplification was indeed identified in a subset of MM, it was suggested that JUN activation can play a major role in the development of the MM cells. To support this idea, JUN amplification was also detected in other types of human sarcoma, which suggested the role of JUN in acquisition of more aggressive and undifferentiated tumor phenotypes.( 47 ) Another recent study indicated a 18q12.1 gain which harbors the CDH2 gene that encodes N‐cadherin.( 48 ) It also showed that 9p21.3 was the most specific for the short‐term recurrence group.( 48 )
Expression profiling using microarray has also identified specific gene expression changes in MM compared with normal mesothelium, effects of fibers/asbestos exposure in mesothelial cells, and several new candidate oncogenes and TSGs of MM.( 49 , 50 , 51 , 52 ) Patient prognosis with MM and predictive resistance to therapy has also been shown to be different based on gene expression profiling.( 53 , 54 )
Aberrant epigenetic events, including DNA hypermethylation‐induced gene silencing, are well‐recognized as important contributors to carcinogenesis. Methylation‐induced TSG silencing has been observed in a number of studies of MM.( 55 , 56 ) Significantly higher lung asbestos burden was shown to be associated with promoter methylation of cell cycle control genes including RASSF1A in MPMs.( 57 ) Epigenetic profiling was also shown to distinguish MPMs from normal pleura.( 58 )
Finally, a recent study using microRNA microarray analysis detected 21 microRNAs which were differentially expressed in the MM tumor samples.( 20 ) Twelve of them were highly expressed in tumor samples, whereas nine were not expressed. The target genes of the microRNAs were predicted by computational approaches. This revealed that known MM‐associated TSGs including CDKN2A and NF2 and known MM‐associated oncogenes including JUN, HGF, and EGF were potential target genes of upregulated and downregulated microRNAs, respectively.( 20 ) Another study suggested that microRNA signatures were significantly different in the histopathological subtypes of MM.( 59 )
Genetic susceptibility to MM
While asbestos is the principal etiological factor of MM, different lines of evidence have suggested a role for genetic background in MM development like in other types of cancers. The evidence that only a minority of asbestos‐exposed subjects develop MM and the frequent report of familial clustering support the role of genetic predisposition to MM, although the interpretation of the latter studies may be complicated by the presence of common environmental exposures.( 60 , 61 ) Many genetic association studies were conducted to show whether polymorphisms in the genes involved in xenobiotic and oxidative metabolism or in DNA repair processes were associated with the etiology and pathogenesis of MM. Under the candidate gene approach, the most commonly studied polymorphism in MM and other asbestos‐associated diseases was glutathione‐S‐transferase (GST) M1 polymorphism, with the GSTM1 null genotype shown to have an increased risk of MM in a subset of studies.( 62 ) Results of the N‐acethyl‐transferase (NAT2) and manganese superoxide dismutase (MnSOD, also reported as SOD2) polymorphisms, remain conflicting due to the relatively small size of study groups.( 62 ) Thus, the use of high‐throughput techniques with larger sample sizes will help to identify the genetic predisposition of MM. Finally, some genetic background has also been indicated to have a role in determining susceptibility to mineral fiber carcinogenesis, specifically to erionite carcinogenesis in Cappadocia, Turkey, where MM was prevalent in certain families and absent in others in three small villages.( 63 )
New therapeutic modalities for MM
Although MM is a highly aggressive tumor and patient prognosis with advanced‐stage MM remains very poor, advances in the systemic treatment of this disease have emerged.( 7 , 64 ) Multicenter trials of neoadjuvant chemotherapy followed by extrapleural pneumonectomy and hemithoracic radiation were conducted recently,( 65 , 66 ) which reported that a radical multimodality approach was feasible and that patients completing all therapy had a more favorable prognosis.
Among a number of new tyrosine kinase inhibitors (TKIs) developed, some have showed dramatic responsiveness in several types of human malignancies including lung cancer and chronic myeloid leukemia. Although several TKIs effective for those malignancies were tested in MM, no satisfactory results were obtained thus far as described above. For example, a phase II study of the EGFR inhibitor, gefitinib, was conducted for 43 patients with previously untreated MM.( 37 ) Although 97% of patients with MM had EGFR overexpression, gefitinib was not active in MM and EGFR expression did not correlate with failure‐free survival. Imatinib, another TKI known to affect both Kit and PDGF‐α and ‐β receptors, showed a limited efficacy for MPM.( 38 ) However, new types of RTK inhibitors or their use in combination therapy may be more promising for treatment of MM patients since multiple RTKs are frequently activated in most MM cells.( 39 )
Meanwhile, other molecular target therapies have also been under investigation for MM treatment.( 7 ) Clinical investigations include inhibitors targeting the VEGF/VEGF receptor pathway, histone deacetylase inhibitors, proteosome inhibitors, and a cytotoxic ribonuclease.( 67 , 68 , 69 , 70 ) A recent microarray analysis found that more aggressive MPM expressed higher levels of Aurora kinases A and B,( 71 ) which are serine/threonine kinases with multiple roles in mitotic progression. Thus, the role of Aurora kinases may be of interest due to the recent development of their small‐molecule inhibitors. Other examples proposed are a small‐molecule inhibitor of TGF‐β type I receptor( 72 ) and all‐trans‐retinoic acid,( 73 ) which were shown to inhibit mesothelioma tumor growth in vivo. Finally, cell surface antigens expressed on MM cells have also been studied for novel cancer immunotherapy for MM. Those include anti‐mesothelin monoclonal antibody (mAb),( 74 , 75 ) recombinant immunotoxin against mesothelin, and anti‐CD26 mAb.( 76 )
Summary
MM is an aggressive malignancy caused by multiple genetic and epigenetic changes. Molecular biological studies have been determining the underlying key events responsible for the development of MPM, some of which may be directly caused by asbestos fibers. While requirement of such a long latency between asbestos exposure and development of MM tumor is still an enigma, it needs to be resolved not only for better understanding of the molecular pathogenesis of MM, but also for development of new prevention and early intervention modalities for individuals susceptible to MM. New animal models of MM and human MM cell lines are also being established to present more useful tools for development of new therapeutic modalities.( 21 , 22 , 77 ) Thus, the knowledge of fundamental abnormalities at the genetic/epigenetic, cellular, and tissue levels in MM cells will be of great help in developing future strategies for potential molecular target therapy as well as other therapeutic modalities such as immunotherapy.
Acknowledgments
This work was supported by a Special Coordination Fund for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science and Technology (H18‐1‐3‐3‐1). The author thanks Dr Hideki Murakami, Dr Makiko Fujii, Dr Yutaka Kondo, and Dr Hirotaka Osada for their helpful comments. The author regrets the lack of citations for many important observations in the text, but their omission is made necessary by restrictions on the preparation of review manuscripts.
References
- 1. Pass HI, Vogelzang N, Hahn S, Carbone M. Malignant pleural mesothelioma. Curr Probl Cancer 2004; 28: 93–174. [DOI] [PubMed] [Google Scholar]
- 2. Robinson BW, Lake RA. Advances in malignant mesothelioma. N Engl J Med 2005; 353: 1591–603. [DOI] [PubMed] [Google Scholar]
- 3. Carbone M, Kratzke RA, Testa JR. The pathogenesis of mesothelioma. Semin Oncol 2002; 29: 2–17. [DOI] [PubMed] [Google Scholar]
- 4. Vogelzang NJ, Rusthoven JJ, Symanowski J et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 2003; 21: 2636–44. [DOI] [PubMed] [Google Scholar]
- 5. Van Meerbeeck JP, Gaafar R, Manegold C et al. Randomized phase III study of cisplatin with or without raltitrexed in patients with malignant pleural mesothelioma: an intergroup study of the European Organisation for Research and Treatment of Cancer Lung Cancer Group and the National Cancer Institute of Canada. J Clin Oncol 2005; 23: 6881–9. [DOI] [PubMed] [Google Scholar]
- 6. Yang H, Testa JR, Carbone M. Mesothelioma epidemiology, carcinogenesis, and pathogenesis. Curr Treat Options Oncol 2008; 9: 147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsao AS, Wistuba I, Roth JA, Kindler HL. Malignant pleural mesothelioma. J Clin Oncol 2009; 27: 2081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu W, Ernst JD, Broaddus VC. Phagocytosis of crocidolite asbestos induces oxidative stress, DNA damage, and apoptosis in mesothelial cells. Am J Respir Cell Mol Biol 2000; 23: 371–8. [DOI] [PubMed] [Google Scholar]
- 9. Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet 2005; 366: 397–408. [DOI] [PubMed] [Google Scholar]
- 10. Faux SP, Houghton CE, Hubbard A, Patrick G. Increased expression of epidermal growth factor receptor in rat pleural mesothelial cells correlates with carcinogenicity of mineral fibres. Carcinogenesis 2000; 21: 2275–80. [DOI] [PubMed] [Google Scholar]
- 11. Kratzke RA, Otterson GA, Lincoln CE et al. Immunohistochemical analysis of the p16INK4 cyclin‐dependent kinase inhibitor in malignant mesothelioma. J Natl Cancer Inst 1995; 87: 1870–5. [DOI] [PubMed] [Google Scholar]
- 12. Xio S, Li D, Vijg J, Sugarbaker DJ, Corson JM, Fletcher JA. Codeletion of p15 and p16 in primary malignant mesothelioma. Oncogene 1995; 11: 511–5. [PubMed] [Google Scholar]
- 13. Cheng JQ, Jhanwar SC, Klein WM et al. p16 alterations and deletion mapping of 9p21‐p22 in malignant mesothelioma. Cancer Res 1994; 54: 5547–51. [PubMed] [Google Scholar]
- 14. Taniguchi T, Karnan S, Fukui T et al. Genomic profiling of malignant pleural mesothelioma with array‐based comparative genomic hybridization shows frequent non‐random chromosomal alteration regions including JUN amplification on 1p32. Cancer Sci 2007; 98: 438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Altomare DA, Menges CW, Pei J et al. Activated TNF‐alpha/NF‐kappaB signaling via down‐regulation of Fas‐associated factor 1 in asbestos‐induced mesotheliomas from Arf knockout mice. Proc Natl Acad Sci U S A 2009; 106: 3420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sekido Y, Pass HI, Bader S et al. Neurofibromatosis type 2 (NF2) gene is somatically mutated in mesothelioma but not in lung cancer. Cancer Res 1995; 55: 1227–31. [PubMed] [Google Scholar]
- 17. Bianchi AB, Mitsunaga SI, Cheng JQ et al. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc Natl Acad Sci U S A 1995; 92: 10854–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baser ME. The distribution of constitutional and somatic mutations in the neurofibromatosis 2 gene. Hum Mutat 2006; 27: 297–306. [DOI] [PubMed] [Google Scholar]
- 19. Thurneysen C, Opitz I, Kurtz S, Weder W, Stahel RA, Felley‐Bosco E. Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer 2009; 64: 140–7. [DOI] [PubMed] [Google Scholar]
- 20. Guled M, Lahti L, Lindholm PM et al. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma‐A miRNA microarray analysis. Genes Chromosomes Cancer 2009; 48: 615–23. [DOI] [PubMed] [Google Scholar]
- 21. Altomare DA, Vaslet CA, Skele KL et al. A mouse model recapitulating molecular features of human mesothelioma. Cancer Res 2005; 65: 8090–5. [DOI] [PubMed] [Google Scholar]
- 22. Jongsma J, Van Montfort E, Vooijs M et al. A conditional mouse model for malignant mesothelioma. Cancer Cell 2008; 13: 261–71. [DOI] [PubMed] [Google Scholar]
- 23. Scoles DR. The merlin interacting proteins reveal multiple targets for NF2 therapy. Biochim Biophys Acta 2008; 1785: 32–54. [DOI] [PubMed] [Google Scholar]
- 24. Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI‐17 through inhibition of a merlin phosphatase. Nature 2006; 442: 576–9. [DOI] [PubMed] [Google Scholar]
- 25. Hamaratoglu F, Willecke M, Kango‐Singh M et al. The tumour‐suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 2006; 8: 27–36. [DOI] [PubMed] [Google Scholar]
- 26. Zhao B, Lei QY, Guan KL. The Hippo‐YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol 2008; 20: 638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yokoyama T, Osada H, Murakami H et al. YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation. Carcinogenesis 2008; 29: 2139–46. [DOI] [PubMed] [Google Scholar]
- 28. Camargo FD, Gokhale S, Johnnidis JB et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 2007; 17: 2054–60. [DOI] [PubMed] [Google Scholar]
- 29. Downward J, Basu S. YAP and p73: a complex affair. Mol Cell 2008; 32: 749–50. [DOI] [PubMed] [Google Scholar]
- 30. Zhao B, Ye X, Yu J et al. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 2008; 22: 1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. López‐Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the Tumor Suppressor NF2, encoding Merlin, Constitutively Activates Integrin‐dependent mTORC1 Signaling. Mol Cell Biol 2009; 29: 4235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jagadeeswaran R, Ma PC, Seiwert TY et al. Functional analysis of c‐Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res 2006; 66: 352–61. [DOI] [PubMed] [Google Scholar]
- 33. Destro A, Ceresoli GL, Falleni M et al. EGFR overexpression in malignant pleural mesothelioma. An immunohistochemical and molecular study with clinico‐pathological correlations. Lung Cancer 2006; 51: 207–15. [DOI] [PubMed] [Google Scholar]
- 34. Altomare DA, You H, Xiao GH et al. Human and mouse mesotheliomas exhibit elevated AKT/PKB activity, which can be targeted pharmacologically to inhibit tumor cell growth. Oncogene 2005; 24: 6080–9. [DOI] [PubMed] [Google Scholar]
- 35. Suzuki Y, Murakami H, Kawaguchi K et al. Activation of the PI3K‐AKT pathway in human malignant mesothelioma cells. Mol Med Rep 2009; 2: 181–8. [DOI] [PubMed] [Google Scholar]
- 36. Thomas RK, Baker AC, Debiasi RM et al. High‐throughput oncogene mutation profiling in human cancer. Nat Genet 2007; 39: 347–51. [DOI] [PubMed] [Google Scholar]
- 37. Govindan R, Kratzke RA, Herndon JE II et al. Gefitinib in patients with malignant mesothelioma: a phase II study by the Cancer and Leukemia Group B. Clin Cancer Res 2005; 11: 2300–4. [DOI] [PubMed] [Google Scholar]
- 38. Mathy A, Baas P, Dalesio O, Van Zandwijk N. Limited efficacy of imatinib mesylate in malignant mesothelioma: a phase II trial. Lung Cancer 2005; 50: 83–6. [DOI] [PubMed] [Google Scholar]
- 39. Kawaguchi K, Murakami H, Taniguchi T et al. Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis 2009; 30: 1097–105. [DOI] [PubMed] [Google Scholar]
- 40. Galffy G, Mohammed KA, Dowling PA, Nasreen N, Ward MJ, Antony VB. Interleukin 8: an autocrine growth factor for malignant mesothelioma. Cancer Res 1999; 59: 367–71. [PubMed] [Google Scholar]
- 41. Ramos‐Nino ME, Blumen SR, Sabo‐Attwood T et al. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra‐1 pathway. Am J Respir Cell Mol Biol 2008; 38: 209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murthy SS, Testa JR. Asbestos, chromosomal deletions, and tumor suppressor gene alterations in human malignant mesothelioma. J Cell Physiol 1999; 180: 150–7. [DOI] [PubMed] [Google Scholar]
- 43. Taguchi T, Jhanwar SC, Siegfried JM, Keller SM, Testa JR. Recurrent deletions of specific chromosomal sites in 1p, 3p, 6q, and 9p in human malignant mesothelioma. Cancer Res 1993; 53: 4349–55. [PubMed] [Google Scholar]
- 44. Balsara BR, Bell DW, Sonoda G et al. Comparative genomic hybridization and loss of heterozygosity analyses identify a common region of deletion at 15q11.1‐15 in human malignant mesothelioma. Cancer Res 1999; 59: 450–4. [PubMed] [Google Scholar]
- 45. Krismann M, Muller KM, Jaworska M, Johnen G. Molecular cytogenetic differences between histological subtypes of malignant mesotheliomas: DNA cytometry and comparative genomic hybridization of 90 cases. J Pathol 2002; 197: 363–71. [DOI] [PubMed] [Google Scholar]
- 46. Heintz NH, Janssen YM, Mossman BT. Persistent induction of c‐fos and c‐jun expression by asbestos. Proc Natl Acad Sci U S A 1993; 90: 3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mariani O, Brennetot C, Coindre JM et al. JUN oncogene amplification and overexpression block adipocytic differentiation in highly aggressive sarcomas. Cancer Cell 2007; 11: 361–74. [DOI] [PubMed] [Google Scholar]
- 48. Ivanov SV, Miller J, Lucito R et al. Genomic events associated with progression of pleural malignant mesothelioma. Int J Cancer 2009; 124: 589–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singhal S, Wiewrodt R, Malden LD et al. Gene expression profiling of malignant mesothelioma. Clin Cancer Res 2003; 9: 3080–97. [PubMed] [Google Scholar]
- 50. Gordon GJ, Rockwell GN, Jensen RV et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large‐scale transcriptional profiling. Am J Pathol 2005; 166: 1827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoang CD, D’Cunha J, Kratzke MG et al. Gene expression profiling identifies matriptase overexpression in malignant mesothelioma. Chest 2004; 125: 1843–52. [DOI] [PubMed] [Google Scholar]
- 52. Gordon GJ, Jensen RV, Hsiao LL et al. Using gene expression ratios to predict outcome among patients with mesothelioma. J Natl Cancer Inst 2003; 95: 598–605. [DOI] [PubMed] [Google Scholar]
- 53. Gray SG, Fennell DA, Mutti L, O’Byrne KJ. In arrayed ranks: array technology in the study of mesothelioma. J Thorac Oncol 2009; 4: 411–25. [DOI] [PubMed] [Google Scholar]
- 54. Roe OD, Anderssen E, Sandeck H, Christensen T, Larsson E, Lundgren S. Malignant pleural mesothelioma: Genome‐wide expression patterns reflecting general resistance mechanisms and a proposal of novel targets. Lung Cancer 2009; doi: 10.1016/jlungcan.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 55. Lee AY, He B, You L et al. Expression of the secreted frizzled‐related protein gene family is downregulated in human mesothelioma. Oncogene 2004; 23: 6672–6. [DOI] [PubMed] [Google Scholar]
- 56. Destro A, Ceresoli GL, Baryshnikova E et al. Gene methylation in pleural mesothelioma: correlations with clinico‐pathological features and patient’s follow‐up. Lung Cancer 2008; 59: 369–76. [DOI] [PubMed] [Google Scholar]
- 57. Christensen BC, Godleski JJ, Marsit CJ et al. Asbestos exposure predicts cell cycle control gene promoter methylation in pleural mesothelioma. Carcinogenesis 2008; 29: 1555–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Christensen BC, Houseman EA, Godleski JJ et al. Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer Res 2009; 69: 227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Busacca S, Germano S, De Cecco L et al. MicroRNA Signature of Malignant Mesothelioma with Potential Diagnostic and Prognostic Implications. Am J Respir Cell Mol Biol 2009; doi: 10.1165/rcmb.2009‐0060OC. [DOI] [PubMed] [Google Scholar]
- 60. Ascoli V, Cavone D, Merler E et al. Mesothelioma in blood related subjects: report of 11 clusters among 1954 Italy cases and review of the literature. Am J Ind Med 2007; 50: 357–69. [DOI] [PubMed] [Google Scholar]
- 61. Ugolini D, Neri M, Ceppi M et al. Genetic susceptibility to malignant mesothelioma and exposure to asbestos: the influence of the familial factor. Mutat Res 2008; 658: 162–71. [DOI] [PubMed] [Google Scholar]
- 62. Neri M, Ugolini D, Dianzani I et al. Genetic susceptibility to malignant pleural mesothelioma and other asbestos‐associated diseases. Mutat Res 2008; 659: 126–36. [DOI] [PubMed] [Google Scholar]
- 63. Carbone M, Emri S, Dogan AU et al. A mesothelioma epidemic in Cappadocia: scientific developments and unexpected social outcomes. Nat Rev Cancer 2007; 7: 147–54. [DOI] [PubMed] [Google Scholar]
- 64. Fennell DA, Gaudino G, O’Byrne KJ, Mutti L, Van Meerbeeck J. Advances in the systemic therapy of malignant pleural mesothelioma. Nat Clin Pract Oncol 2008; 5: 136–47. [DOI] [PubMed] [Google Scholar]
- 65. Weder W, Stahel RA, Bernhard J et al. Multicenter trial of neo‐adjuvant chemotherapy followed by extrapleural pneumonectomy in malignant pleural mesothelioma. Ann Oncol 2007; 18: 1196–202. [DOI] [PubMed] [Google Scholar]
- 66. Krug LM, Pass HI, Rusch VW et al. Multicenter Phase II Trial of Neoadjuvant Pemetrexed Plus Cisplatin Followed by Extrapleural Pneumonectomy and Radiation for Malignant Pleural Mesothelioma. J Clin Oncol 2009; 27: 3007–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kindler HL. Systemic treatments for mesothelioma: standard and novel. Curr Treat Options Oncol 2008; 9: 171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ramalingam SS, Belani CP, Ruel C et al. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J Thorac Oncol 2009; 4: 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sartore‐Bianchi A, Gasparri F, Galvani A et al. Bortezomib inhibits nuclear factor‐kappaB dependent survival and has potent in vivo activity in mesothelioma. Clin Cancer Res 2007; 13: 5942–51. [DOI] [PubMed] [Google Scholar]
- 70. Beck AK, Pass HI, Carbone M, Yang H. Ranpirnase as a potential antitumor ribonuclease treatment for mesothelioma and other malignancies. Future Oncol 2008; 4: 341–9. [DOI] [PubMed] [Google Scholar]
- 71. Lopez‐Rios F, Chuai S, Flores R et al. Global gene expression profiling of pleural mesotheliomas: overexpression of aurora kinases and P16/CDKN2A deletion as prognostic factors and critical evaluation of microarray‐based prognostic prediction. Cancer Res 2006; 66: 2970–9. [DOI] [PubMed] [Google Scholar]
- 72. Suzuki E, Kim S, Cheung HK et al. A novel small‐molecule inhibitor of transforming growth factor beta type I receptor kinase (SM16) inhibits murine mesothelioma tumor growth in vivo and prevents tumor recurrence after surgical resection. Cancer Res 2007; 67: 2351–9. [DOI] [PubMed] [Google Scholar]
- 73. Tabata C, Tabata R, Hirayama N et al. All‐trans‐retinoic acid inhibits tumour growth of malignant pleural mesothelioma in mice. Eur Respir J 2009; doi: 10.1183/09031936.00195708. [DOI] [PubMed] [Google Scholar]
- 74. Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer 2008; 44: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feng Y, Xiao X, Zhu Z et al. A novel human monoclonal antibody that binds with high affinity to mesothelin‐expressing cells and kills them by antibody‐dependent cell‐mediated cytotoxicity. Mol Cancer Ther 2009; 8: 1113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Inamoto T, Yamada T, Ohnuma K et al. Humanized anti‐CD26 monoclonal antibody as a treatment for malignant mesothelioma tumors. Clin Cancer Res 2007; 13: 4191–200. [DOI] [PubMed] [Google Scholar]
- 77. Usami N, Fukui T, Kondo M et al. Establishment and characterization of four malignant pleural mesothelioma cell lines from Japanese patients. Cancer Sci 2006; 97: 387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Seidel C, Schagdarsurengin U, Blumke K et al. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol Carcinog 2007; 46: 865–71. [DOI] [PubMed] [Google Scholar]
- 79. Tapon N, Harvey KF, Bell DW et al. Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002; 110: 467–78. [DOI] [PubMed] [Google Scholar]
- 80. Kawahara M, Hori T, Chonabayashi K, Oka T, Sudol M, Uchiyama T. Kpm/Lats2 is linked to chemosensitivity of leukemic cells through the stabilization of p73. Blood 2008; 112: 3856–66. [DOI] [PubMed] [Google Scholar]
- 81. Strazisar M, Mlakar V, Glavac D. LATS2 tumour specific mutations and down‐regulation of the gene in non‐small cell carcinoma. Lung Cancer 2009; 64: 257–62. [DOI] [PubMed] [Google Scholar]
- 82. Xu MZ, Yao TJ, Lee NP et al. Yes‐associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 2009; doi: 10.1002/cncr.24495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Steinhardt AA, Gayyed MF, Klein AP et al. Expression of Yes‐associated protein in common solid tumors. Hum Pathol 2008; 39: 1582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chan SW, Lim CJ, Guo K et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res 2008; 68: 2592–8. [DOI] [PubMed] [Google Scholar]