

Abstract
The role of nuclear factor‐κB (NF‐κB) activation in cancer cell apoptosis appears to be tailored specifically for each cell type and the type of NF‐κB inducer. The present study aimed to determine whether or not NF‐κB activation is associated with chemosensitivity to doxorubicin (DOX) using the DOX‐sensitive SNU‐601 and DOX‐resistant SNU‐216 gastric cancer cell lines. The effect of NF‐κB activation on DOX (1 µg/mL) sensitivity was analyzed after the suppression of NF‐κB activation using transfection of the super‐suppressive mutant form of IκBα (mIκBα) or pretreatment with pyrrolidine dithiocarbamate. In addition, the association between NF‐κB and manganese superoxide dismutase (MnSOD) in relation to DOX sensitivity was analyzed after the modulation of MnSOD expression. The NF‐κB activity was much higher in DOX‐resistant SNU‐216 cells than in DOX‐sensitive SNU‐601 cells before and after DOX treatment. Overexpression of mIκBα or pyrrolidine dithiocarbamate pretreatment decreased the DOX resistance in SNU‐601 cells with low MnSOD expression, but not in SNU‐216 cells with high MnSOD expression. In comparison, the overexpression of MnSOD, which also suppressed NF‐κB activation in both cell lines, increased DOX resistance in SNU‐601 cells. Blocking of MnSOD expression using RNA interference techniques increased DOX sensitivity in SNU‐216 cells, which was further augmented by the additional inhibition of NF‐κB activity. Our results showed that whether NF‐κB contributes to DOX sensitivity in gastric cancer cells is determined by the level of MnSOD expression. Thus, targeting both MnSOD and NF‐κB may be helpful for increasing the efficacy of DOX treatment of DOX‐resistant SNU gastric cancer cells. (Cancer Sci 2008; 99: 1117–1124)
- Abbreviations: DAPI
4′,6′‐diamidino‐2‐phenylindole
- DHR
dihydrorhodamine 123
- DOX
doxorubicin
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EMSA
electrophoretic mobility shift assay
- mIκBα
super‐suppressive mutant form of IκBα
- MnSOD
manganese superoxide dismutase
- NF‐κB
nuclear factor‐κB
- PARP
poly (ADP ribose) polymerase
- PBS
phosphate‐buffered saline
- PDTC
pyrrolidine dithiocarbamate
- PMSF
phenylmethylsulfonyl fluoride
- RNAi
RNA interference
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- TNF
tumor necrosis factor
Gastric cancer is one of the most common malignancies =worldwide and the major cause of cancer death in Asia.( 1 ) In an effort to improve the survival of patients with gastric cancer, the use of chemotherapy is increasing. Unfortunately, most gastric cancers are still resistant to chemotherapy.
Because resistance to anticancer drugs presents a complex problem to both clinical and molecular oncologists, a greater understanding of the molecular mechanisms underlying drug resistance should help us predict treatment outcomes and improve the efficacy of conventional anticancer chemotherapies.
Nuclear factor‐κB is a transcription factor that regulates various genes involved in cell proliferation, angiogenesis, metastasis, and suppression of apoptosis in cancer cells.( 2 ) Among the various functions of NF‐κB, the control of apoptosis has drawn the most attention in recent years because apoptosis is an important aspect of the cytotoxicity induced by anticancer drugs.
Many malignant tumors display constitutive NF‐κB activation that allows malignant cells to escape apoptosis induced by anticancer drugs.( 2 ) Transfection of mIκBα, which inhibits constitutive NF‐κB activation, increases the gemcitabine sensitivity of lung cancer cells,( 3 ) the etoposide or doxorubicin sensitivity of pancreatic carcinoma cells,( 4 ) and the cisplatin sensitivity of ovarian cancer cells.( 5 ) Notably, NF‐κB induction by several chemotherapeutic agents can lower their efficacy even when NF‐κB is not constitutively active.( 2 , 6 ) Thus, inhibition of NF‐κB activity seems to be a promising means to enhance the cytotoxic effects of anticancer agents.( 7 ) Indeed, clinical trials using several NF‐κB inhibitors are currently in progress with promising results for the treatment of lung cancer and myeloma.( 8 , 9 ) In contrast, pro‐apoptotic function of active NF‐κB in cancer cells has been reported. In a subset of breast cancer cells, stable transfection of antisense IκBα increases paclitaxel‐induced apoptosis.( 10 ) In N‐type neuroblastoma cells, inhibition of NF‐κB activity decreases DOX‐induced apoptosis.( 11 ) These data indicate that the role of NF‐κB activation in cancer cell apoptosis appears to be tailored specifically for each cell type and the type of NF‐κB inducer.
With respect to gastric cancer, we reported previously that constitutive activation of NF‐κB was found in 18% of gastric cancers.( 12 ) However, the role of NF‐κB in the chemoresistance of gastric cancer cells is not well known. At the present time, we know of only one published report on the role of NF‐κB activation in anticancer drug‐induced apoptosis in gastric cancer cells. Camp et al. reported that inducible NF‐κB activation in gastric cancer cells contributes to chemoresistance against 5‐fluorouracil and irinotecan.( 13 )
It has been proposed that NF‐κB contributes to the cytoprotection against several redox‐mediated therapeutic agents.( 14 ) The chemotherapeutic agent DOX, a quinone‐containing anthracycline analog, is one of the most extensively studied anticancer drugs used as a single agent or in combination chemotherapy for the treatment of gastric cancer.( 15 , 16 ) Although DOX generates superoxide in gastric cancer cells,( 17 ) the relationship between NF‐κB activation and DOX resistance in gastric cancer cells is not known.
The present study investigated whether NF‐κB activity is involved in the resistance of gastric cancer cells to DOX. For this, we suppressed NF‐κB activation by using a retroviral vector expressing mIκBα or pretreatment with the NF‐κB inhibitor PDTC. In addition, we examined the association between NF‐κB and MnSOD, a superoxide scavenger and known regulator and target of NF‐κB in cancer cells,( 18 , 19 , 20 , 21 , 22 ) in relation to DOX sensitivity in gastric cancer cells.
Materials and Methods
Cell lines and cultures. Two human gastric cancer cell lines, DOX‐resistant SNU‐216 and DOX‐sensitive SNU‐601,( 17 ) were purchased from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in RPMI‐1640 (Life Technologies, Grand Island, NY, USA) containing 10% fetal bovine serum, 2 mg/mL sodium bicarbonate, 100 U/mL penicillin, and 100 µg/mL streptomycin (Life Technologies) at 37°C in a humidified 95% air and 5% CO2 atmosphere.
Chemosensitivity assay. A crystal violet assay was used to assess cell numbers as described previously.( 23 ) Cells were seeded in 24‐well plates at a density of 2.5 × 104 cells/well for SNU‐216 cells or 3.5 × 104 cells/well for SNU‐601 cells 1 day prior to the experiment, then treated with DOX at various concentrations and for the various incubation periods. To determine the viability of cancer cells, attached cells were stained with 0.2% crystal violet in 20% methanol for 10 min, dissolved in 10% SDS, transferred to 96‐well plates, and the absorbance was measured at 570 nm using an enzyme‐linked immunosorbent assay reader (Bio‐Rad, Hercules, CA, USA). Absorbance values were normalized to the values obtained for the medium control group cells to determine the survival percentage.
Western blotting. Cell lysates were prepared in 100–200 µL of 1× SDS lysis buffer (125 mM Tris‐HCl [pH 6.8], 4% SDS, 0.004% bromophenol blue, and 20% glycerol). Protein content was measured using BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). Equal amounts of protein were loaded onto a 10% discontinuous SDS and polyacrylamide gel and transferred electrophoretically to nitrocellulose membranes blocked with 5% non‐fat dry milk in PBS–Tween‐20 (0.1%, v/v) for 1 h. The membranes were then incubated with either rabbit antihuman MnSOD (1:1000) (StressGen, Victoria, BC, Canada), rabbit anti‐IκBα (New England Biolabs, Beverly, MA, USA), mouse anti‐PARP (1:1000) (Biomol Research Laboratory, Plymouth Meeting, PA, USA), or mouse anti‐β‐actin (Sigma, St Louis, MO, USA) at 4°C overnight with or without 2 h incubation at room temperature. Horseradish peroxidase‐conjugated antirabbit IgG (1:2000) (Zymed, San Francisco, CA, USA) or antimouse IgG (1:2500) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a secondary antibody. Enhanced chemiluminescence was used to detect the immunoreactive proteins. Equal protein loading was confirmed by β‐actin.
4′,6′‐Diamidino‐2‐phenylindole staining. Apoptosis was evaluated by DAPI staining as described previously.( 24 ) Briefly, cells were fixed with 4% paraformaldehyde for 30 min, washed three times with PBS, and stained with DAPI (1 µg/mL) in the dark for 30 min and then examined under a fluorescence microscope. Cells were considered apoptotic if their nuclei were condensed or fragmented.
Luciferase reporter assay. The NF‐κB–luciferase reporter plasmid (pNF‐κB‐luciferase) (Stratagene, La Jolla, CA, USA) contains a 5 × NF‐κB response element fused to luciferase. SNU cells were cotransfected transiently with 0.4 µg pNF‐κB‐luciferase and 0.4 µg β‐galactosidase vector, an internal control, using Lipofectamine Plus (Life Technologies). Twenty‐four hours after transfection, SNU cells were treated with DOX for 2–9 h and assays for luciferase and β‐galactosidase were carried out using a Dual‐Luciferase Reporter Assay System (Promega, Madison, WI, USA) and measured on an AutoLumat LB 9505c luminometer (Berthold Analytical Instruments, Nashua, Germany). NF‐κB luciferase activity was normalized by β‐galactosidase activity, and luciferase activity in pNF‐κB‐luciferase‐transfected SNU‐601 cells without DOX treatment was arbitrarily set to 1.
Preparation of nuclear and cytoplasmic extracts. Cell lines were scraped into 1 mL cold PBS, washed with PBS, centrifuged, and cell pellets were suspended in cold lysis buffer A (10 mM Tris [pH 8.0], 60 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1% Nonidet P‐40, 1 mM PMSF), incubated on ice for 10 min, centrifuged, and cytoplasmic extracts in the supernatant fractions were transferred into fresh tubes. For nuclear extracts, cell pellets were resuspended in cold lysis buffer B (200 mM HEPES [pH 7.9], 0.75 mM spermidine, 0.15 mM spermine, 0.2 mM EDTA, 2 mM ethylene glycol bis(beta‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid, 2 mM DTT, 20% glycerol, 1 mM PMSF, 0.4 M NaCl), incubated on ice for 10 min with occasional vortexing and then centrifuged at 170 g at 4°C for 2 min.
Electrophoretic mobility shift assay. Double‐stranded oligonucleotides containing the NF‐κB consensus sequence (sense strand) 5′‐AGTTGAGGGGACTTTCCCAGGC‐3′ were labeled with [α‐32P]‐ATP (Amersham, Arlington Heights, IL, USA) using a T4 polynucleotide kinase (Promega). Nuclear proteins (10 µg) were preincubated in a reaction buffer containing 10 mM Tris‐HCl, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 5% glycerol, and 1 µg poly dI‐dC (Amersham) for 10 min. Thereafter, binding reactions were carried out for 30 min with 2 µL (>20 000 cpm) of end‐labeled κB‐oligonucleotide probes in a final volume of 20 µL. Protein–DNA complexes were then separated from the free probe on 5% polyacrylamide gels in 1× Tris‐Glycine‐EDTA buffer. The gels were then dried and exposed to radiography film at –80°C for 6–18 h.
Infection with retroviral vectors expressing the IκBα super repressor. The retroviral vectors MFG.EGFP.IRES.puro and MFG.IκBαM. IRES.puro, which encodes mIκBα, were generated and infected into cells as described previously.( 25 ) Pooled puromycin‐resistant cells were used for subsequent analysis.
Treatment with PDTC. PDTC (Sigma) was prepared in PBS (pH 7.4) as a 100‐mM stock solution and used at a final concentration of 100 µM. For NF‐κB‐inhibition experiments, the cells were preincubated with PDTC (100 µM) for 1 h.
Transient transfection with a MnSOD‐expressing plasmid. The effect of MnSOD overexpression on DOX sensitivity was examined. An expression vector, pcDNA3 containing a human MnSOD cDNA insert (780 bp), was kindly provided by Dr B. Palazzotti (Catholic University of Italy, Rome, Italy). This construct (0.4 µg) or empty pcDNA3 vector (0.4 µg) was transfected into 105 cells/well in 24‐well plates using Lipofectamine Reagent according to the manufacturer's instructions.
Small interfering RNA design and delivery. To transiently silence the expression of human MnSOD, siRNA corresponding to the MnSOD gene was designed and synthesized by Proligo (La Jolla, CA, USA). The sequences of the sense and antisense strands of the 21‐mer oligonucleotides used were 5′‐GGUCAUAUCAAUCAUAGCATT‐3′ and 5′‐UGCUAUGAUUGAUAUGACCTT‐3′, respectively, corresponding to position 283–301 relative to the start codon of human MnSOD mRNA (GenBank accession no. NM_000636). No other mammalian mRNA in the National Center for Biotechnology Information database contained this sequence. For control siRNA, the sequence of the same region of MnSOD mRNA was scrambled. Cells were transfected with siRNA duplex using Lipofectamine. Two days after transfection, cell extracts were used for western blotting or chemosensitivity assays.
Infection with lentivirus expressing shRNA. The shRNA‐expressing lentiviral vectors shLenti2.4Z/MnSOD and shLenti2.4Z/scramble were prepared by Macrogen (Seoul, Korea). In brief, the shLenti2.4Z lentiviral vector was designed to produce shRNA promoted from the U6 promoter and to express lacZ protein from the hCMV promoter. The shLenti2.4Z/MnSOD virus, which is capable of generating siRNA specific for MnSOD, was constructed by inserting the synthetic double‐stranded oligonucleotides described above into the EcoR|–Xba| restriction enzyme sites of the shLenti2.4Z lentiviral vector. The nucleotide sequence of the constructs was verified by sequencing. As a control vector, scrambled shRNA sequences were inserted into the shLenti2.4Z lentiviral vector (shLenti2.4Z/scramble virus) as described above. The viral infection was carried out by incubating SNU‐216 cells with lentivirus for 4 h in the presence of 4 µg/mL Polybrene (Sigma). Pooled puromycin (2 µg/mL)‐resistant cells were harvested and stored for further analysis.
Detection of H2O2 using a fluorogenic probe. Conversion of H2O2 from O2 − after DOX treatment was detected by a method reported by Negre‐Salvayre et al.( 26 ) with slight modifications. Generation of cellular H2O2 was observed by using DHR (Sigma), which reacts with H2O2 but not with O2 − produced in the mitochondria. Twenty‐four hours after plating, cells were exposed to DOX (1 µg/mL) for 36 h. Control cells were not treated with DOX. DOX‐treated cells were loaded with DHR (5 µM) for 30 min. The fluorescence intensity of treated cells and control cells was observed under a fluorescence microscope.
Statistical analysis. The results are presented as mean ± SD. Data were analyzed by one‐way ANOVA, and differences were considered significant when P‐values were <0.05 in the Newman–Keuls multiple comparison test. Graphpad Prism 2.00 for Windows XT (GraphPad Software, San Diego, CA, USA) was used to conduct the analysis.
Results
Sensitivity to DOX in human gastric cancer cell lines is cell specific. First, we evaluated the sensitivity of gastric cancer cell lines to DOX treatment. We found that SNU‐216 cells are resistant to DOX treatment whereas SNU‐601 cells are sensitive in a time‐ and dose‐dependent manner (Fig. 1a,b, respectively).
Figure 1.
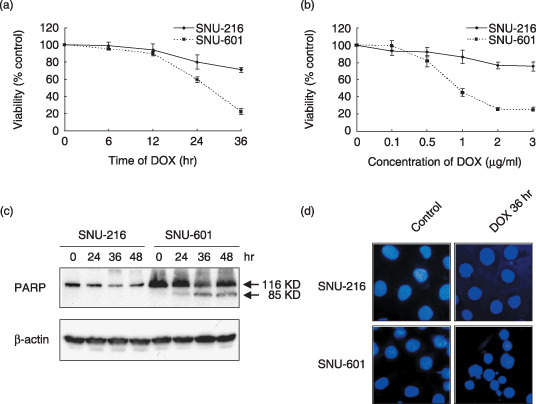
Sensitivity of gastric cancer cell lines to doxorubicin (DOX). (a) SNU‐216 and SNU‐601 cells were treated with DOX (1 µg/mL) for 6–36 h. (b) Cells were treated with various concentrations (0–3 µg/mL) of DOX for 36 h. Cell survival represents the mean percentage survival ± SD compared to untreated cells. Each assay was carried out in triplicate in three independent experiments. (c) Cells were incubated with DOX (1 µg/mL) for 24–48 h. Poly (ADP ribose) polymerase (PARP) cleavage was determined by western blotting. (d) 4′,6′‐Diamidino‐2‐phenylindole staining and fluorescence microscopy showing morphological changes in SNU cells after DOX (1 µg/mL) treatment for 36 h. SNU‐601 cells show more frequent nuclear chromatin condensation and fragmentation than SNU‐216 cells (×400).
To examine whether DOX‐induced cell death occurs via apoptosis, we observed PARP (a caspase substrate) cleavage. After DOX (1 µg/mL) treatment, a characteristic 85‐kDa fragment was evident within 36 h in SNU‐601 cells, but not in SNU‐216 cells (Fig. 1c). The presence of apoptosis was confirmed by DAPI staining, which showed frequent peripheral chromatin condensation and nuclear fragmentation in DOX‐treated SNU‐601 cells (Fig. 1d).
Nuclear factor‐κB activity is different in gastric cancer cell lines. We compared the NF‐κB activity in SNU cell lines by luciferase reporter assay (Fig. 2a,b) and EMSA (Fig. 2c). We found that NF‐κB activity varies in the individual gastric cancer cell lines (Fig. 2a). The NF‐κB activity was much higher in DOX‐resistant SNU‐216 cells than in DOX‐sensitive SNU‐601 cells before and after DOX treatment, although SNU‐601 cells showed inducible NF‐κB activity after DOX treatment for 6 h (P < 0.001 vs the basal level) (Fig. 2b). These results were confirmed by EMSA (Fig. 2c).
Figure 2.
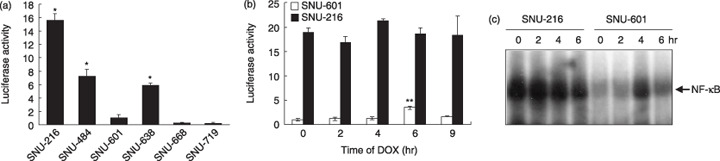
Comparison of nuclear factor‐κB (NF‐κB) activity in SNU cell lines. (a) The NF‐κB luciferase activities of gastric cancer cells were measured after transient cotransfection with pNF‐κB‐luciferase and a β‐galactosidase vector. (b) Transiently cotransfected cells were treated with doxorubicin (DOX) (1 µg/mL) for 2–9 h. The luciferase activity in pNF‐κB‐luciferase‐transfected SNU‐601 cells without DOX‐treatment was normalized by β‐galactosidase activity and was arbitrarily set to 1 and the luciferase activities of other cells were adjusted accordingly. *Cell line with high NF‐κB activity. **P < 0.001 compared with SNU‐601 cells treated with DOX for 4 h. (c) NF‐κB binding activity was measured by electrophoretic mobility shift assay after DOX treatment for 2–6 h.
Effect of NF‐κB suppression on DOX‐induced cytotoxicity is cell specific. In order to suppress the NF‐κB activation, we first produced stable gastric cancer cell lines overexpressing mIκBα acting as a IκBα super‐repressor (Fig. 3a,b insets). The luciferase reporter assay showed that mIκBα overexpression reduced the NF‐κB transcription activity in both SNU cell lines by more than 90% (P < 0.001) (Fig. 3a,b). Then, the cytotoxicity test showed that mIκBα overexpression increased DOX‐induced cell death in SNU‐601 cells (P < 0.001 vs vector control; Fig. 3c), whereas SNU‐216 cells were not affected by NF‐κB suppression (Fig. 3d).
Figure 3.
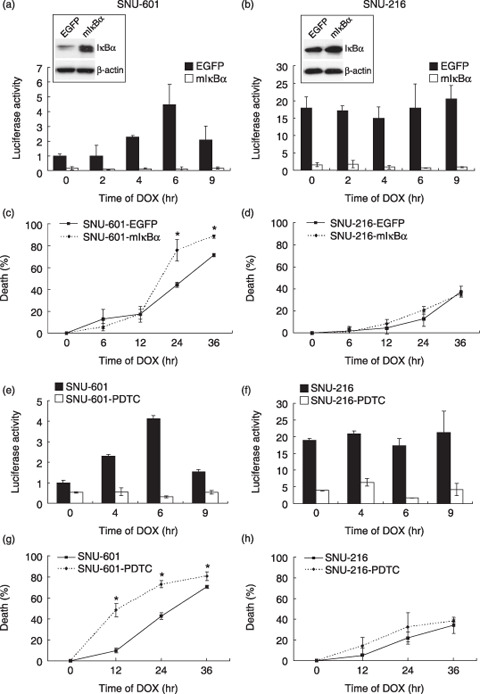
Effect of nuclear factor‐κB (NF‐κB) activation on doxorubicin (DOX) (1 µg/mL)‐induced cytotoxicity in SNU‐601 cells (left panels) and SNU‐216 cells (right panels). (a,b,e,f) NF‐κB activity was determined by luciferase reporter assay and normalized by β‐galactosidase activity as described above after DOX treatment for 0–9 h. (c,d,g,h) DOX‐induced cytotoxicity was represented as the percentage of cell death versus untreated cells after DOX treatment for 0–36 h. (a,b) Overexpression of the super‐suppressive mutant form of IκBα (mIκBα) in stable cell lines infected with either MFG.EGFP.IRES.puro or MFG.IκBαM.IRES.puro retroviral vector was detected by western blotting (insets). (c) Cells were treated with DOX for (c) 2–9 h or (d) 6–36 h. *P < 0.001 compared with SNU‐601 cells overexpressing enhanced green fluorescent protein. (e,f) Cells were pretreated with pyrrolidine dithiocarbamate (PDTC) (100 µM) for 1 h followed by treatment with DOX for 0–9 h (g,h) PDTC‐pretreated cells were treated with DOX for 0–36 h. *P < 0.001 compared with SNU‐601 cells without PDTC‐pretreatment.
To further confirm the effect of NF‐κB activation on DOX sensitivity, SNU cells were pretreated with PDTC (100 µM), a NF‐κB inhibitor, for 1 h. The NF‐κB transcriptional activities of both cell lines decreased (Fig. 3e,f) and DOX‐induced cell death in SNU‐601 cells subsequently increased (P < 0.001 vs control; Fig. 3g), but not in SNU‐216 cells (Fig. 3h). Taken together, these results indicate that DOX‐induced cytotoxicity contributes to the level of constitutive NF‐κB activation in SNU‐601 cells, but not in SNU‐216 cells.
Manganese superoxide dismutase overexpression suppresses NF‐κB activation. Previously, we showed that MnSOD overexpression reduces DOX sensitivity in SNU‐601 cells.( 17 ) To determine whether MnSOD is involved in the association between NF‐κB activation and DOX sensitivity, MnSOD was overexpressed by transient transfection of human MnSOD cDNA (Fig. 4a,b insets). We found that MnSOD overexpression decreased the DOX‐induced cell death of SNU‐601 cells although NF‐κB transcriptional activity in both SNU‐601 cells and SNU‐216 cells was suppressed (Fig. 4a,b).
Figure 4.
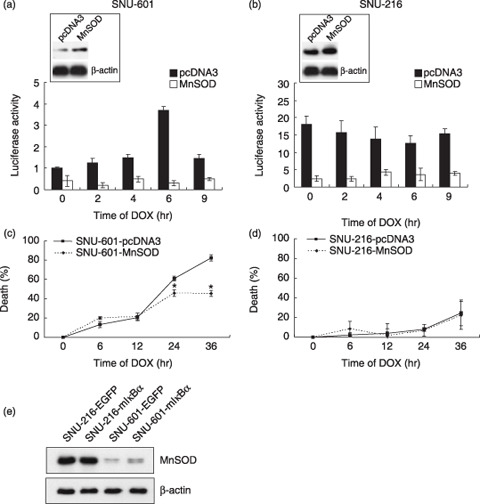
(a–d) Effect of manganese superoxide dismutase (MnSOD) overexpression on nuclear factor‐κB (NF‐κB) activation and doxorubicin (DOX)‐induced cell death. SNU‐601 cells (left panels) and SNU‐216 cells (right panels) were transiently transfected with either pcDNA3 or MnSOD cDNA. (a,b) MnSOD protein expression in transfected cells was analyzed by western blotting (insets). Cells were incubated with DOX (1 µg/mL) for 2–9 h and the NF‐κB activities were determined by luciferase reporter assay and normalized by β‐galactosidase activity as described above. (c,d) Cells were treated with DOX (1 µg/mL) for 6–36 h. DOX‐induced cytotoxicity is represented as the percentage of cell death versus untreated cells. *P < 0.001 compared with SNU‐601‐pcDNA3. (e) Effect of the super‐suppressive mutant form of IκBα overexpression on MnSOD expression was determined by western blotting.
As previous studies have shown that MnSOD is a target of NF‐κB transcription in Ewing sarcoma and prostate cancer cells,( 21 , 22 ) we further examined whether cross‐talk between constitutive NF‐κB activation and MnSOD expression exists in gastric cancer cells. We investigated MnSOD expression after infecting cells with a retrovirus overexpressing mIκBα (Fig. 4e) and found that mIκBα overexpression did not change the MnSOD expression in either cell line.
Manganese superoxide dismutase suppression enhances DOX‐induced cytotoxicity. As DOX sensitivity in DOX‐resistant SNU‐216 cells, which show a high level of MnSOD expression, was not controlled by NF‐κB activation, we examined whether MnSOD suppression alters the effect of NF‐κB activation on DOX sensitivity in SNU‐216 cells. First, we confirmed that transfection of MnSOD siRNA into SNU‐216 cells leads to a decrease in MnSOD protein expression (Fig. 5a). The luciferase reporter assay showed that mIκBα overexpression reduced NF‐κB transcription activity in SNU‐216 cells regardless of the transfection of MnSOD siRNA (Fig. 5c). MnSOD siRNA overexpression increased DOX cytotoxicity in SNU‐216 cells (P < 0.001, SNU‐216E‐siMnSOD vs SNU‐216E‐control; Fig. 5e). Moreover, DOX‐induced cell death was further enhanced by mIκBα overexpression when MnSOD siRNA was transfected into SNU‐216 cells (P < 0.001, SNU‐216α‐siMnSOD vs SNU‐216α‐control; Fig. 5e). Consistent results were shown when cells were pretreated with PDTC (P < 0.001; data not shown).
Figure 5.
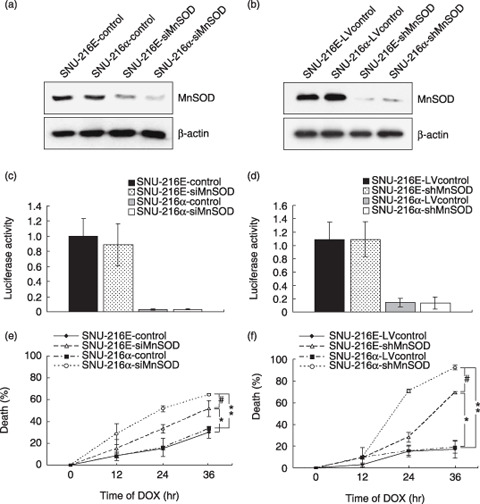
Effect of manganese superoxide dismutase (MnSOD) suppression induced by small interfering RNA (siRNA) (left panels) or lentivirus‐delivered short hairpin RNA (shRNA) (right panels) in SNU‐216 cells. Cells overexpressing either enhanced green fluorescent protein (SNU‐216E) or the super‐suppressive mutant form of IκBα (mIκBα) (SNU‐216α) were transfected with either MnSOD siRNA (SNU‐216E‐siMnSOD or SNU‐216α‐siMnSOD, respectively) or control siRNA (SNU‐216E‐control or SNU‐216α‐control, respectively). Cells were also infected with either shLenti2.4Z/MnSOD virus (SNU‐216E‐shMnSOD or SNU‐216α‐shMnSOD, respectively) or shLenti2.4Z/scramble virus (SNU‐216E‐LVcontrol or SNU‐216α‐LVcontrol, respectively). (a,b) Expression levels of MnSOD and β‐actin proteins were determined by western blotting. (c,d) Nuclear factor‐κB (NF‐κB) activity was determined by luciferase reporter assay. (e,f) Cells were treated with doxorubicin (DOX) (1 µg/mL) for 12–36 h. DOX cytotoxicity is represented as the percentage of cell death versus untreated cells. *P < 0.001 compared with SNU‐216E‐control or SNU‐216E‐LVcontrol. # P < 0.001 compared with SNU‐216E‐siMnSOD or SNU‐216E‐shMnSOD. **P < 0.001 compared with SNU‐216α‐control or SNU‐216α‐LVcontrol.
Next, we further confirmed the above data by using lentivirus‐mediated expression of MnSOD shRNA. Infection of SNU‐216 cells with lentivirus shLenti2.4Z/MnSOD or shLenti2.4Z/scramble (control virus) showed results consistent with those above and was more efficient than transfection with MnSOD siRNA (Fig. 5b,d,f). Taken together, these results indicate that the association between NF‐κB activity and DOX sensitivity in SNU‐216 cells is regulated by MnSOD expression.
Modulation of MnSOD expression determines DOX‐induced ROS generation. Our data shown in Figure 6 confirmed that DOX treatment causes the generation of superoxide, which is converted to H2O2 by mitochondrial MnSOD. MnSOD overexpression brought about a marked increase in the DOX‐induced fluorescence intensity of SNU‐601 cells (Fig. 6a). However, MnSOD suppression caused a marked decrease in the DOX‐induced fluorescence intensity of SNU‐216 cells (Fig. 6b).
Figure 6.
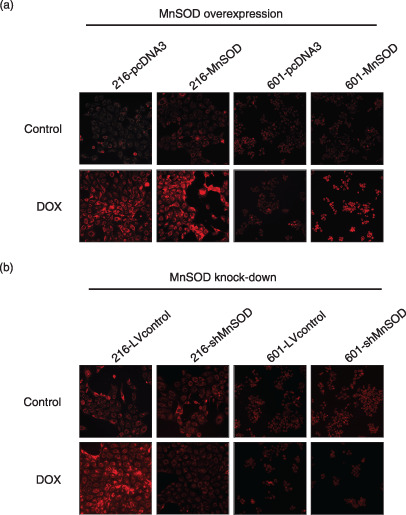
Fluorescence microphotographs obtained after loading dihydrorhodamine 123 showing H2O2 generation in gastric cancer cell lines after treatment with doxorubicin (DOX) for 36 h. Compared with the untreated control of each cell line, DOX‐treated SNU‐216 cells with a high manganese superoxide dismutase (MnSOD) content showed increased fluorescence intensity, but not in SNU‐601 cells with a low MnSOD content. (a) MnSOD overexpression markedly increased the DOX‐induced fluorescence intensity in SNU‐601 cells (601‐MnSOD vs 601‐pcDNA3), (b) whereas MnSOD suppression markedly decreased the fluorescence intensity in SNU‐216 cells (216‐shMnSOD vs 216‐LVcontrol).
Discussion
Because the role of NF‐κB activation in anticancer drug treatment is cell type and drug specific,( 3 , 4 , 10 , 11 ) it needs to be elucidated whether gastric cancer is suitable for NF‐κB inhibition for successful treatment. The present study is the first report on the role of NF‐κB in the DOX resistance of gastric cancer cells.
In the present study, we confirmed that SNU‐216 cells are DOX resistant whereas SNU‐601 cells are DOX sensitive and show apoptotic characteristics. As NF‐κB activation was much higher in the DOX‐resistant SNU‐216 cells than in the DOX‐sensitive SNU‐601 cells before and after DOX treatment, we initially speculated that NF‐κB activity contributes to the DOX resistance of gastric cancer cells. However, although suppression of NF‐κB activation by overexpression of mIκBα or pretreatment with PDTC dramatically reduced basal and DOX‐induced NF‐κB activation in both SNU cell lines compared with the vector controls (P < 0.001), paradoxical results were shown in DOX‐induced apoptosis. The suppression of NF‐κB increased DOX cytotoxicity in SNU‐601 cells, but DOX cytotoxicity in SNU‐216 cells was not affected by NF‐κB inhibition. These data indicate that NF‐κB activation differentially contributes to DOX resistance in gastric cancer cells, probably depending on the upstream molecules.
Nuclear factor‐κB is a representative redox‐sensitive transcription factor induced by ROS, such as the superoxide and hydroxyl radicals. Although both the mitochondrial antioxidant enzyme MnSOD and NF‐κB have been reported to have a protective function against oxidative stress‐induced cell death, their relationship varies in different cancer cells. In breast cancer cells, NF‐κB activation induced by TNF or ionizing radiation was inhibited by MnSOD expression.( 18 , 19 , 20 ) Thus, MnSOD appears to be a negative regulator of NF‐κB activation. In contrast, NF‐κB has antioxidant activity via an increase in MnSOD levels in Ewing sarcoma cells treated with TNF or in prostate cancer cells treated with radiation.( 21 , 22 ) Thus, MnSOD might be either an upstream signaling molecule in the redox‐sensitive NF‐κB pathway or a target gene of NF‐κB in the cellular adaptive responses to ROS.
With respect to gastric cancer cells, we know of no published report on the relationship between NF‐κB activation and MnSOD so far. In the present study, MnSOD overexpression resulted in a marked inhibition of constitutive NF‐κB activation (by approximately 75%) in both SNU cell lines and cells containing an empty vector construct. However, MnSOD expression was not changed by mIκBα overexpression in either SNU cell line. Thus, it appears that MnSOD changes the metabolic state of gastric cancer cells through the removal of superoxide in the mitochondria,( 17 ) and influences the transcriptional activity of NF‐κB, but not vice versa.
Our results showed that NF‐κB is constitutively active in SNU‐216 cells but not in SNU‐601 cells, although the expression of MnSOD, which suppressed NF‐κB activation in both cell lines, is more abundant in SNU‐216 cells than in SNU‐601 cells. This may be explained by the fact that the NF‐κB to MnSOD activity ratio varies in the individual gastric cancer cell lines. Previously, we reported that MnSOD expression and activity is different in gastric cancer cell lines.( 17 ) In the present study, we measured the NF‐κB luciferase activity and compared the NF‐κB to MnSOD activity ratio in several gastric cancer cell lines. Of the SNU cell lines expressing high levels of MnSOD (SNU‐216, SNU‐638, and SNU‐668), SNU‐216 and SNU‐638 showed a high level of NF‐κB activity whereas SNU‐668 showed a low level of NF‐κB activity. However, of the SNU cell lines expressing low levels of MnSOD (SNU‐484, SNU‐601, and SNU‐719), SNU‐484 showed a high level of NF‐κB activity whereas SNU‐601 and SNU‐719 showed a low level of NF‐κB activity. These results demonstrated different NF‐κB to MnSOD activity ratios in the individual gastric cancer cell lines. We speculate that this variation is, at least in part, due to the activities of molecules other than MnSOD. Thus, in order to investigate the association between the activities of MnSOD and NF‐κB in each cell line, we used a transfection technique to directly modulate MnSOD protein expression in each cell line instead of simply comparing the MnSOD levels and NF‐κB activities in different cell lines.
We previously reported that constitutive MnSOD expression plays a critical role in the chemoresistance of gastric cancer cells against ROS‐generating drugs, such as DOX and mitomycin C.( 17 ) We also demonstrated that the activity of MnSOD in gastric cancer cells correlates with the level of MnSOD protein.( 17 ) In the present study, NF‐κB activation was suppressed by MnSOD overexpression in both cell lines. In SNU‐601 cells showing a low endogenous MnSOD expression, MnSOD overexpression increased DOX resistance despite a decrease in NF‐κB activation. In contrast, in SNU‐216 cells showing high endogenous MnSOD expression, MnSOD overexpression did not affect DOX resistance although NF‐κB activation was decreased. Thus, it appears that MnSOD rather than NF‐κB plays a more crucial role in the DOX cytotoxicity of gastric cancer cells with high MnSOD expression.
Recent advances in biology demonstrate that RNAi is a powerful tool for tumor biology. In the present study, we inhibited MnSOD expression in two different ways: transfection of siRNA with Lipofectamine and infection with a shRNA‐expressing lentivirus. Our results showed that both methods were effective at demonstrating MnSOD‐dependent drug resistance in SNU‐216 cells. Furthermore, lentivirus‐delivered shRNA silenced MnSOD expression and resensitized SNU‐216 cells to doxorubicin more effectively than siRNA. As the lentiviral vector system appears to provide more efficient gene delivery and stable expression, it could be used in the future as a gene therapy tool for overcoming drug resistance in gastric cancer chemotherapy. However, the overexpression of MnSOD suppressed NF‐κB activity in SNU‐216 cells. Thus, it was expected that the blocking of MnSOD would enhance NF‐κB activation in this cell line. However, despite the effective knock down of MnSOD, NF‐κB activity was not increased in SNU‐216 cells treated with either siRNA or shRNA. Given our observation that the basal level of NF‐κB activity in SNU‐216 cells is substantially high, we speculate that NF‐κB activity is already upregulated to the saturation level, and thus cannot be further induced by MnSOD suppression.
In the present study we used H2O2 content as a surrogate marker of the DOX resistance and found that MnSOD overexpression brought about a marked increase in the DOX‐induced fluorescence intensity as well as a significant decrease in DOX‐sensitivity in SNU‐601 cells having a low level of MnSOD. In contrast, MnSOD suppression caused a marked decrease in the DOX‐induced fluorescence intensity as well as a significant increase in the DOX sensitivity of SNU‐216 cells having a high level of MnSOD. These data indicate the involvement of ROS in the sensitivity to DOX. Furthermore, we found that NF‐κB inhibition sensitizes SNU‐601 cells to DOX cytotoxicity, but it does not affect SNU‐216 cells. However, after silencing the MnSOD gene, the DOX sensitivity of SNU‐216 cells was enhanced significantly by NF‐κB inhibition. Taken together, as the mechanism by which DOX sensitization by NF‐κB inhibition depends on MnSOD, we propose that MnSOD provides first‐line resistance and NF‐κB provides second‐line resistance. DOX, a redox‐cycling compound, generates ROS in the mitochondrial inner membrane, and ROS can be removed by MnSOD in the mitochondrial matrix. When ROS overcome the radical‐scavenging ability of MnSOD, cancer cells suffer from oxidative stress and finally die.( 27 ) In SNU‐601 cells, DOX‐induced ROS that are not effectively removed in the mitochondria are released into the cytoplasm and activate NF‐κB, which in turn mediates the acquisition of drug resistance. In SNU‐216 cells, DOX‐induced ROS are removed in the mitochondrial matrix before they are released out of the mitochondria, which gives NF‐κB no opportunity to participate in the acquisition of drug resistance. However, when MnSOD expression is inhibited, DOX‐induced ROS activate NF‐κB and drug resistance is induced. Therefore, inhibition of both MnSOD and NF‐κB is required for the effective sensitization of gastric cancer cells to DOX. This hypothesis is summarized in Figure 7.
Figure 7.
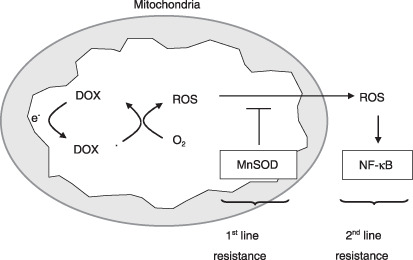
A hypothetical mechanism underlying doxorubicin (DOX) resistance in gastric cancer cells. Manganese superoxide dismutase (MnSOD) first removes mitochondrial reactive oxygen species (ROS) generated by DOX as the first‐line resistance, and then nuclear factor‐κB (NF‐κB) secondly enables cancer cells to survive under oxidative stress conditions as the second‐line resistance.
In conclusion, our results indicate that the NF‐κB dependency of the DOX resistance of gastric cancer cells is determined by the extent of MnSOD expression. Thus, blocking of MnSOD expression followed by inhibition of NF‐κB activity may be helpful for increasing the efficacy of the DOX treatment of DOX‐resistant SNU gastric cancer cells.
Acknowledgments
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF‐2006‐531‐E00008). S. J. Cho and Y. S. Ko were supported by the second‐stage Brain Korea 21 Project in 2006. We thank Dr Bernardetta Palazzotti of Catholic University for his generous gifts of human MnSOD cDNA.
References
- 1. Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of eighteen major cancers in 1985. Int J Cancer 1993; 54: 594–606. [DOI] [PubMed] [Google Scholar]
- 2. Orlowski RZ, Baldwin AS Jr. NF‐κB as a therapeutic target in cancer. Trends Mol Med 2002; 8: 385–9. [DOI] [PubMed] [Google Scholar]
- 3. Jones DR, Broad RM, Madrid LV et al . Inhibition of NF‐κB sensitizes non‐small cell lung cancer cells to chemotherapy‐induced apoptosis. Ann Thorac Surg 2000; 70: 930–7. [DOI] [PubMed] [Google Scholar]
- 4. Arlt A, Vorndamm J, Breitenbroich M et al . Inhibition of NF‐κB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene 2001; 20: 859–68. [DOI] [PubMed] [Google Scholar]
- 5. Mabuchi S, Ohmichi M, Nishio Y et al . Inhibition of NFκB increases the efficacy of cisplatin in vitro and in vivo ovarian cancer models. J Biol Chem 2004; 279: 23 477–85. [DOI] [PubMed] [Google Scholar]
- 6. Kucharczak J, Simmons MJ, Fan Y et al . To be, or not to be: NF‐κB is the answer – role of Rel/NF‐κB in the regulation of apoptosis. Oncogene 2003; 22: 8961–82. [DOI] [PubMed] [Google Scholar]
- 7. Nakanishi C, Toi M. Nuclear factor‐κB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 2005; 5: 297–309. [DOI] [PubMed] [Google Scholar]
- 8. Aghajanian C, Soignet S, Dizon DS et al . A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin Cancer Res 2002; 8: 2505–11. [PubMed] [Google Scholar]
- 9. Richardson PG, Barlogie B, Berenson J et al . A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003; 348: 2609–17. [DOI] [PubMed] [Google Scholar]
- 10. Huang Y, Johnson KR, Norris JS et al . Nuclear factor‐κB/IκB signaling pathway may contribute to the mediation of paclitaxel‐induced apoptosis in solid tumor cells. Cancer Res 2000; 60: 4426–32. [PubMed] [Google Scholar]
- 11. Bian X, McAllister‐Lucas LM, Shao F et al . NF‐κB activation mediates doxorubicin‐induced cell death in N‐type neuroblastoma cells. J Biol Chem 2001; 276: 48 921–9. [DOI] [PubMed] [Google Scholar]
- 12. Lee BL, Lee HS, Jung J et al . Nuclear factor‐κB activation correlates with better prognosis and Akt activation in human gastric cancer. Clin Cancer Res 2005; 11: 2518–25. [DOI] [PubMed] [Google Scholar]
- 13. Camp ER, Li J, Minnich DJ et al . Inducible nuclear factor‐κB activation contributes to chemotherapy resistance in gastric cancer. J Am Coll Surg 2004; 199: 249–58. [DOI] [PubMed] [Google Scholar]
- 14. Grimes KR, Warren GW, Fang F et al . Cyclooxygenase‐2 inhibitor, nimesulide, improves radiation treatment against non‐small cell lung cancer both in vitro and in vivo . Oncol Rep 2006; 16: 771–6. [PubMed] [Google Scholar]
- 15. Scartozzi M, Galizia E, Verdecchia L et al . Chemotherapy for advanced gastric cancer: across the years for a standard of care. Expert Opin Pharmacother 2007; 8: 797–808. [DOI] [PubMed] [Google Scholar]
- 16. Shin SJ, Jeung HC, Ahn JB et al . Capecitabine and doxorubicin combination chemotherapy as salvage therapy in pretreated advanced gastric cancer. Cancer Chemother Pharmacol 2007; 61: 157–65. [DOI] [PubMed] [Google Scholar]
- 17. Hur GC, Cho SJ, Kim CH et al . Manganese superoxide dismutase expression correlates with chemosensitivity in human gastric cancer cell lines. Clin Cancer Res 2003; 9: 5768–75. [PubMed] [Google Scholar]
- 18. Li JJ, Oberley LW, Fan M et al . Inhibition of AP‐1 and NF‐κB by manganese‐containing superoxide dismutase in human breast cancer cells. FASEB J 1998; 12: 1713–23. [DOI] [PubMed] [Google Scholar]
- 19. Manna SK, Zhang HJ, Yan T et al . Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor‐induced apoptosis and activation of nuclear transcription factor‐κB and activated protein‐1. J Biol Chem 1998; 273: 13 245–54. [DOI] [PubMed] [Google Scholar]
- 20. Ozeki M, Tamae D, Hou DX et al . Response of cyclin B1 to ionizing radiation: regulation by NF‐κB and mitochondrial antioxidant enzyme MnSOD. Anticancer Res 2004; 24: 2657–63. [PMC free article] [PubMed] [Google Scholar]
- 21. Djavaheri‐Mergny M, Javelaud D, Wietzerbin J et al . NF‐κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNF α‐treated Ewing sarcoma cells. FEBS Lett 2004; 578: 111–15. [DOI] [PubMed] [Google Scholar]
- 22. Josson S, Xu Y, Fang F et al . RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene 2006; 25: 1554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim WH, Schnaper HW, Nomizu M et al . Apoptosis in human fibrosarcoma cells is induced by a multimeric synthetic Tyr‐Ile‐Gly‐Ser‐Arg (YIGSR)‐containing polypeptide from laminin. Cancer Res 1994; 54: 5005–10. [PubMed] [Google Scholar]
- 24. Wang X, Gorospe M, Huang Y et al . p27Kip1 overexpression causes apoptotic death of mammalian cells. Oncogene 1997; 15: 2991–7. [DOI] [PubMed] [Google Scholar]
- 25. Nam SY, Jung GA, Hur GC et al . Upregulation of FLIP(S) by Akt, a possible inhibition mechanism of TRAIL‐induced apoptosis in human gastric cancers. Cancer Sci 2003; 94: 1066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Negre‐Salvayre A, Auge N, Duval C et al . Detection of intracellular reactive oxygen species in cultured cells using fluorescent probes. Meth Enzymol 2002; 352: 62–71. [DOI] [PubMed] [Google Scholar]
- 27. Jung K, Reszka R. Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev 2001; 49: 87–105. [DOI] [PubMed] [Google Scholar]