

Abstract
Many advanced cancers receive cisplatin‐based chemotherapy. However, cisplatin resistance is a major obstacle for cancer chemotherapy. Foxo3a is a member of the Foxo transcription factor family, which modulates the expression of genes involved in DNA damage repair, apoptosis, and other cellular processes. In this study, we found that cisplatin‐resistant cells were more sensitive to the anticancer agent mithramycin than their parental cells, and had a decreased level of Foxo3a expression. Foxo3a knockdown increased cell proliferation and resistance to cisplatin. On the other hand, mithramycin stimulated Foxo3a expression through reactive oxygen species production and sensitized cells to cisplatin, which was abolished by Foxo3a knockdown, while the acetylation status of Foxo3a was decreased in response to cisplatin treatment and was lower in cisplatin‐resistant cells. Knockdown of Foxo3a‐associated acetyltransferase p300 promoted cancer‐cell growth and cisplatin resistance. In addition, non‐acetylation‐mimicking Foxo3a overexpression decreased cancer cell growth and sensitized cells to cisplatin less than wild‐type Foxo3a overexpression. The current work may contribute to the evaluation of the therapeutic potential of inducing the Foxo3a pathway and acetylating the Foxo3a transcription factor, and lead to the reevaluation of cancer treatments based on mithramycin.
(Cancer Sci 2010; 101: 1177–1185)
Many advanced cancer patients receive cisplatin‐based chemotherapy. Although cisplatin‐based treatment decreases patients’ tumor burdens and increases life expectancy, there are many cases of non‐response or relapse during cisplatin treatment. This phenomenon is referred to as cisplatin resistance, and is a major obstacle to effective cancer treatment. Therefore, it is critical that cancer chemotherapy can overcome cisplatin resistance. We have previously investigated the mechanisms of resistance of anticancer drugs including cisplatin, vincristine, etoposide, and doxorubicin.( 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 ) Several molecules that are associated with the acquisition of cisplatin resistance have been identified, including detoxifying enzymes, drug‐efflux pumps, DNA repair enzymes, and apoptosis‐related genes.( 9 , 10 )
Foxo3a is a member of the Foxo family of transcription factors. Foxo transcription factors, which belong to the “other” class of the Fox superfamily, are involved in multiple signaling pathways and play critical roles in a number of physiological and pathological processes.( 11 ) It has recently been reported that the Forkhead transcription factor Foxo3a regulates the DNA damage response and stimulates the DNA repair pathway.( 12 , 13 ) Moreover, Foxo transcription factors regulate oxidative detoxification and cell cycle‐related genes.( 14 ) On the other hand, Foxo transcription factors regulate apoptosis‐related genes, such as Bim,( 15 ) tumor necrosis factor‐receptor ligand,( 16 ) p27Kip1,( 17 ) growth arrest and DNA damage‐inducible protein 45α (GADD45α),( 12 ) and Fas ligand.( 18 ) Recently, Foxo transcription factors have been shown to be acetylated or deacetylated by p300 and Sirtuinl (Sirt1), respectively, resulting in alterations of their DNA‐binding and transcriptional abilities.( 11 )
Mithramycin is a classical drug, which has been used to treat several types of cancer including testicular cancer, chronic myeloid leukemia, and acute myeloid leukemia, as well as hypercalcemia in patients with metastatic bone regions and Paget’s disease. Mithramycin binds to GC‐rich regions in chromatin and interferes with the transcription of genes that bear GC‐rich motifs in their promoters.( 19 , 20 ) Its mechanism of action involves a reversible interaction with double‐stranded DNA with GC‐base specificity.
Previously, it has been reported that inhibiting the phosphorylation of Foxo transcription factor sensitized human ovarian cancer cells to cisplatin.( 21 ) More recently, it was shown that in cisplatin‐sensitive colon‐cancer cells, cisplatin induced Foxo3a dephosphorylation and nuclear translocation, and the expression of its target genes, whereas the effect of cisplatin on Foxo3a was incomplete in cisplatin‐resistant cells.( 22 ) In this study, we found that expression and acetylation of Foxo3a were involved in the proliferation and sensitivity of cancer cells to cisplatin. In addition, mithramycin increased the effectiveness of cisplatin‐based chemotherapy by modulating the expression level of Foxo3a.
Materials and Methods
Cell culture. Human bladder cancer T24, KK47, T24/DDP10, and KK47/DDP20 cells were cultured in Eagle’s minimal essential medium, which was purchased from Invitrogen (San Diego, CA, USA) and contained 10% fetal bovine serum. As previously described, cisplatin‐resistant T24/DDP10 and KK47/DDP20 cells were established from T24 and KK47 cells, respectively.( 1 , 4 ) Stable transfectants, that is KK47‐AcGFP (#1 and #2), KK47‐Foxo3a‐GFP (#1 and #2), and KK47‐Foxo3a‐GFP KR (#1 and #2) cells which were derived from KK47 cells and stably expressed the corresponding proteins were established as described previously.( 8 ) Briefly, KK47 cells were transfected with AcGFP, Foxo3a‐GFP, or Foxo3a‐GFP KR expression plasmid described in the following section, and cultured for 2 weeks with selection medium containing 1000 μg/mL of geneticin (Nacalai tesque, Kyoto, Japan). Protein expressions of obtained clones were verified using western blotting and fluorescence microscope (Biozero; Keyence, Tokyo, Japan). Isolated clones were maintained in the presence of 1000 μg/mL of geneticin. The cell lines were maintained in 5% CO2 atmosphere at 37°C.
Antibodies and plasmids. Antibodies against p21 (sc‐397), Sp1 (sc‐420), p300 (sc‐585), and pan‐AcetylLys (sc‐8663) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐Foxo3a and anti‐Sirt1 antibodies were purchased from Epitomics (Burlingame, CA, USA). Anti‐GFP antibody (AB513) was obtained from Evrogen (Moscow, Russia). Anti‐β‐actin and anti‐Lamin B1 antibodies were purchased from Sigma (St. Louis, MO, USA). Foxo3a‐GFP plasmid expressing C‐terminally GFP‐tagged Foxo3a protein was purchased from OriGene (Rockville, MD, USA). To construct non‐acetylation‐mimicking Foxo3a‐GFP expression plasmid (Foxo3a‐GFP KR), mutation was introduced into the lysines 242 and 245 of Foxo3a which were substituted into arginine using a KOD Mutagenesis Kit (Toyobo, Osaka, Japan), the Foxo3a‐GFP plasmid as a template, and the following primer pairs: 5′‐AGGAGCGGA‐AGAGCCCCCCGGCGGCGGGCTGTCTCC‐3′ and 5′‐CCCC‐CCATCAGGGTTGATGATCCACCAAGAGCTCTTGC‐3′. Underlined nucleotides indicate mutated sequences.
Western blot analysis. Whole‐cell lysates (30 μg) and nuclear extracts (30 μg) were prepared and western blot analysis was performed as previously described.( 23 , 24 , 25 , 26 , 27 )
Knockdown analysis using siRNAs. Knockdown analysis using siRNAs was performed as previously described.( 23 , 24 , 25 , 26 , 27 ) Briefly, the following double‐stranded RNA 25‐base‐pair oligonucleotides were commercially generated: 5′‐UAGAAUUGGUGCGUGAACGGAAGUC‐3′ (sense) and 5′‐GACUUCCGUUCACGCACCAAUUCUA‐3′ (antisense) for Foxo3a siRNA #1 (Invitrogen); 5′‐UAUACGGGAAGCUAGAGCUC‐CGCUG‐3′ (sense) and 5′‐CAGCGGAGCUCUAGCUUC‐CCGUAUA‐3′ (antisense) for Foxo3a siRNA #2 (Invitrogen); 5′‐AUUAUAGGCUGACGUGGCAUUCCUA‐3′ (sense) and 5′‐CCUGCCCGGUGAACUCUCCUAUAAU‐3′ (antisense) for p300 siRNA #1 (Invitrogen); 5′‐UUAAACAGCCAUCACAGACGAAUCC‐3′ (sense), 5′‐GGAUUCGUCUGUGAUGGCUGUUAA‐3′ (antisense) for p300 siRNA #2 (Invitrogen); and 5′‐CUACUACUACCACCAGGAATT‐3′ (sense), 5′‐UUGCUGGUGGUAGUAGUAGTT‐3′ (antisense) for Sp1 siRNA (Sigma). T24 and KK47 cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer’s instructions.
Cytotoxicity analysis. Cytotoxicity analysis was performed as previously described.( 7 , 24 , 25 ) Briefly, T24, KK47, and their cisplatin‐resistant cells (2.5 × 103); KK47 cells (2.5 × 103) transfected with 40 nm of the indicated siRNA as described above; stable transfectants of KK47 cells (2.5 × 103); or KK47 cells (2.5 × 103) with media containing vehicle, 2 nm of mithramycin, 2 nm of doxorubicin, or 2 μm of 5‐fluorouracil (5‐FU) were seeded into 96‐well plates. The following day, various concentrations of the indicated anticancer drugs were applied. After 48 h, the surviving cells were stained using the alamarBlue assay (TREK Diagnostic Systems, Cleveland, OH, USA) for 180 min at 37°C. Absorbance of each well was measured using a plate reader (ARVO MX; Perkin Elmer, Waltham, MA, USA).
Cell proliferation assay. The cell proliferation assay was performed as previously described.( 7 , 8 , 23 , 25 , 26 , 27 ) Briefly, KK47 cells (2.5 × 104) transfected with 40 nm of the indicated siRNA or stable transfectants of KK47 cells (2.5 × 104) were seeded into 12‐well plates. The time point of 12 h after plating was set as 0 h. The cells were harvested with trypsin and counted daily using a cell counter (Beckman Coulter, Fullerton, CA, USA). The results were normalized by the cell counts at 0 h, and are representative of at least three independent experiments.
RNA isolation, reverse transcription, and quantitative real‐time PCR. These procedures were performed as previously described.( 25 , 26 , 27 ) Quantitative real‐time PCR was performed with the following: TaqMan Gene Expression Assay for Foxo3a (assay identification number Hs00921424_m1) and GAPDH (assay identification number Hs02758991_g1) (Applied Biosystems, Foster City, CA, USA) and TaqMan Gene Expression Master Mix (Applied Biosystems), or the following primer pairs: 5′‐TTTGGGGAAAGGGAGGAAGA‐3′ (forward) and 5′‐AGCCAAGAGAGGCACCACAG‐3′ (reverse) for Sp1 and 5′‐GGAACGGTGAAGGTGACAGC‐3′ (forward) and 5′‐AATCAAAGTCCTCGGCCACA‐3′ (reverse) for β‐actin and SYBR Premix Ex Taq II (Takara Bio, Shiga, Japan) using ABI 7900HT.
Measurement of intracellular reactive oxygen species (ROS). Measurement of intracellular ROS was performed as previously described.( 25 ) Briefly, KK47 cells (2.5 × 103) seeded into 96‐well palates were incubated with vehicle, mithramycin, doxorubicin, cisplatin, 5‐FU, and/or N‐acetyl‐L‐cysteine (NAC) for 48 h. Intracellular ROS levels were measured using CM‐H2DCFDA (Invitrogen) according to the manufacturer’s protocol. The fluorescence intensities of the wells were measured using the ARVO MX plate reader. At the same time, surviving cells were measured using the alamarBlue assay. The intracellular ROS levels were corrected by the corresponding results of the alamarBlue assay. The results are representative of at least three independent experiments.
Immunoprecipitation and western blot analysis. Immuno‐precipitation and western blot analysis was performed as previously described.( 27 ) Briefly, whole‐cell extracts (500 μg) were prepared and incubated with 20 μL of A/G agarose (Santa‐Cruz Biotechnology) for 1 h at 4°C. After centrifugation at 2400g at 4°C, the supernatants were incubated with 2.0 μg of rabbit IgG, anti‐Foxo3a, or anti‐GFP antibody with 20 μL of A/G agarose for overnight at 4°C. After washing three times with X‐buffer, the immunoprecipitated samples were applied to SDS‐PAGE, and western blotting was performed as previously described.
Results
Cisplatin‐resistant cells are more sensitive to mithramycin than their parental cells and show decreased expression of Foxo3a transcription factor. We have been researching the mechanisms of cisplatin resistance. We have previously established cisplatin‐resistant cells of bladder cancer.( 1 , 4 ) To identify anticancer agents that are effective against cisplatin‐resistant cells, we carried out a cytotoxicity assay to compare drug sensitivity between cisplatin‐resistant cells and their parental cells using 5‐FU, actinomycin D, methotrexate, gemcitabine, docetaxel, and paclitaxel. We have previously reported that cisplatin‐resistant cells were cross‐resistant to doxorubicin, etoposide, vincristine, mitomycin C, and camptothecin.( 1 , 4 ) In addition, cisplatin‐resistant cells were cross‐resistant with actinomycin D, methotrexate, gemcitabine, docetaxel, and paclitaxel (Supporting Fig. S1a). However, to our surprise, both cisplatin‐resistant cell lines (T24/DDP10 and KK47/DDP20) are significantly more sensitive to mithramycin than their parental cells by about 3‐fold (Fig. 1a). Previously, Foxo transcription factors have been shown to be involved in cisplatin resistance.( 21 , 22 ) Therefore, we next investigated the expression level of Foxo3a transcription factor. Both mRNA and protein levels of Foxo3a were lower in cisplatin‐resistant cells compared with those in their parental cells (Fig. 1b).
Figure 1.
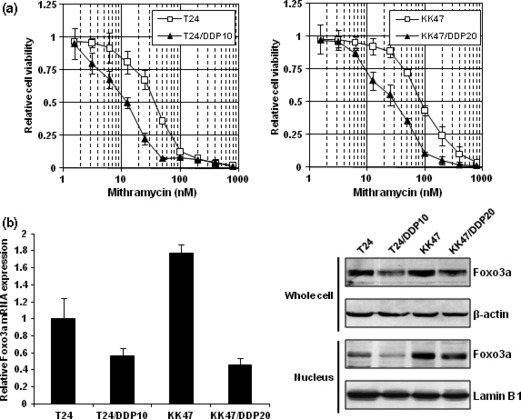
Cisplatin‐resistant cells are more sensitive to mithramycin than their parental cells and show decreased expression of Foxo3a transcription factor. (a) T24, T24/DDP10, KK47, and KK47/DDP20 cells were seeded into 96‐well plates. The following day, various concentrations of mithramycin were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of mithramycin corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD. (b) After extraction of total RNA from T24, T24/DDP10, KK47, and KK47/DDP20 cells and synthesis of cDNA, quantitative real‐time PCR was performed using the primers and probes for Foxo3a and GAPDH. The Foxo3a transcript level was corrected for the corresponding GAPDH transcript level. All values represent at least three independent experiments. The level of Foxo3a transcript from T24 cells was defined as 1. Boxes, mean; bars, ±SD. Whole‐cell extracts and nuclear extracts of T24, T24/DDP10, KK47, and KK47/DDP20 cells were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblots for β‐actin and Lamin B1 are shown as loading controls.
Foxo3a knockdown augments cancer cell growth and cisplatin resistance. Because Foxo transcription factors have been shown to be implicated in cancer cell growth,( 11 , 28 ) we next examined whether Foxo3a was involved in cancer cell growth in addition to cisplatin resistance. As shown in Figure 2(a), two kinds of Foxo3a‐specific siRNA, referred to as Foxo3a siRNA #1 and Foxo3a siRNA #2, suppressed Foxo3a expression at both the mRNA and protein levels. When KK47 cells were transfected with Foxo3a‐specific siRNAs, they grew more rapidly (Fig. 2b). In addition, as speculated from a finding that Foxo3a expression was decreased in cisplatin‐resistant cells, Foxo3a knockdown made KK47 cells more than 3‐fold resistant to cisplatin (Fig. 2c).
Figure 2.
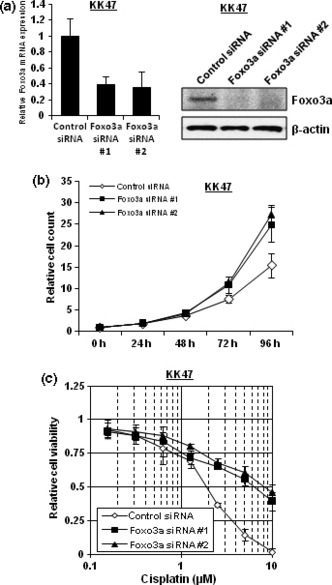
Foxo3a knockdown augments cancer cell growth and cisplatin resistance. (a) KK47 cells were transfected with 40 nm of control siRNA, Foxo3a siRNA #1 or Foxo3a siRNA #2. After extraction of total RNA and synthesis of cDNA, quantitative real‐time PCR was performed using the primers and probes for Foxo3a and GAPDH. The Foxo3a transcript level was corrected for the corresponding GAPDH transcript level. All values represent at least three independent experiments. The level of Foxo3a transcript from KK47 cells transfected with control siRNA was defined as 1. Boxes, mean; bars, ±SD. Whole‐cell extracts of KK47 cells transfected with 40 nm of control siRNA, Foxo3a siRNA #1, or Foxo3a siRNA #2 were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblot for β‐actin is shown as a loading control. (b) KK47 cells transfected with 40 nm of control siRNA, Foxo3a siRNA #1, or Foxo3a siRNA #2 were seeded into 12‐well plates and incubated. The number of cells was counted at the indicated times. The results were normalized to the number of cells at 0 h. All values represent at least three independent experiments. Boxes, mean; bars, ±SD. (c) KK47 cells transfected with 40 nm of control siRNA, Foxo3a siRNA #1, or Foxo3a siRNA #2 were seeded into 96‐well plates. The following day, various concentrations of cisplatin were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of cisplatin corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD.
Mithramycin increases the sensitivity of cancer cells to cisplatin through Foxo3a induction by oxidative stress. Foxo transcription factors are known to be implicated in oxidative stress.( 29 ) We compared intracellular ROS levels after the application of vehicle, mithramycin, doxorubicin, cisplatin, and 5‐FU. Mithramycin could induce intracellular ROS most effectively whereas doxorubicin and cisplatin induced little oxidative stress at least in our experimental setting. This ROS‐inducible effect of mithramycin was reduced by NAC addition (Fig. 3a). Foxo transcription factors are also known to be a stress‐inducible factor.( 30 ) Indeed, hydrogen peroxide and mithramycin enhanced the expression of Foxo3a transcription factor more than doxorubicin, cisplatin, and 5‐FU. However, Foxo3a induction by mithramycin was blunted by NAC addition, suggesting that Foxo3a induction may be dependent on oxidative stress by mithramycin (Fig. 3b). Since mithramycin is known to be an inhibitor of Sp1 binding to its cognate site, an effect of mithramycin on the induction of Foxo3a related to Sp1 inhibition was suggested. When Sp1 was suppressed using Sp1‐specific siRNA, Foxo3a expression was not affected at both mRNA and protein levels. Because it was suggested that Foxo3a is involved in cisplatin resistance as shown in 1, 2, we next carried out a cytotoxicity assay to investigate whether mithramycin could sensitize cancer cells to cisplatin. Although doxorubicin and 5‐FU could slightly sensitize KK47 cells to cisplatin, mithramycin increased the sensitivity of KK47 cells to cisplatin by more than 3‐fold and was the most effective of these anticancer agents (Fig. 3d). To investigate the mechanism by which mithramycin increases the sensitivity of cells to cisplatin, we performed a cytotoxicity assay after Foxo3a knockdown. After KK47 cells were transfected with Foxo3a‐specific siRNAs and 2 nm of mithramycin was added, the cells were exposed to various concentrations of cisplatin. As shown in Figure 3(e), Foxo3a knockdown abolished the favorable effects of mithramycin associated with augmentation of cisplatin cytotoxicity, suggesting that augmentation of cisplatin cytotoxicity by mithramycin involves the upregulation of Foxo3a.
Figure 3.
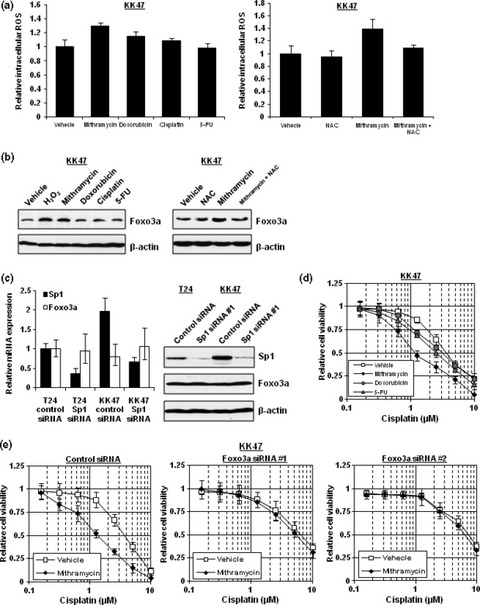
Mithramycin increases sensitivity of cancer cells to cisplatin through Foxo3a induction by oxidative stress. (a) KK47 cells were seeded into 96‐well plates and incubated. On the following day, the media were replaced with media containing vehicle, 2 nm of mithramycin, 2 nm of doxorubicin, 0.2 μm of cisplatin, or 2 μm of 5‐fluorouracil (5‐FU) and/or 5 mm of NAC. After 48 h, the cells were stained with CM‐H2DCFDA and measured for their florescence intensities. All values are representative of at least three independent experiments. The fluorescence intensity of KK47 cells with vehicle was set as 1. Boxes, mean; bars, ±SD. (b) Whole‐cell extracts of KK47 incubated with media containing vehicle, 2 μm of hydrogen peroxide, 2 nm of mithramycin, 2 nm of doxorubicin, 0.2 μm of cisplatin, or 2 μm of 5‐FU and/or 5 mm of NAC for 48 h were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblot for β‐actin is shown as a loading control. (c) T24 and KK47 cells were transfected with 40 nm of control siRNA or Sp1 siRNA. After extraction of total RNA and synthesis of cDNA, quantitative real‐time PCR was performed using the primers and probes for Sp1, Foxo3a, β‐actin, and GAPDH. The Sp1 and Foxo3a transcript levels were corrected for the corresponding β‐actin and GAPDH transcript levels, respectively. All values represent at least three independent experiments. The level of Sp1 and Foxo3a transcript from T24 cells transfected with control siRNA was defined as 1. Boxes, mean; bars, ±SD. Whole‐cell extracts of T24 and KK47 cells transfected with 40 nm of control siRNA or Sp1 siRNA were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblot for β‐actin is shown as a loading control. (d) KK47 cells were seeded into 96‐well plates with media containing vehicle, 2 nm of mithramycin, 2 nm of doxorubicin, or 2 μm of 5‐FU. The following day, various concentrations of cisplatin were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of cisplatin corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD. (e) KK47 cells transfected with 40 nm control siRNA, Foxo3a siRNA #1, or Foxo3a siRNA #2 were seeded into 96‐well plates with media containing vehicle or 2 nm of mithramycin. The following day, various concentrations of cisplatin were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of cisplatin corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD.
Deacetylation of Foxo3a is induced by cisplatin treatment and the acetylated‐Foxo3a level is decreased in cisplatin‐resistant cells. It has been reported that the acetylation status of Foxo transcription factor could influence its transcriptional activity and selection of the target gene.( 18 ) Sirt1 and p300 are known to deacetylate and acetylate Foxo transcription factors, respectively.( 30 , 31 , 32 , 33 , 34 ) First, we investigated whether cisplatin affects the expression of the Foxo3a‐associated deacetylase and acetyltransferase. As shown in Figure 4(a), Sirt1 expression increased according to the duration of cisplatin treatment up to 24 h, although p300 decreased. Consequently, the acetylated‐Foxo3a level decreased according to the duration of cisplatin treatment (Fig. 4b). Based on the above finding that cisplatin induces Foxo3a deacetylation, we analyzed the expression levels of Sirt1 and p300 in cisplatin‐resistant cells. As shown in Figure 4(c), both cisplatin‐resistant cell lines showed increased expression of Sirt1 but decreased expression of p300. As a result, both cisplatin‐resistant cell lines had a lower acetylation status of Foxo3a, suggesting that the acetylation status of Foxo3a is also involved in the cisplatin‐resistant phenotype (Fig. 4d).
Figure 4.
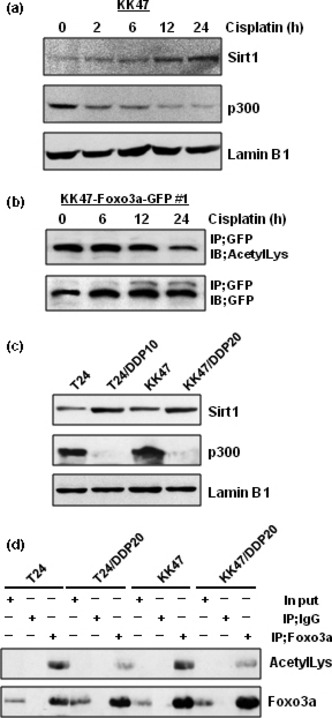
Deacetylation of Foxo3a is induced by cisplatin treatment and the acetylated‐Foxo3a level is decreased in cisplatin‐resistant cells. (a) KK47 cells were treated with 1 μm of cisplatin for the indicated times. The cells were then harvested and nuclear extracts were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblotting for Lamin B1 is shown as a loading control. (b) KK47‐Foxo3a‐GFP #1 cells were seeded into six‐well plates. The following day, the cells were treated with 1 μm of cisplatin for the indicated times. Whole‐cell extracts were immunoprecipitated with 2.0 μg of anti‐GFP antibody and 20 μL of A/G agarose. After washing three times, the supernatants were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. (c) Nuclear extracts of T24, T24/DDP10, KK47, and KK47/DDP20 cells were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblot for Lamin B1 is shown as a loading control. (d) Whole‐cell extracts (500 μg) of T24, T24/DDP10, KK47, and KK47/DDP20 cells were immunoprecipitated with antibody against Foxo3a and A/G agarose. After washing three times, the supernatants were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies.
Silencing of the Foxo3a‐associated acetyltransferase p300 promotes cell growth and increases cellular resistance to cisplatin. Because Foxo3a is implicated in tumor cell growth as shown in Figure 2(b), we examined whether the acetylation status of Foxo3a is also implicated in cell growth. In KK47 cells, p300 knockdown using p300‐specific siRNAs induced more rapid cell proliferation (Fig. 5a). In addition, T24 cells also grew more rapidly when p300 expression was silenced (Fig. 5b). To investigate whether the acetylation status of Foxo3a influences cisplatin resistance, we performed a cytotoxicity assay after suppressing p300 expression. We found that p300 knockdown rendered the T24 and KK47 cells resistant to cisplatin, but not to 5‐FU (Figs 5c,d).
Figure 5.
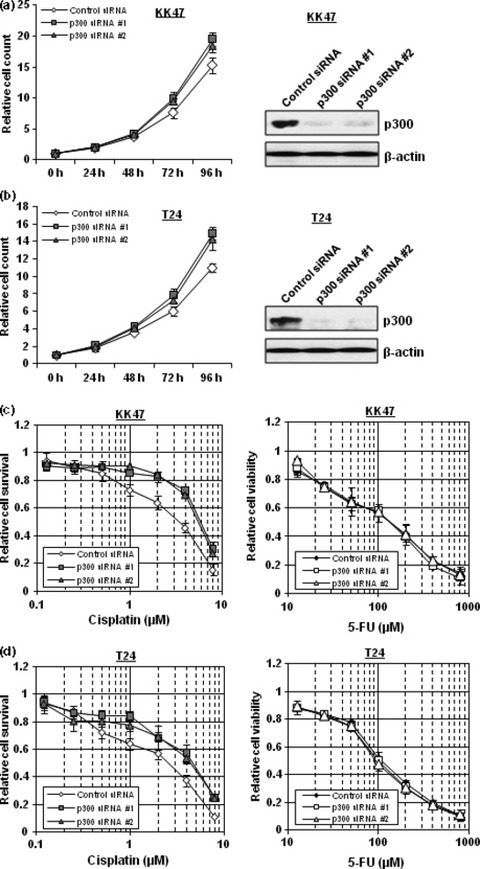
Silencing of the Foxo3a‐associated acetyltransferase p300 promotes cell growth and renders cells resistant to cisplatin. (a, b) KK47 (a) and T24 (b) cells transfected with 40 nm control siRNA, p300 siRNA #1, or p300 siRNA #2 were seeded into 12‐well plates and incubated. The number of cells was counted at the indicated times. The results were normalized to the number of cells at 0 h. All values represent at least three independent experiments. Boxes, mean; bars, ±SD. Whole‐cell extracts of KK47 (a) and T24 (b) cells transfected with 40 nm of control siRNA, p300 siRNA #1 or p300 siRNA #2 were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblot for β‐actin is shown as a loading control. (c,d) KK47 (c) and T24 (d) cells transfected with 40 nm control siRNA, p300 siRNA #1, or p300 siRNA #2 were seeded into 96‐well plates. The following day, various concentrations of cisplatin or 5‐fluorouracil (5‐FU) were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of cisplatin or 5‐FU corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD.
Non‐acetylation‐mimicking Foxo3a overexpression suppresses cancer‐cell growth and cisplatin resistance to a lesser extent compared with wild‐type Foxo3a overexpression. Because expression and acetylation levels of Foxo3a were suggested to be implicated in cancer cell growth and cisplatin resistance, we investigated effects on cancer cell growth and drug resistance by wild‐type and non‐acetylation‐mimicking Foxo3a overexpressions. Then, we prepared Foxo3a‐GFP and Foxo3a‐GFP KR expression plasmids. Foxo3a‐GFP KR plasmid expresses non‐acetylation‐mimicking Foxo3a in which lysines 242 and 245 were substituted into arginines. In addition, stable transfectants of KK47 cells expressing GFP, Foxo3a‐GFP, or Foxo3a‐GFP KR protein were established. As shown in Figure 6(a), these stable transfectants expressed almost equal amounts of exogeneous GFP‐tagged proteins and endogeneous Foxo3a proteins. Further, mutated Foxo3a protein (Foxo3a‐GFP KR) was not acetylated (Fig. 6b). KK47‐Foxo3a‐GFP (#1 and #2) cells prominently grew slowly compared with KK47‐AcGFP (#1 and #2) cells. However, KK47‐Foxo3a‐GFP KR (#1 and #2) cells grew more rapidly than KK47‐Foxo3a‐GFP (#1 and #2) cells although more slowly than KK47‐AcGFP (#1 and #2) cells (Fig. 6c). In addition, Foxo3a overexpression made KK47 cells more than 4‐fold sensitive to cisplatin. However, KK47‐Foxo3a‐GFP KR (#1 and #2) cells were less sensitive to cisplatin than KK47‐Foxo3a‐GFP (#1 and #2) cells (Fig. 6d).
Figure 6.
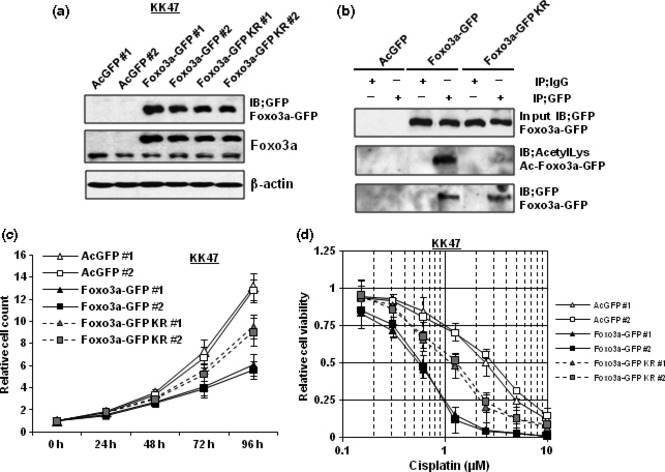
Non‐acetylation‐mimicking Foxo3a overexpression suppresses cancer cell growth and cisplatin resistance to a lesser extent compared with wild‐type Foxo3a overexpression. (a) Whole‐cell extracts of the indicated stable transfectants of KK47 cells were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblot for β‐actin is shown as a loading control. (b) Whole‐cell extracts (500 μg) of the indicated stable transfectants of KK47 cells were immunoprecipitated with antibody against GFP and A/G agarose. After washing three times, the supernatants were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. (c) The indicated stable transfectants of KK47 cells were seeded into 12‐well plates and incubated. The number of cells was counted at the indicated times. The results were normalized to the number of cells at 0 h. All values represent at least three independent experiments. Boxes, mean; bars, ±SD. (d) The indicated stable transfectants of KK47 cells were seeded into 96‐well plates. The following day, various concentrations of cisplatin were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of cisplatin corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD.
Discussion
Foxo3a is a member of the Foxo family of transcription factors which, on exposure to oxidative stress, translocate to the nucleus and activate transcription by specifically binding to the consensus sequence TTGTTTAC in the promoters of target genes.( 35 ) Overexpression of Foxo3a protects cells from oxidative damage.( 29 ) Furthermore, it has been reported that ionizing radiation induces Foxo3a expression.( 13 ) In addition, this study revealed that hydrogen peroxide and mithramycin increased Foxo3a expression through ROS production. On the other hand, no apparent Foxo3a induction by doxorubicin and cisplatin was observed although doxorubicin and cisplatin produced little ROS (Fig. 3). These findings may indicate that Foxo3a expression can be induced by oxidative stress although Foxo3a expression may be modulated also by DNA‐ and RNA‐binding functions of anticancer agents and by other factors, accounting for inconsistency between ROS production and Foxo3a expression by doxorubicin and cisplatin treatments. On the other hand, cisplatin‐resistant cells showed decreased expressions of Foxo3a mRNA and protein, and Foxo3a knockdown decreased the sensitivity to cisplatin (Figs 1,2). This is consistent with the finding that inhibiting the phosphorylation of Foxo3a sensitizes ovarian cancer cells to cisplatin( 21 ) and that cisplatin induces Foxo3a dephosphorylation and nuclear translocation, and expression of its target genes in cisplatin‐sensitive cells.( 22 ) Furthermore, these results are supported by another finding that Foxo transcription factors induce cell cycle arrest and apoptosis in response to genotoxic stresses such camptothecin and ionizing radiation.( 13 ) Cisplatin also exhibits DNA damage stress and leads to cell cycle arrest or apoptosis via DNA damage signaling. Because Foxo3a can induce cell cycle arrest in response to DNA damage and the DNA repair pathway, cellular apoptosis by cisplatin might be dependent on Foxo3a. On the other hand, it was reported that Foxo3a protects cells from oxidative stress.( 29 ) Although cisplatin is also known to induce oxidative stress,( 36 ) the stress level produced by cisplatin was less than that induced by either doxorubicin or mithramycin. Therefore, it seems that cytotoxicity induced by cisplatin is less dependent on the protective function of Foxo3a against oxidative stress compared with other cytotoxic anticancer agents that produce greater oxidative stress. In addition, Foxo3a transcription factor might affect cisplatin resistance by inducing known or unknown Foxo3a‐target genes. We have previously shown that both Twist1 and YB‐1 are implicated in cisplatin resistance.( 7 , 8 ) Therefore, Foxo3a might be involved in cisplatin resistance by regulating Twist1, YB‐1, and other targets.
Cisplatin sensitivity is well known to have a close association with cell growth. Generally, rapid‐growing cells are more sensitive to cisplatin. Inversely, cancer stem cells which are thought to be resistant to conventional therapy such as chemotherapy have been revealed to grow slowly. As shown in Supporting Figure S1(b), cisplatin‐resistant cells grew more slowly compared with their parental cells. This finding seemed to be inconsistent with the results that Foxo3a downregulation increased cell growth and that Foxo3a expression decreased in cisplatin‐resistant cells. However, intriguingly, overexpression of YB‐1, which is a molecule promoting cisplatin resistance, has recently been revealed to trigger an epithelial–mesenchymal transition, but to suppress cell growth in breast epithelial cells.( 37 ) This phenomenon may be one of causes of slow growth in cisplatin‐resistant cells. In addition, it is known that cisplatin resistance is gained by multiple mechanisms. It may be accounted for, at least in part, by the fact that both cisplatin‐resistant cells more abundantly expressed p21 which is a famous inhibitor of cyclin‐dependent kinase, resulting in suppression of cell growth.
Although phosphorylation and intracellular localization of Foxo transcription factors has been reported to be involved in cisplatin resistance, intracellular localisation of Foxo3a in cisplatin‐resistant cells seemed not to be affected compared with their parental cells in this study (Fig. 1b). Acetylation of Foxo3a, which affects its DNA‐binding and transcriptional ability, but not intracellular localization, might be a key factor of cisplatin resistance in our settings. Several researchers have previously reported that the acetylation status of Foxo transcription factors could influence its transcriptional activity ( 30 ) and Foxo transcription factors interact with Sirt1 and p300, which deacetylate and acetylate Foxo3a, respectively.( 18 , 31 , 32 , 33 , 34 ) In this study, cisplatin treatment induced the deacetylation of Foxo3a, and cisplatin‐resistant cells were revealed to possess a reduced amount of the acetylated form of Foxo3a compared with their parental cells. As shown in Figure 5, p300 affected cellular sensitivity to cisplatin; this functional effect may be obtained through a modulation of Foxo3a activity by p300.( 34 ) It has previously been shown that Sirt1 suppressed the ability of Foxo3a to induce the transcription of the proapoptotic factor Bim.( 38 ) In addition, it was reported that the transcription of Fas ligand is decreased by Sirt1.( 18 ) On the other hand, the DNA repair‐related gene GADD45α and the antioxidant protein MnSOD were induced by Sirt1 overexpression.( 18 ) In addition to our results that p300 knockdown increases resistance to cisplatin, Sirt1 was recently shown to be involved in cisplatin resistance.( 38 ) Treatment with a Sirt1 inhibitor, sirtinol, was shown to increase sensitivity to camptothecin and cisplatin.( 39 ) Moreover, others have shown that reducing Sirt1 expression by Sirt1 siRNA sensitized cisplatin‐resistant cells to cisplatin, and that Sirt1 overexpression increased cisplatin resistance in cisplatin‐sensitive cells by altering mitochondrial metabolism.( 40 ) Thus, in addition to the decreased Foxo3a protein level, the deacetylation status of Foxo3a also contributed to cell survival after genotoxic stress (Fig. 5). These findings indicate that Foxo3a may be a promising molecular target for overcoming cisplatin resistance. Moreover, we showed that p300 knockdown increases cancer cell proliferation, which is supported by the findings that p300 depletion leads to c‐Myc upregulation and premature G1 exit.( 41 ) Therefore, examining the expression level and acetylation status of Foxo3a in addition to Foxo3a‐associated acetyltransferases and deacetylases in clinical cancer might be useful to predict the effect of cisplatin‐based chemotherapy and prognosis of cancer. In addition, this study indicated that we could increase cisplatin cytotoxicity by manipulating the expression and acetylation status of Foxo3a.
Mithramycin is currently rarely used in cancer treatment. However, our findings suggest that mithramycin is very effective when used for cisplatin‐resistant cell or in combination with cisplatin. In addition, the effectiveness of mithramycin and cisplatin combination therapy was due to the induction of Foxo3a by mithramycin (Fig. 3). Thalidomide was initially used in Europe as a sedative, tranquilizer, and as an antiemetic for emesis gravidarum, but was withdrawn from the market in 1961 because of reports of congenital defects when taken during gestation. More recently, thalidomide has been shown to prevent neoangiogenesis in human malignancies and to exert immunomodulatory and anti‐inflammatory properties. Accordingly, thalidomide was first used 10 years ago to treat patients with multiple myeloma.( 42 ) Mithramycin, which is a strong Foxo3a inducer, might also become a re‐emerging anticancer drug similar to thalidomide. Furthermore, other molecules, which might more strongly induce Foxo transcription factors and acetylate Foxo transcription factors should be identified and developed. Accordingly, studies are needed to investigate whether mithramycin or other unknown Foxo‐inducing agents could suppress cancer cell growth and cisplatin resistance when used in combination with or without cisplatin in vivo.
Acknowledgments
This work was supported by Health Sciences Research Grants for Clinical Research for Evidenced‐based Medicine and Grants‐in‐Aid for Cancer Research (016), from the Ministry of Health, Labor and Welfare of Japan. We would like to thank Dr. Dongchon Kang (Kyushu University, Fukuoka, Japan) for helping with quantitative real‐time PCR, and Noriko Hakoda and Seiko Kamori for their technical assistance. The authors would like to acknowledge the technical expertise of the Support Center for Education and Research, Kyushu University.
Supporting information
Fig. S1. (a) T24, KK47, and their cisplatin‐resistant cells were seeded into 96‐well plates. The following day, various concentrations of the indicated anticancer drugs were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of anticancer drug corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD. (b) T24, KK47, and their cisplatin‐resistant cells were seeded into 12‐well plates and incubated. The number of cells was counted at the indicated times. The results were normalized to the number of cells at 0 h. All values represent at least three independent experiments. Boxes, mean; bars, ±SD. (c) Nuclear extracts of T24, T24/DDP10, KK47, and KK47/DDP20 cells were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblots for Lamin B1 are shown as loading controls.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
References
- 1. Kotoh S, Naito S, Yokomizo A et al. Increased expression of DNA topoisomerase I gene and collateral sensitivity to camptothecin in human cisplatin‐resistant bladder cancer cells. Cancer Res 1994; 54: 3248–52. [PubMed] [Google Scholar]
- 2. Hasegawa S, Abe T, Naito S et al. Expression of multidrug resistance‐associated protein (MRP), MDR1 and DNA topoisomerase II in human multidrug‐resistant bladder cancer cell lines. Br J Cancer 1995; 71: 907–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yokomizo A, Ono M, Nanri H et al. Cellular levels of thioredoxin associated with drug sensitivity to cisplatin, mitomycin C, doxorubicin, and etoposide. Cancer Res 1995; 55: 4293–6. [PubMed] [Google Scholar]
- 4. Kotoh S, Naito S, Yokomizo A, Kohno K, Kuwano M, Kumazawa J. Enhanced expression of gamma‐glutamylcysteine synthetase and glutathione S‐transferase genes in cisplatin‐resistant bladder cancer cells with multidrug resistance phenotype. J Urol 1997; 157: 1054–8. [PubMed] [Google Scholar]
- 5. Tada Y, Wada M, Kuroiwa K et al. MDR1 gene overexpression and altered degree of methylation at the promoter region in bladder cancer during chemotherapeutic treatment. Clin Cancer Res 2000; 6: 4618–27. [PubMed] [Google Scholar]
- 6. Tsunoda T, Koga H, Yokomizo A et al. Inositol 1,4,5‐trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 2003; 24: 1396–402. [DOI] [PubMed] [Google Scholar]
- 7. Shiota M, Izumi H, Onitsuka T et al. Twist promotes tumor cell growth through YB‐1 expression. Cancer Res 2008; 68: 98–105. [DOI] [PubMed] [Google Scholar]
- 8. Shiota M, Izumi H, Tanimoto A et al. Programmed cell death protein 4 down‐regulates Y‐box binding protein‐1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res 2009; 69: 3148–56. [DOI] [PubMed] [Google Scholar]
- 9. Naito S, Yokomizo A, Koga H. Mechanisms of drug resistance in chemotherapy for urogenital carcinoma. Int J Urol 1999; 6: 427–39. [DOI] [PubMed] [Google Scholar]
- 10. Naito S, Koga H, Yokomizo A et al. Molecular analysis of mechanisms regulating drug sensitivity and the development of new chemotherapy strategies for genitourinary carcinomas. World J Surg 2000; 24: 1183–6. [DOI] [PubMed] [Google Scholar]
- 11. Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene 2008; 27: 2312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tran H, Brunet A, Grenier JM et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002; 296: 530–4. [DOI] [PubMed] [Google Scholar]
- 13. Tsai WB, Chung YM, Takahashi Y, Xu Z, Hu MC. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol 2008; 10: 460–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci 2007; 120: 2479–87. [DOI] [PubMed] [Google Scholar]
- 15. Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol 2003; 162: 613–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor‐related apoptosis inducing ligand expression. Implication for PTEN mutation in prostate cancer. J Biol Chem 2002; 277: 47928–37. [DOI] [PubMed] [Google Scholar]
- 17. Dijkers PF, Medema RH, Pals C et al. Forkhead transcription factor FKHR‐L1 modulates cytokine‐dependent transcriptional regulation of p27KIP1 . Mol Cell Biol 2000; 20: 9138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brunet A, Sweeney LB, Sturgill JF et al. Stress‐dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303: 2011–5. [DOI] [PubMed] [Google Scholar]
- 19. Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J Clin Invest 1991; 88: 1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Majee S, Dasgupta D, Chakrabarti A. Interaction of the DNA‐binding antitumor antibiotics, chromomycin and mithramycin with erythroid spectrin. Eur J Biochem 1999; 260: 619–26. [DOI] [PubMed] [Google Scholar]
- 21. Arimoto‐Ishida E, Ohmichi M, Mabuchi S et al. Inhibition of phosphorylation of a forkhead transcription factor sensitizes human ovarian cancer cells to cisplatin. Endocrinology 2004; 145: 2014–22. [DOI] [PubMed] [Google Scholar]
- 22. Fernández de Mattos S, Villalonga P, Clardy J, Lam EW. Foxo3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol Cancer Ther 2008; 7: 3237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shiota M, Izumi H, Onitsuka T et al. Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 2008; 27: 5543–53. [DOI] [PubMed] [Google Scholar]
- 24. Shiota M, Izumi H, Miyamoto N et al. Ets regulates peroxiredoxin1 and 5 expressions through their interaction with the high mobility group protein B1. Cancer Sci 2008; 99: 1950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shiota M, Yokomizo A, Tada Y et al. Castration resistance of prostate cancer cells caused by castration‐induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene 2010; 29: 237–50. [DOI] [PubMed] [Google Scholar]
- 26. Shiota M, Yokomizo A, Tada Y et al. Peroxisome proliferator activated receptor γ coactivator‐1α interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol Endocrinol 2010; 24: 114–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shiota M, Yokomizo A, Masubuchi D et al. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate doi: 10.1002/pros.21088 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 28. You H, Yamamoto K, Mak TW. Regulation of transactivation‐independent proapoptotic activity of p53 by FOXO3a. Prog Natl Acad Sci U S A 2006; 103: 9051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc‐dependent signaling pathway. Science 2002; 295: 2450–2. [DOI] [PubMed] [Google Scholar]
- 30. Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends in Cell Biol 2004; 14: 408–12. [DOI] [PubMed] [Google Scholar]
- 31. Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 vai Sirt‐dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 2005; 280: 20589–95. [DOI] [PubMed] [Google Scholar]
- 32. Kitamura YI, Kitamura T, Kruse JP et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab 2005; 2: 153–63. [DOI] [PubMed] [Google Scholar]
- 33. Perrot V, Recfler MM. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1‐induced transcription. Mol Endocrinol 2005; 19: 2283–98. [DOI] [PubMed] [Google Scholar]
- 34. Van Der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300‐mediated acetylation. Trends in Biochem Sci 2005; 30: 81–6. [DOI] [PubMed] [Google Scholar]
- 35. Pierrou S, Hellqvist M, Samuelsson L, Enerbäck S, Carlsson P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J 1994; 13: 5002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 2008; 73: 994–1007. [DOI] [PubMed] [Google Scholar]
- 37. Evdokimova V, Tognon C, Ng T et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB‐1 promotes an epithelial‐mesenchymal transition. Cancer Cell 2009; 15: 402–15. [DOI] [PubMed] [Google Scholar]
- 38. Motta MC, Divecha N, Lemieux M et al. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004; 116: 551–63. [DOI] [PubMed] [Google Scholar]
- 39. Kojima K, Ohhashi R, Fujita Y et al. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun 2008; 373: 423–8. [DOI] [PubMed] [Google Scholar]
- 40. Liang XJ, Finkel T, Shen DW, Yin JJ, Aszalos A, Gottesman MM. SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol Cancer Res 2008; 6: 1499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kolli S, Buchmann AM, Williams J, Weitzmen S, Thimmapaya B. Antisense‐mediated depletion of p300 in human cells leads to premature G1 exit and up‐regulation of c‐MYC . Prog Natl Acad Sci U S A 2001; 98: 4646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palumbo A, Facon T, Sonneveld P et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood 2008; 111: 3968–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (a) T24, KK47, and their cisplatin‐resistant cells were seeded into 96‐well plates. The following day, various concentrations of the indicated anticancer drugs were applied. After incubation for 48 h, cell survival was analyzed by a cytotoxicity assay. Cell survival in the absence of anticancer drug corresponds to 1. All values are representative of at least three independent experiments. Boxes, mean; bars, ±SD. (b) T24, KK47, and their cisplatin‐resistant cells were seeded into 12‐well plates and incubated. The number of cells was counted at the indicated times. The results were normalized to the number of cells at 0 h. All values represent at least three independent experiments. Boxes, mean; bars, ±SD. (c) Nuclear extracts of T24, T24/DDP10, KK47, and KK47/DDP20 cells were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. Immunoblots for Lamin B1 are shown as loading controls.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item