

Abstract
Vascular endothelial growth factor (VEGF)‐D induces lymphangiogenesis by activating VEGF receptor (VEGFR)‐3, which is expressed mainly by lymphatic endothelial cells. VEGFR‐3 has also been detected in several types of malignant cells, but the significance of VEGFR‐3 expression by malignant cells remains unclear. We examined the expression and function of VEGF‐D/VEGFR‐3 in human gastric carcinoma cells. Expression of VEGF‐D and VEGFR‐3 was analyzed in three human gastric carcinoma cell lines and 29 surgical specimens. cDNA microarray analysis was used to examine the effect of VEGF‐D on the expression of genes associated with disease progression in VEGFR‐3‐expressing KKLS cells. VEGF‐D‐transfected cells and control cells were transplanted into the gastric wall of nude mice. In 10 of the 29 (34%) gastric carcinoma specimens and two of the three cell lines, cancer cells expressed both VEGF‐D and VEGFR‐3. In vitro treatment of KKLS cells with exogenous VEGF‐D increased expression of cyclin D1 and Bcl‐2 and stimulated cell proliferation. VEGF‐D transfection into KKLS cells resulted in stimulation of angiogenesis, lymphangiogenesis, and cell proliferation, and in inhibition of apoptosis. VEGF‐D may participate in the progression of human gastric carcinoma by acting via autocrine and paracrine mechanisms. (Cancer Sci 2010)
Gastric cancer is one of the most frequently occurring malignancies in the world.( 1 ) Acquisition of metastatic potential by tumor cells results in a poor prognosis for patients with gastric carcinoma. The extent of lymph node metastasis is one of the most important prognostic factors and determines the course of cancer therapy.( 2 )
Lymphangiogenesis and angiogenesis are regulated by members of the vascular endothelial growth factor (VEGF) family and their receptors.( 3 , 4 ) VEGF‐A is a major inducer of angiogenesis and vessel permeability.( 5 , 6 ) The VEGF family includes VEGF‐A, ‐B, ‐C, ‐D, ‐E, ‐F, and placental growth factor (PlGF).( 7 ) VEGF‐D, also known as c‐fos‐induced growth factor,( 8 ) is secreted as a preprotein that undergoes proteolytic processing.( 9 ) Full‐length VEGF‐D displays high affinity for VEGFR‐3, whereas its affinity for VEGFR‐2 is increased through progressive proteolytic cleavage.( 9 ) In animal studies, VEGF‐D has been shown to enhance tumor angiogenesis and lymphangiogenesis, thereby promoting metastatic spread of tumor cells via the lymphatics.( 10 , 11 , 12 ) In clinical studies, correlation between expression of VEGF‐D by tumor cells and lymph node metastasis has been reported.( 13 , 14 , 15 , 16 , 17 , 18 ) In contrast, some clinical studies have shown that the level of VEGF‐D expression is lower in tumor tissue than in the corresponding normal tissue.( 19 , 20 , 21 , 22 , 23 ) Therefore, it remains controversial whether VEGF‐D is the main regulator of lymphangiogenesis. Because of its structural similarity to VEGF‐C, VEGF‐D is thought to have a similar biologic effect. Recently, we studied the expression and role of the VEGF‐C/VEGFR‐3 axis in human gastric carcinoma.( 24 ) We showed that the tumor cells expressed not only VEGF‐C but also VEGFR‐3. In vitro and in vivo experiments showed that VEGF‐C acts as a progressive growth factor, in addition to acting as a lymphangiogenic and angiogenic factor. However, the autocrine role of VEGF‐D in gastric carcinoma cells has not been characterized. Thus, we analyzed the role of the VEGF‐D/VEGFR‐3 axis in human gastric carcinoma cells.
Materials and Methods
Surgical specimens of gastric carcinoma. Paraffin‐embedded archival specimens of invasive gastric carcinoma obtained from 29 patients who underwent surgical resection at Hiroshima University Hospital were studied by immunohistochemistry. All tumors had invaded beyond the submucosa. The criteria for staging and histologic classification were those proposed by the Japanese Research Society for Gastric Cancer.( 25 ) The patient group comprised 27 men and two women, with a median age of 66 years (range, 34–81 years). The tissues were immediately snap‐frozen and stored in liquid nitrogen for semiquantitative RT‐PCR.
Cell cultures. Three cell lines established from human gastric carcinomas were maintained in RPMI‐1640 medium (Nissui, Tokyo, Japan) with 10% FBS (MA BioProducts, Walkersville, MD, USA). The TMK‐1 cell line (derived from poorly differentiated adenocarcinoma) was provided by Dr E. Tahara (Hiroshima University, Hiroshima, Japan). The KKLS cell line (derived from undifferentiated carcinoma) was provided by Dr Y. Takahashi (Chiba University, Chiba, Japan). The MKN‐1 cell line (derived from adenosquamous carcinoma) was obtained from the Health Science Research Resources Bank (Osaka, Japan).
Cell proliferation assay. In vitro growth was measured with a Cell Proliferation Biotrak ELISA System, version 2 (Amersham Biosciences, Piscataway, NJ, USA) according to the manufacturer’s instructions to determine whether recombinant human VEGF‐D (rhVEGF‐D) (R&D Systems, Minneapolis, MN, USA) would stimulate proliferation of KKLS cells and TMK‐1 cells. Cells (1 × 104) were seeded on a 96‐well plate, and BrdU (final concentration, 10 μM) was added to the culture medium with or without rhVEGF‐D (2, 20, or 100 ng/mL) for 24 h. Cell proliferation was quantified in a plate reader (Microplate Manager 5.2.1; Bio‐Rad, Hercules, CA, USA) at 450 nm.
Microarray analysis. We performed microarray analysis using the Human Cancer CHIP (version 4; Takara Shuzo, Kyoto, Japan), in which 886 human cancer‐related genes are spotted on glass plates. KKLS cells were cultured in RPMI‐1640 medium without FBS for 6 h and then cultured with or without rhVEGF‐D (20 ng/mL) for 8 h. A fluorescent probe synthesized by reverse transcription of 1 μg of the mRNA with 50 U AMV reverse transcriptase (Takara Shuzo) was added to each reaction mixture. Cy3‐ and Cy5‐labeled probes were prepared by using mRNAs isolated from control cells and rhVEGF‐D cells, and both were mixed in the reaction buffer (6× SSC/0.2% SDS and 5× Denhardt’s solution, 0.8 mg/mL poly [dA], and 1 mg/mL yeast tRNA). The mixture was hybridized to the cDNA CHIP at 65°C overnight. The CHIP was washed twice with 2× SSC/0.2% SDS solution at 55°C for 30 min and then with the same solution at 65°C for 5 min. Finally, the CHIP was washed with 0.05 × SSC at room temperature for 10 min. Signals on the hybridized CHIP were visualized and quantified with a Scan‐Array 5000 laser scanner (GSI Lomonids) and normalized to the averaged signals of housekeeping genes. Genes were excluded from further investigation when the intensities of both Cy3 and Cy5 were below 1000 fluorescence units. Those with Cy3/Cy5 signal ratios >2.0 were regarded as up‐regulated.
Semiquantitative reverse transcription‐polymerase chain reaction (RT‐PCR) and quantitative real‐time PCR. Total RNA was extracted from the gastric cancer cell lines with an RNeasy Kit (Qiagen, Tokyo, Japan) according to the manufacturer’s instructions. RT‐PCR was performed with the isolated RNA (1 μg). cDNA was generated from 1 μg of total RNA with a First‐Strand cDNA Synthesis Kit (Amersham Biosciences, Buckinghamshire, UK). The primers and annealing temperatures for VEGF‐D, VEGFR‐2, VEGFR‐3, β‐actin, GAPDH, and Bcl‐2 are given in Table 1. The primers were designed with specific Primer analysis software (Primer Designer; Scientific and Educational Software, Arlington, MA, USA), and the specificities of the sequences were confirmed by FASTA analysis (EMBL Nucleotide Sequence Database). Semiquantitative RT‐PCR was performed with an AmpliTaq Gold Kit (Roche, Mannheim, Germany), and quantitative real‐time PCR was performed with a LightCycler‐FastStart DNA Master SYBR‐Green I Kit (Roche) according to the manufacturer’s instructions. Quantitative real‐time PCR was used to monitor gene expression and was performed with a LightCycler system and Light Cycler Data Analysis Software version 3.5 (Roche) according to standard procedures. To correct for differences in both RNA quality and quantity between samples, the data were normalized to those for β‐actin.
Table 1.
Application of PCR
| Gene | Primer sequences | Number of cycles | Annealing temperature | Product size (bp) |
|---|---|---|---|---|
| VEGF‐D | F: 5′‐GTATGGACTCTCGCTCAGCAT‐3′ | 32 | 59 | 226 |
| R: 5′‐AGGCTCTCTTCATTGCAACAG‐3′ | ||||
| VEGFR‐2 | F: 5′‐GCATCTCATCTGTTACAGC‐3′ | 32 | 62 | 331 |
| R: 5′‐CTTCATCAATCTTTACCCC‐3′ | ||||
| VEGFR‐3 | F: 5′‐GGTTCCTCCAGGATGAAGAC‐3′ | 32 | 62 | 505 |
| R: 5′‐CAAGCAGTAACGCCAGTGTC‐3′ | ||||
| β‐Actin | F: 5′‐GGACTTCGAGCAAGAGATGG‐3′ | 35 | 55 | 234 |
| R: 5′‐AGCACTGTGTTGGCGTACAG‐3′ | ||||
| GAPDH | F: 5′‐ATCATCCCTGCCTCTACTGG‐3′ | 28 | 55 | 188 |
| R: 5′‐CCCTCCGACGCCTGCTTCAC‐3′ | ||||
| Bcl‐2 | F: 5′‐GGTGGAGGAGCTCTTCCAGG‐3′ | 35 | 60 | 206 |
| R: 5′‐ACAGTTCCACAAAGGCATCC‐3′ |
VEGF‐D, vascular endothelial growth factor D; VEGFR‐2, VEGF receptor 2.
Immunofluorescence staining for pVEGFR‐3. KKLS cells were cultured in RPMI‐1640 medium without FBS for 24 h, then treated with or without rhVEGF‐D (20 ng/mL) for 10 min. pVEGFR‐2,3 (1:1000; Calbiochem, San Diego, CA, USA) was used to stain pVEGFR‐2,3 in tumor cells.
Western blot analysis. Western blotting was used to evaluate the expression of cyclin D1, Akt and MAPK, p38, and JNK phosphorylation. To evaluate Akt, MAPK, p38, and JNK phosphorylation of KKLS cells, cells were plated at 1.5 × 105 cells/mL per well in a six‐well plate and incubated overnight. Cells were then starved for 24 h in serum‐free medium and stimulated with rhVEGF‐D (20 ng/mL) or rhVEGF‐C (20 ng/mL) (R&D Systems) for 10 min at 37°C. After stimulation, cells were washed three times in cold PBS containing 1 mmol/L sodium. Cell lysates were prepared with M‐PER Mammalian Protein Extraction Reagent (Pierce, Rockford, IL, USA). Samples of cell lysates (20 μg protein) were separated by SDS‐PAGE and transferred to nitrocellulose transfer membranes (Whatman, Dassel, Germany). Blots were blocked for 1 h at room temperature in TBS containing 1% skim milk and 0.1% Tween 20. Membranes were incubated overnight at 4°C with primary antibodies against polyclonal mouse antihuman cyclin D1 (Dako, Glostrup, Denmark), polyclonal rabbit antibody to phospho‐Akt (phosphorylated at Ser473; Cell Signaling Technology, Beverly, MA, USA), phospho‐MAPK, p38, JNK (Promega, Madison, WI, USA), and β‐actin (Sigma, St. Louis, MO, USA). Membranes were then washed three times in TBS containing 0.1% Tween 20 and incubated with secondary antibody for 1 h at room temperature. Immune complexes were visualized by enhanced chemiluminescence with an ECL Plus Kit (Amersham Biosciences).
Gene transfection and cloning of transfected cell lines. Full‐length human VEGF‐D cDNA was inserted into the EcoRI‐EcoRI site of pBR322 (Invitrogen, Carlsbad, CA, USA). The resultant plasmid was digested with XbaI‐BamHI and cloned into the XbaI‐BamHI site of the pcDNA4/myc‐His vector (Invitrogen) to yield VEGF‐D expression. Expression of VEGF‐D cDNA was under the control of the cytomegalovirus promoter. KKLS cells were transfected with either the full‐length VEGF‐D cDNA or empty pcDNA4/myc‐His vector by using Lipofectin (Life Technologies, Gaithersburg, MD, USA) according to the manufacturer’s instructions. After transfection, cells were grown in selective medium (10% FBS‐RPMI‐1640 containing 400 μg/mL Zeocin). Zeocin‐resistant colonies were lifted from culture dishes and grown individually to establish stable VEGF‐D‐overexpressing clonal lines.
Enzyme‐linked immunosorbent assay (ELISA) for VEGF‐D protein. KKLS cells were plated at 1.5 × 105 cells/mL per well in a six‐well plate (Becton Dickinson Labware, Franklin Lakes, NJ, USA) and cultured in RPMI‐1640 containing 10% FBS. After 48 h, the supernatants were collected and used to measure VEGF‐D protein. We used the Quantikine Human VEGF‐D Immunoassay (R&D Systems) according to the manufacturer’s instructions to measure VEGF‐D levels.
In vitro cell growth. Cells (1 × 104) were seeded in a six‐well plate and cultured in RPMI‐1640 containing 0.5% FBS. The medium was changed every 48 h. Cell counts were determined with a Countess Automated Cell Counter (Invitrogen) from triplicate cultures.
Animal models. Male athymic BALB/c nude mice were obtained from Charles River Japan (Tokyo, Japan). The mice were maintained under specific pathogen‐free conditions and used when 5 weeks old. Experiments were done according to the animal experimental guidelines of Hiroshima University.
Orthotopic (gastric mucosa) xenograft model. To produce gastric tumors, KKLS/VEGF‐D and KKLS/control cells growing in culture were harvested from subconfluent cultures by brief treatment with 0.25% trypsin and 0.02% EDTA. Cells (1 × 106) were resuspended in HANKS and implanted into the gastric wall of nude mice under a zoom stereo microscope. After 4 weeks, the mice were killed and tumors were resected for study.
Immunohistochemistry. Formalin‐fixed, paraffin‐embedded serial sections (4 μm) were deparaffinized and rehydrated. Sections were immunostained for VEGF‐D, VEGFR‐3, Ki‐67, and Lyve1. Antigen retrieval was performed in Dako REAL Target Retrieval Solution (Dako) in a microwave oven for 10 min. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in 100% methanol for 10 min. Sections were washed with PBS and blocked with 3% normal horse serum for 10 min. Frozen sections (8 μm) were immunostained for CD31 and fixed in cold acetone for 10 min. VEGF‐D was detected with goat antihuman polyclonal antibody (1:200; R&D Systems), and VEGFR‐3 was detected with goat antihuman polyclonal antibody (1:50; R&D Systems). Ki‐67 was detected with mouse antihuman polyclonal antibody (MIB‐1, 1:25; Dako), and Lyve1 was detected with rat antimouse monoclonal antibody (1:20; R&D Systems). CD31 was detected with rat antimouse polyclonal antibody (1:200; Pharmingen, San Diego, CA, USA). Sections were incubated overnight at 4°C with the primary antibodies. Sections were then washed with PBS three times and incubated for 1 h with peroxidase‐conjugated secondary antibody. A positive reaction was detected by exposure to stable 3,3′‐diaminobenzidine. The slides were counterstained with hematoxylin.
Quantification of Ki‐67 labeling index, lymphatic vessel density (LVD), and Microvessel density (MVD). MVD was determined from the counts of CD31‐positive vessels. Vessel density was assessed by light microscopy of the intratumoral and peritumoral lesions containing the greatest number of capillaries and small venules. Highly vascular areas were identified by scanning tumor sections at low power (×40 and ×100). After the six areas of greatest neovascularization were identified, a vessel count was performed at ×400, and the mean count of six fields was calculated and defined as the mean MVD. Identification of a vessel lumen was not necessary for a structure to be defined as a vessel.( 26 ) Lymphatic activity was evaluated according to the area of lymphatic vessels stained with anti‐Lyve1 antibody. For quantification of the lymphatic vessel area, six random fields at ×400 magnification were captured for each tumor, and the outline of each lymphatic vessel including a lumen was manually traced. The area was then calculated with the use of NIH ImageJ software. Ki‐67 staining was assessed by determining the Ki‐67 labeling index by means of light microscopy at the sites of the greatest number of Ki‐67‐positive cells. Cells were counted in ten fields at ×40 magnification, and the mean percentage of stained cells was calculated. The mean percentage of Ki‐67‐positive cells was taken as the Ki‐67 labeling index.
Histochemical detection of apoptosis and determination of apoptotic index. Apoptotic cells in tissue sections were detected by terminal deoxynucleotide transferase‐mediated TUNEL with the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA) according to manufacturer’s instructions. The apoptotic index was expressed as the ratio of positively stained tumor cells and bodies to all tumor cells, given as a percentage for each case. Five random fields in a section were selected by light microscopy; at least 1000 cells were counted under ×400 magnification.
Statistical analysis. The chi‐squared test was used for analysis of categoric data, and the Mann–Whitney U‐test was used for analysis of continuous variables. The significance level was set at 5% for all analysis.
Results
Immunohistochemistry for VEGF‐D and VEGFR‐3 in human gastric carcinoma tissues. We examined expression of VEGF‐D and VEGFR‐3 protein in 29 human gastric carcinoma tissues by immunohistochemistry. VEGFR‐3 expression was observed on lymphatic endothelial cells. Of the 29 specimens of gastric carcinoma, 14 (48%) showed intense VEGF‐D immunoreactivity (Fig. 1a,b), and 15 (52%) showed intense VEGFR‐3 immunoreactivity (Fig. 1c,d) on tumor cells. In 10 of the 29 (34%) gastric carcinoma specimens, tumor cells expressed both VEGF‐D and VEGFR‐3. When the VEGF‐D expression was analyzed in relation to histological types, positivity for VEGF‐D in the diffuse (undifferentiated) type (7/10, 70%) was significantly higher than that in the intestinal (differentiated) type (7/19, 36%).
Figure 1.
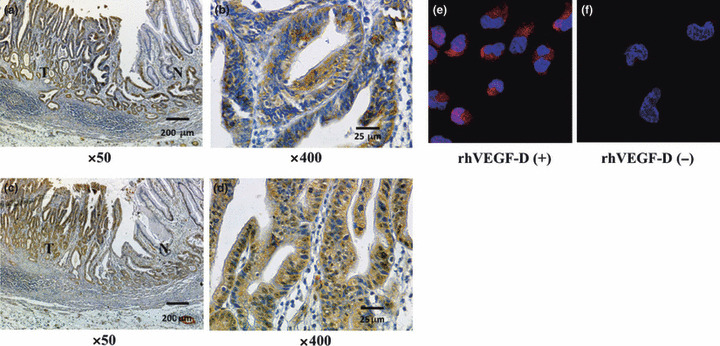
Immunohistochemical staining for (a,b) VEGF‐D and (c,d) VEGFR‐3 in human gastric carcinoma tissues. Immunoreactivities for VEGF‐D and VEGFR‐3 were detected in tumor cells (T) but not in normal gastric mucosa (N). Scale bars: (a,c) 200 μm, (b,d) 25 μm. Immunofluorescence staining for phosphorylation of VEGFR‐3 after stimulation with (e) or without (f) VEGF‐D in KKLS cells. Tumor cells stained positively for pVEGFR‐2,3 (red).
Expression of VEGF‐D, VEGFR‐2, VEGFR‐3 mRNA in gastric carcinoma cell lines. We examined expression of VEGF‐D, VEGFR‐2, VEGFR‐3 mRNA in gastric cancer cell lines by semiquantitative RT‐PCR. All gastric carcinoma cell lines constitutively expressed VEGF‐D mRNA. Expression of VEGFR‐2 mRNA was not observed in any of the cell lines examined. Two of the three cell lines expressed VEGFR‐3 mRNA (Fig. 2a). KKLS cells expressed VEGFR‐3 at a high level; therefore, we used KKLS cells for further studies. Treatment of KKLS cells with VEGF‐D induced phosphorylation of VEGFR‐3 (Fig. 1e,f) and its downstream signaling molecule, Akt (Fig. 2b). Furthermore, treatment of KKLS cells with VEGF‐D resulted in stimulation of cell proliferation. Treatment with VEGF‐D did not stimulate proliferation of TMK‐1 cells (VEGFR‐3 negative) (Fig. 2c).
Figure 2.
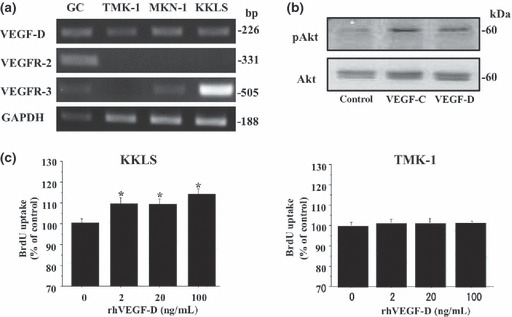
Expression of VEGF‐D, VEGFR‐2, and VEGFR‐3 in gastric carcinoma cell lines and effect of VEGF‐D on KKLS cells. (a) VEGF‐D, VEGFR‐2, and VEGFR‐3 expression by gastric carcinoma cell lines was examined by RT‐PCR. Gastric cancer tissue (GC) was used for control. (b) Western blot for phosphorylation of Akt. Treatment with VEGF‐C or VEGF‐D induced Ser473 phosphorylation of Akt in KKLS cells. (c) KKLS and TMK‐1 cells were incubated with rhVEGF‐D (2, 20, or 100 ng/mL) for 24 h, and cell proliferation was measured with a cell proliferation ELISA system. *P < 0.05; bars, SE.
VEGF‐D up‐regulated expression of several genes associated with disease progression in KKLS cells. To investigate the various cancer‐related genes up‐regulated by VEGF‐D, we performed microarray analysis. Various mRNA expression levels were compared between control KKLS cells and cells treated with rhVEGF‐D for 8 h. Microarray analysis revealed up‐regulation of 52 genes by VEGF‐D stimulation. Representative genes that were up‐regulated in cells treated with rhVEGF‐D are shown in Table 2. Among these genes, we confirmed increased expression of Bcl‐2 by quantitative real‐time PCR (Fig. 3a) and of cyclin D1 (Fig. 3b) by western blotting. Treatment with VEGF‐D increased expression of Bcl‐2 mRNA and cyclin D1 protein.
Table 2.
cDNA microarray analysis of KKLS cells treated with VEGF‐D
| Gene function | Gene symbol | Gene name | Fold change* |
|---|---|---|---|
| Cell cycle regulator | CCND1 | Cyclin D1 | 3 |
| CDK4 | Cyclin‐dependent kinase 4 | 2.8 | |
| Apoptosis | BCL2 | B‐cell CLL/lymphoma 2 | 6.3 |
| DAPK2 | Death‐associated protein kinase 1 | 5.9 | |
| NDUFA13 | Cell death‐regulatory protein GRIM19 | 2.8 | |
| Angiogenesis | THBS1 | Thrombospondin 1 | 7.3 |
| ANG | Angiogenin, ribonuclease, RNase A family, 5 | 4.2 | |
| Cell adhesion | CDH1 | Cadherin 1, type 1, E‐cadherin (epithelial) | 5.1 |
| ITGA7 | Integrin, alpha 7 | 3.8 | |
| CTNNA2 | Catenin (cadherin‐associated protein), alpha 2 | 3.6 | |
| VIM | Vimentin | 3.3 | |
| Motility, invasion | PLG | Plasminogen | 10.8 |
| PLAUR | Plasminogen activator, urokinase receptor | 4 | |
| AMFR | Autocrine motility factor receptor | 2.9 |
*Fold change = the degree of upregulation in comparison to expression in control (untreated) cells.
Figure 3.
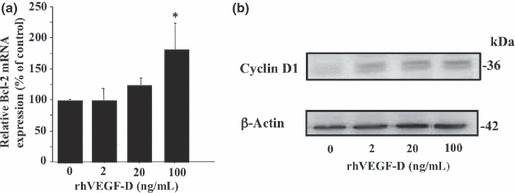
KKLS cells were treated with rhVEGF‐D (2, 20, or 100 ng/mL) for 8 h. Expression of (a) Bcl‐2 and (b) cyclin D1 was examined by real‐time PCR and western blot analysis, respectively. PCR were carried out in triplicate. Data for real‐time PCR were normalized to those of β‐actin. *P < 0.05; bars, SE.
Establishment of VEGF‐D‐overexpressing clonal cell line. To examine the biological function of VEGF‐D, VEGF‐D expression vector or the empty vector was stably transfected into KKLS cells. Overexpression of VEGF‐D mRNA and protein was confirmed by quantitative real‐time PCR (Fig. 4a) and ELISA (Fig. 4b), respectively.
Figure 4.
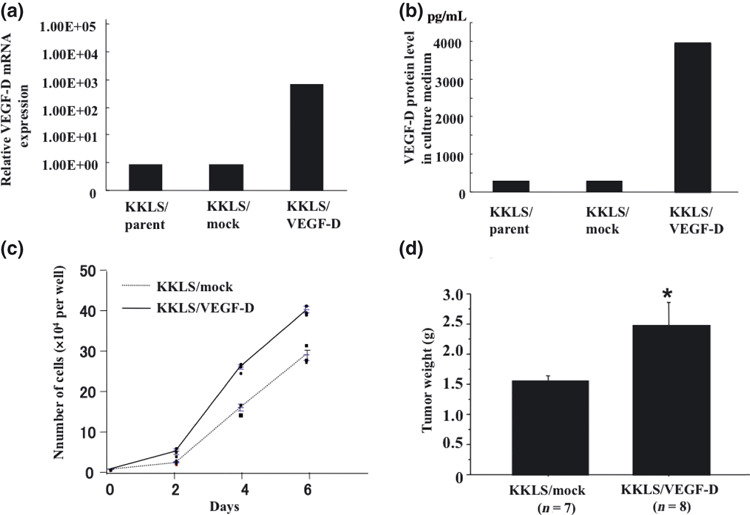
Establishment of clonal cell line overexpressing VEGF‐D, and in vitro and in vivo growth of VEGF‐D‐transfected cells. (a) Expression of VEGF‐D mRNA was examined by real‐time PCR. (b) VEGF‐D protein level in culture medium was examined by ELISA. (c) In vitro cell growth of VEGF‐D‐transfected KKLS cells. Cells (1 × 104) were seeded in a six‐well plate and cultured in RPMI‐1640 containing 0.5% FBS. The number of cells was determined in triplicate cultures. (d) Orthotopic (gastric mucosa) xenograft model. Tumor weight at 4 weeks after transplantation of the VEGF‐D‐overexpressing clone (KKLS/VEGF‐D) and corres‐ponding vector control (KKLS/control). *P < 0.05; bars, SE.
In vitro and in vivo proliferation of VEGF‐D‐transfected KKLS cells. The effect of overexpression of VEGF‐D on the ability of KKLS cells to grow in vitro was evaluated. Transfection with VEGF‐D increased in vitro cell growth of KKLS cells (Fig. 4c). To examine the effect of VEGF‐D overexpression in an animal model, we transplanted KKLS/VEGF‐D and KKLS/mock cells into the gastric wall of nude mice. The mice were killed after 4 weeks, and tumors derived from KKLS/VEGF‐D cells were significantly larger than KKLS/mock tumors (Fig. 4d). VEGF‐D secreted by the tumor did not promote lymphatic metastasis.
Immunohistochemistry for Ki‐67, CD31, and Lyve1 and in situ detection of apoptotic cells in KKLS cells growing in the gastric wall of nude mice. We evaluated MVD, LVD, and the Ki‐67 labeling index based on immunodetection of CD31, Lyve1, and Ki‐67. Indeed, statistically significant increases in MVD (Fig. 5a) and the Ki‐67 labeling index (Fig. 5b) were noted in KKLS/VEGF‐D tumors in comparison to those in KKLS/mock tumors. The number of lymphatic vessels was not increased in response to VEGF‐D transfection (data not shown); however, the luminal area of lymphatic vessels was significantly enlarged (Fig. 5c). The apoptotic index was also evaluated. A significant decrease in the apoptotic index was noted in KKLS/VEGF‐D tumors in comparison to that in KKLS/mock tumors (Fig. 5d).
Figure 5.
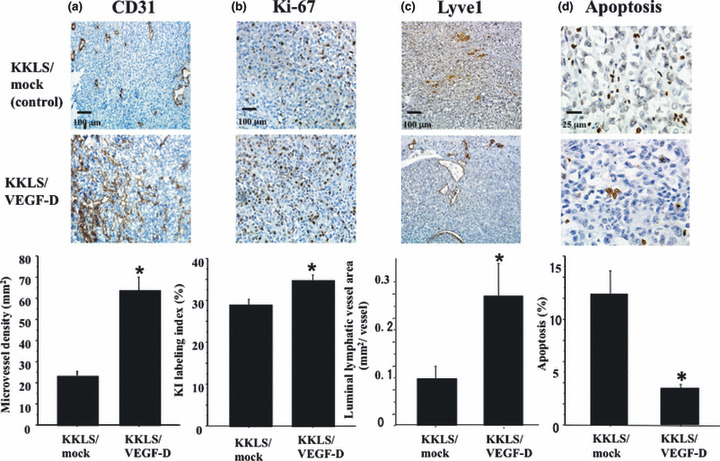
Immunohistochemistry for (a) CD31, (b) Ki‐67, and (c) Lyve1, and (d) in situ detection of apoptotic cells in KKLS cells growing in the gastric wall of nude mice. Lower panels, quantification of CD31‐positive vessels, Ki‐67‐positive cells, Lyve1‐positive vessels, and apoptotic cells. Scale bars: (a,c) 200 μm. *P < 0.05; bars, SE.
Discussion
VEGF‐C and VEGF‐D are specific ligands for VEGFR‐3 and VEGFR‐2.( 27 , 28 ) They have been shown to stimulate lymphangiogenesis and angiogenesis both in vitro and in vivo. ( 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ) It has been reported that VEGF‐D induced lymphangiogenesis in a mouse tumor model.( 10 , 29 ) The expression of VEGF‐D has been correlated with tumor lymphangiogenesis and lymph node metastasis in many human carcinomas, including breast,( 14 ) lung,( 19 ) gastric,( 18 ) and colorectal cancers.( 13 ) We previously examined expression of VEGF‐C and VEGF‐D by immunohistochemistry in 140 archival surgical specimens of submucosally invasive gastric carcinoma. VEGF‐C immunoreactivity was associated with histologic type, lymphatic invasion, lymph node metastasis, and MVD. There was no association between VEGF‐D immunoreactivity and clinicopathologic features. These results suggest that VEGF‐C is the dominant regulator of lymphangiogenesis in early stage human gastric carcinoma.( 22 )
VEGFR‐3 has also been detected in several types of malignant cells,( 30 , 31 ) although it is mainly expressed by lymphatic endothelial cells. In various types of cancer cells, autocrine VEGF‐A/VEGFR‐2 loops on tumor cells regulate their growth and survival.( 32 , 33 , 34 ) We and others have reported the existence of autocrine stimulation of tumor growth via the VEGF‐C/VEGFR‐3 axis in lung,( 30 ) breast,( 31 ) and gastric cancers.( 24 ) In the present study, we examined the expression of VEGF‐D and VEGFR‐3 in human gastric carcinomas and the role of the VEGF‐D/VEGFR‐3 axis in tumor cells. In 10 of 29 (34%) gastric carcinoma specimens, tumor cells expressed both VEGF‐D and VEGFR‐3. Treatment of KKLS cells (VEGFR‐3‐expressing cell line) with recombinant VEGF‐D induced tyrosine phosphorylation of VEGFR‐3 and Akt, indicating that VEGFR‐3 on tumor cells is functional. Microarray analysis showed up‐regulation of cyclin D1 (cell cycle regulator) and Bcl‐2 (anti‐apoptotic protein) by treatment with VEGF‐D. We reported previously that treatment with VEGF‐C induced phosphorylation of VEGFR‐3 and Akt and increased expression of cyclin D1, PlGF, autocrine motility factor (AMF), and AMF receptor by KKLS cells.( 24 ) Makinen et al. ( 35 ) showed that VEGFR‐3 signaling induces protein kinase C (PKC)‐dependent p42/p44 MAPK activation and wortmannin‐sensitive phosphorylation of Akt. These findings suggest that VEGF‐C and VEGF‐D are associated with cell proliferation and survival via these signaling cascades in tumor cells.
To stimulate VEGF‐D/VEGFR‐3 signaling in an autocrine manner, VEGF‐D expression vector was transfected into KKLS cells (KKLS/VEGF‐D cells), and these cells were then transplanted into the gastric wall of nude mice. It has been reported that binding of VEGF‐D to VEGFR‐3 on the lymphatic endothelial cells resulted in dilatation of existing lymphatic vessels as well as in vegetation of new vessels.( 36 ) In the present study, the luminal area of lymphatic vessels was increased in KKLS/VEGF‐D tumors in comparison to that in control (KKLS/mock) tumors, indicating that the KKLS/VEGF‐D cells produce functional VEGF‐D protein. However, VEGF‐D secreted by the tumor did not promote lymphatic metastasis. In contrast, Stacker et al. ( 10 ) reported that expression of VEGF‐D in tumor cells led to spread of the tumor to lymph nodes. These discrepancies may be due to differences in cell lines and experimental animal models. Lymphatic metastasis is the consequence of a complex metastatic process, which includes lymphangiogenesis,( 37 , 38 ) dissemination, transport, settlement, and growth in the lymphatic system. These involve not only VEGF‐D but many other growth factors such as VEGF‐C, angiopoietins, fibroblast growth factor, and platelet‐derived growth factor.( 39 )
In vitro treatment with VEGF‐D increased expression of cyclin D1 and stimulated proliferation of KKLS cells. The Ki‐67 labeling index and tumor weight increased in orthotopic KKLS/VEGF‐D tumors in comparison to those in orthotopic KKLS/mock tumors. These increases were associated with increased MVD, indicating that VEGF‐D transfection into KKLS cells stimulates angiogenesis. In addition, we found that in vitro treatment with VEGF‐D increased expression of Bcl‐2, a key regulator of apoptotic inhibition. The number of apoptosis cells was lower in KKLS/VEGF‐D tumors than in KKLS/mock tumors. Consistent with our results, Akahane et al. ( 40 ) reported that overexpression of VEGF‐D in breast cancer cell lines resulted in up‐regulation of the Bcl‐2 gene.
In conclusion, human gastric carcinoma cells express VEGF‐D as well as VEGFR‐3. VEGF‐D may be involved in the progression of human gastric carcinoma not only by stimulating angiogenesis and lymphangiogenesis via a paracrine mechanism, but also by regulating cell proliferation and apoptosis via an autocrine mechanism. Further studies are needed to identify precise signaling mechanisms responsible for the autocrine role of VEGF‐D.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was carried out with the kind cooperation of the Analysis Center of Life Science, Hiroshima University (Hiroshima, Japan) and was supported, in part, by Grants‐in‐Aid for Cancer Research from the Ministry of Education, Culture, Science, Sports and Technology of Japan, and from the Ministry of Health, Labor and Welfare of Japan.
References
- 1. Smith MG, Hold GL, Tahara E, El‐Omar EM. Cellular and molecular aspects of gastric cancer. World J Gastroenterol 2006; 12: 2979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. Lymphangiogenesis and cancer metastasis. Nat Rev Cancer 2002; 2: 573–83. [DOI] [PubMed] [Google Scholar]
- 3. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2002; 2: 795–803. [DOI] [PubMed] [Google Scholar]
- 4. Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 2002; 1: 219–27. [DOI] [PubMed] [Google Scholar]
- 5. Berse B, Brown LF, Van de Water L, Dvorak HF, Senger DR. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell 1992; 3: 211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferrara N, Houck K, Jakeman L, Leung DW. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev 1992; 13: 18–32. [DOI] [PubMed] [Google Scholar]
- 7. Otrock ZK, Makarem JA, Shamseddine AI. Vascular endothelial growth factor family of ligands and receptors: review. Blood Cells Mol Dis 2007; 38: 258–68. [DOI] [PubMed] [Google Scholar]
- 8. Orlandini M, Marconcini L, Ferruzzi R, Oliviero S. Identification of a c‐fos‐induced gene that is related to the platelet‐derived growth factor/vascular endothelial growth factor family. Proc Natl Acad Sci USA 1996; 93: 11675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stacker SA, Stenvers K, Caesar C et al. Biosynthesis of vascular endothelial growth factor‐D involves proteolytic processing which generates non‐covalent homodimers. J Biol Chem 1996; 274: 32127–36. [DOI] [PubMed] [Google Scholar]
- 10. Stacker SA, Caesar C, Baldwin ME et al. VEGF‐D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 2001; 7: 186–91. [DOI] [PubMed] [Google Scholar]
- 11. Thelen A, Scholz A, Benckert C et al. VEGF‐D promotes tumor growth and lymphatic spread in a mouse model of hepatocellular carcinoma. Int J Cancer 2008; 122: 2471–81. [DOI] [PubMed] [Google Scholar]
- 12. Von Marschall Z, Scholz A, Stacker SA et al. Vascular endothelial growth factor‐D induces lymphangiogenesis and lymphatic metastasis in models of ductal pancreatic cancer. Int J Oncol 2005; 27: 669–79. [PubMed] [Google Scholar]
- 13. White JD, Hewett PW, Kosuge D et al. Vascular endothelial growth factor‐D expression is an independent prognostic marker for survival in colorectal carcinoma. Cancer Res 2002; 62: 1669–75. [PubMed] [Google Scholar]
- 14. Nakamura Y, Yasuoka H, Tsujimoto M et al. Prognostic significance of vascular endothelial growth factor D in breast carcinoma with long‐term follow‐up. Clin Cancer Res 2003; 9: 716–21. [PubMed] [Google Scholar]
- 15. Kurahara H, Takao S, Maemura K, Shinchi H, Natsugoe S, Aikou T. Impact of vascular endothelial growth factor‐C and ‐D expression in human pancreatic cancer: its relationship to lymph node metastasis. Clin Cancer Res 2004; 10: 8413–20. [DOI] [PubMed] [Google Scholar]
- 16. Yokoyama Y, Charnock‐Jones DS, Licence D et al. Vascular endothelial growth factor‐D is an independent prognostic factor in epithelial ovarian carcinoma. Br J Cancer 2003; 88: 237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yokoyama Y, Charnock‐Jones DS, Licence D et al. Expression of vascular endothelial growth factor (VEGF)‐D and its receptor, VEGF receptor 3, as a prognostic factor in endometrial carcinoma. Clin Cancer Res 2003; 9: 1361–9. [PubMed] [Google Scholar]
- 18. Jüttner S, Wissmann C, Jöns T et al. Vascular endothelial growth factor‐D and its receptor VEGFR‐3: two novel independent prognostic markers in gastric adenocarcinoma. J Clin Oncol 2006; 24: 228–40. [DOI] [PubMed] [Google Scholar]
- 19. Niki T, Iba S, Tokunou M, Yamada T, Matsuno Y, Hirohashi S. Expression of vascular endothelial growth factors A, B, C, and D and their relationships to lymph node status in lung adenocarcinoma. Clin Cancer Res 2000; 6: 2431–9. [PubMed] [Google Scholar]
- 20. O‐charoenrat P, Rhys‐Evans P, Eccles SA. Expression of vascular endothelial growth factor family members in head and neck squamous cell carcinoma correlates with lymph node metastasis. Cancer 2001; 92: 556–68. [DOI] [PubMed] [Google Scholar]
- 21. George ML, Tutton MG, Janssen F et al. VEGF‐A, VEGF‐C, and VEGF‐D in colorectal cancer progression. Neoplasia 2001; 3: 420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Onogawa S, Kitadai Y, Tanaka S, Kuwai T, Kimura S, Chayama K. Expression of VEGF‐C and VEGF‐D at the invasive edge correlates with lymph node metastasis and prognosis of patients with colorectal carcinoma. Cancer Sci 2004; 95: 32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kitadai Y, Kodama M, Cho S et al. Quantitative analysis of lymphangiogenic markers for predicting metastasis of human gastric carcinoma to lymph nodes. Int J Cancer 2005; 115: 388–92. [DOI] [PubMed] [Google Scholar]
- 24. Kodama M, Kitadai Y, Tanaka M et al. Vascular endothelial growth factor C stimulates progression of human gastric cancer via both autocrine and paracrine mechanism. Clin Cancer Res 2008; 14: 7205–14. [DOI] [PubMed] [Google Scholar]
- 25. Japanese Research Society for Gastric Cancer . Japanese Classification of Gastric Carcinoma. Kanehara & Co., Tokyo, 1999. [Google Scholar]
- 26. Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis: correlation in invasive breast carcinoma. N Engl J Med 1991; 324: 1–8. [DOI] [PubMed] [Google Scholar]
- 27. Joukov V, Pajusola K, Kaipainen A et al. A novel vascular endothelial growth factor, VEGF‐C, is a ligand for the Flt4 (VEGFR‐3) and KDR (VEGFR‐2) receptor tyrosine kinases. EMBO J 1996; 15: 290–8. [PMC free article] [PubMed] [Google Scholar]
- 28. Achen MG, Jeltsch M, Kukk E et al. Vascular endothelial growth factor D (VEGF‐D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA 1998; 95: 548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kopfstein L, Veikkola T, Djonov VG et al. Distinct roles of vascular endothelial growth factor‐D in lymphangiogenesis and metastasis. Am J Pathol 2007; 170: 1348–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tanno S, Ohsaki Y, Nakanishi K, Toyoshima E, Kikuchi K. Human small cell lung cancer cells express functional VEGF receptors, VEGFR‐2 and VEGFR‐3. Lung Cancer 2004; 46: 11–19. [DOI] [PubMed] [Google Scholar]
- 31. Timoshenko AV, Rastogi S, Lala PK. Migration‐promoting role of VEGF‐C and VEGF‐C binding receptors in human breast cancer cells. Br J Cancer 2007; 97: 1090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Von Marschall Z, Cramer T, Höcker M et al. De novo expression of vascular endothelial growth factor in human pancreatic cancer: evidence for an autocrine mitogenic loop. Gastroenterology 2000; 119: 1358–72. [DOI] [PubMed] [Google Scholar]
- 33. Bachelder RE, Wendt MA, Mercurio AM. Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res 2002; 62: 7203–6. [PubMed] [Google Scholar]
- 34. Jackson MW, Roberts JS, Heckford SE et al. A potential autocrine role for vascular endothelial growth factor in prostate cancer. Cancer Res 2002; 62: 854–9. [PubMed] [Google Scholar]
- 35. Mäkinen T, Veikkola T, Mustjoki S et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF‐C/D receptor VEGFR‐3. EMBO J 2002; 20: 4762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Veikkola T, Jussila L, Makinen T et al. Signalling via vascular endothelial growth factor receptor‐3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J 2001; 20: 1223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Achen MG, McColl BK, Stacker SA. Focus on lymphangiogenesis in tumor metastasis. Cancer Cell 2005; 7: 121–7. [DOI] [PubMed] [Google Scholar]
- 38. Karpanen T, Alitalo K. Lymphatic vessels as targets of tumor therapy? J Exp Med 2001; 194: F37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Folkman J. Looking for a good endothelial address. Cancer Cell 2002; 1: 113–15. [DOI] [PubMed] [Google Scholar]
- 40. Akahane M, Akahane T, Matheny SL, Shah A, Okajima E, Thorgeirsson UP. Vascular endothelial growth factor‐D is a survival factor for human breast carcinoma cells. Int J Cancer 2006; 118: 841–9. [DOI] [PubMed] [Google Scholar]